Fabrication of a mesoporous Co(OH)2/ITO nanowire composite electrode and its application in supercapacitors
Duc Tai
Dam
,
Xin
Wang
and
Jong-Min
Lee
*
School of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore 637459, Singapore. E-mail: jmlee@ntu.edu.sg; Tel: +65 651 381 29
First published on 5th September 2012
Abstract
A novel mesoporous α-Co(OH)2/ITO NWs heterostructure has been successfully synthesized via a new method of combining chemical vapor deposition (CVD) on a titanium substrate and electrodeposition through a hexagonal lyotropic liquid crystalline (LLC) phase for electrochemical energy storage application. Electrochemical measurements demonstrate the improved ionic conductivity of an electrode and enhanced supercapacitive behavior, as compared to a mesoporous cobalt hydroxide film on a titanium current collector without indium tin oxide (ITO) NWs as “electron highways”. Galvanostatic charge–discharge measurements in 1 M KOH solution prove that the electrode of the as-prepared heterostructure possesses excellent electrochemical capacitance, in the potential range from −0.1 V to 0.5 V with a maximum specific capacitance of 1128 F g−1 at a discharge current density of 1 A g−1. The unique mesoporous α-Co(OH)2/ITO NWs heterostructure provides a high capacitance retention ratio at high discharge current densities and long-term electrochemical stability in an alkaline solution.
Introduction
In recent years, electrochemical supercapacitors (ECs) have been extensively studied as attractive energy storage devices. They have potential applications in portable electronics and electric vehicles because of their high power energy densities and long cyclic life.1,2 Based on the nature of the charge-storage mechanism and active materials, electrochemical capacitors can be classified into two types: electrochemical double layer capacitors (EDLCs) and redox supercapacitors (pseudocapacitors). EDLCs utilizing carbon-based active materials, such as activated carbon (AC) and carbon nanotubes (CNTs) with charge stored at the electrode–electrolyte interface, are currently the most commonly used devices.3 On the other hand, pseudocapacitors or redox capacitors use fast and reversible faradaic surface reactions for charge storage. Conducting polymers4,5 and transition metal oxides and hydroxides6–8 have been investigated as possible electrode materials for redox capacitors. Redox capacitors have drawn much more attention than EDLCs due to their high theoretical specific capacitance (SC) values,9,10 which are determined by specific surface area and the faradaic redox characteristics of materials.11Initially, J. P. Zheng et al. reported the crystal structure, particle size and prominent electrochemical properties of hydrous ruthenium oxide formed by a sol–gel process.8 In this study, specific capacitance as high as 720 F g−1 was measured for RuO2 powder annealed at 150 °C. In addition, Hu and Chen obtained the maximum capacitance of 1580 F g−1 from an AC–RuOx/RuOx/Au/SS composite electrode which was very close to the theoretical value.12 It is essential to develop alternative inexpensive electrode materials with high capacitive performance such as NiO,13 CoOx,14 MnO2,15 Ni(OH)2,16 Co(OH)2,17etc. Of all the candidates of redox electrode materials, Co(OH)2 is of particular interest as a high-performance EC electrode material because of its layered structure with large interlayer spacing and great reaction reversibility.18,19 Tremendous efforts have been devoted to fabricate cobalt hydroxide electrodes. The precipitation methodology was initially used to synthesize Co(OH)2 for a supercapacitor.20,21 However, cobalt hydroxide prepared by this technique demonstrated very low specific capacitance (SC), in a range of 200–400 F g−1. Because the major limitation of Co(OH)2 is a low conductivity, S. Chen et al. initiated an idea of fabricating graphene–Co(OH)2 nanocomposites by the use of some reducing agents such as HS− and H2S.22 As a result, this research group managed to achieve a specific capacitance of 972.5 F g−1 which is much higher than each of the individual counterparts. However, in the above study, a uniform distribution of Co(OH)2 is believed to be the main challenge. On the other hand, most of the reports were more concerned about the enhancement of SC values. Insufficient attention was paid to capacitance degradations of metal oxide/hydroxide materials at high current densities.23 Our group previously reported the utilization of a lyotropic liquid crystal (LLC) template in the potentiostatic electrodeposition of Co(OH)2 which led to a significant improvement in electrochemical performance.24 On the other hand, indium-tin oxide nanowire is known to possess excellent electrical properties and thermal stability25,26 which allows great applications in the field of electronics.27,28 In this study, we first prepared indium-tin oxide nanowires (ITO NWs) by chemical vapor deposition (CVD) on a titanium substrate, and then deposited mesoporous Co(OH)2 nanoflakes (NFs) onto the indium-tin oxide nanowires (ITO NWs) by electrodeposition through a lyotropic liquid crystal (LLC) template. In addition, a mesoporous Co(OH)2 film, directly deposited on pure Ti substrate, was prepared for comparison.
Experimental
Synthesis of indium-tin oxide nanowires
ITO NWs were synthesized by the carbothermal reduction method with graphite as a reducing agent and Au as a catalyst in a horizontal double-tube system.29–31 A slender quartz tube (inner diameter 1.5 cm, length 25 cm, one sealed end and one opened end) was used for nanowire growth. In2O3, SnO2 and graphite powder (weight ratio 0.375![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.0415:0.0830) were thoroughly mixed and placed at the sealed end of slender quartz tube as the material source. Titanium substrates coated with 5 nm Au film were placed downstream in the slender quartz tube to collect the products. The slender quartz tube was then loaded into the large furnace tube (inner diameter 3.3 cm, length 75 cm) with the source material at the high temperature region and closed end facing the argon gas flow. Before heating, the tube furnace was purged with 200 sccm of argon gas for at least 10 min. Subsequently, the central temperature of the furnace was increased to 1000 °C at the rate of 15 °C min−1 and kept for 120 min under a constant Ar flow of 50 sccm (standard cubic centimetre per minute). The titanium substrates coated with Au film were located at the temperature region of 400–600 °C during growth. A series of experiments had been preliminary carried out to determine an optimal condition for nanowire growth in terms of purity and size of nanowires. The pressure during nanowire synthesis was maintained in the range of 5–6 mbar. After growth, the furnace was gradually cooled to room temperature under continuous Ar gas flow.
:0.0415:0.0830) were thoroughly mixed and placed at the sealed end of slender quartz tube as the material source. Titanium substrates coated with 5 nm Au film were placed downstream in the slender quartz tube to collect the products. The slender quartz tube was then loaded into the large furnace tube (inner diameter 3.3 cm, length 75 cm) with the source material at the high temperature region and closed end facing the argon gas flow. Before heating, the tube furnace was purged with 200 sccm of argon gas for at least 10 min. Subsequently, the central temperature of the furnace was increased to 1000 °C at the rate of 15 °C min−1 and kept for 120 min under a constant Ar flow of 50 sccm (standard cubic centimetre per minute). The titanium substrates coated with Au film were located at the temperature region of 400–600 °C during growth. A series of experiments had been preliminary carried out to determine an optimal condition for nanowire growth in terms of purity and size of nanowires. The pressure during nanowire synthesis was maintained in the range of 5–6 mbar. After growth, the furnace was gradually cooled to room temperature under continuous Ar gas flow.
Electrodeposition of mesoporous Co(OH)2
Brij56 (polyoxyethylene(10) cetyl ether, C16[EO]10) and Co2+ electrolyte (0.9 M Co(NO3)2 and 0.075 M NaNO3) with a weight ratio of 1:1 were mixed thoroughly to prepare the lyotropic liquid crystalline (LLC) template. This template was used as an electrolyte for electrodeposition. A standard three-electrode cell was used for electrodeposition with an ITO NWs/Ti working electrode, a large surface area platinum counter electrode and a standard Ag/AgCl electrode. The potentiostatic electrodeposition was performed with a voltage of −0.75 V (vs. Ag/AgCl) at 50 °C using a CHI660d potentiostat and the total passed charge was 0.6 C cm−2 for the same amount of electroactive Co(OH)2. The mechanism of electrodeposition could include the electrochemical reaction and precipitation as follows: | NO3− + 7H2O + 8e− → NH4+ + 10OH− | (1) |
| Co2+ + 2OH− → Co(OH)2 | (2) |
After the electrodeposition, the electrode was immersed into ethanol to remove Brij56. The ethanol was replaced every 2 h for at least 4 times in order to completely remove the surfactant. Subsequently, the electrode was immersed in 2-propanol for another 2 h, rinsed by DI water and annealed under vacuum at 120 °C for 2 h. A similar procedure was used to deposit Co(OH)2 on pure Ti substrate for comparative studies.
Characterization
Morphologies of the products were characterized using field emission scanning electron microscopy (JEOL-JSM-6700F microscope) operated at 5 kV. The crystalline structures of the specimens were characterized by using X-ray diffraction measurements (Bruker D8 Advance X-Ray Diffractometer) with Cu-Kα radiation (λ = 0.15406 nm) in the range from 10° to 45°. Detailed structure analyses were performed by transmission electron microscopy (TEM, JEOL 2010) operated at 300 kV. For the TEM analysis, the products were dispersed in ethanol, sonicated and dropped on a copper grid. The samples were then dried in a vacuum oven before the analysis. The chemical compositions were analyzed using energy-dispersive X-ray spectroscopy (EDX) attached to the FESEM. In order to analyze chemical bonding states of the surface, Raman measurements were performed using a Renishaw inVia Raman Microscope equipped with a charge couple device (CCD) detector. An Ar+ ion laser with the wavelength of 514 nm was used as an excitation source. The laser intensity at the sample surface was maintained at around 1.5 mW for all measurements. The electrochemical measurements of all samples were conducted by a CHI660d electrochemical workstation. Electrochemical impedance analysis was carried out by Autolab PGSTAT302 potentiostat.Results and discussion
Fig. 1 illustrates the fabrication procedures of the α-Co(OH)2/ITO NWs composite electrodes. The electrodes were prepared by a new method of combining CVD and potentiostatic electrodeposition. One side of the titanium current collector was used for ITO growth and α-Co(OH)2 electrodeposition. The loading mass of α-Co(OH)2 was controlled by varying the total passed charge. For comparison, a mesoporous α-Co(OH)2 film was also directly deposited onto bare Ti substrate.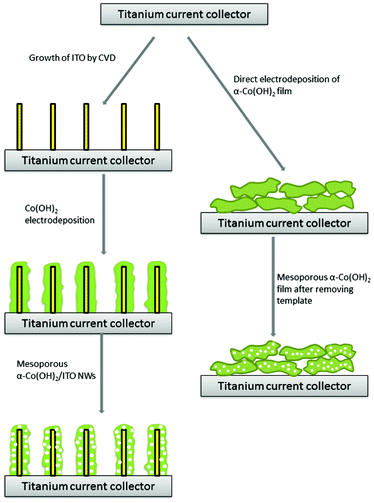 | ||
| Fig. 1 Schematic illustration of the experimental procedure for fabrication of the α-Co(OH)2/ITO NWs and α-Co(OH)2/Ti electrodes via a new method of combining chemical vapor deposition and potentiostatic electrodeposition. | ||
Fig. 2a shows the wide-angle XRD pattern of the mesoporous α-Co(OH)2/ITO NWs heterostructure. The eight peaks detected can be indexed as ITO(221), ITO(222), ITO(321), ITO(400), ITO(411), ITO(420), ITO(332) and ITO(422) (JCPDS 06-0416). The other two diffraction peaks at 10.8° and 22.2° can be assigned to the (003) and (006) planes of layered α-phase Co(OH)2,32 which is usually observed in Co(OH)2 nanostructures prepared by electrodeposition.33,34 The sharp and dominant ITO peak at 30.58° indicates that the majority of ITO nanowires grew along the [111] direction while only a small amount of nanowires were oriented in other directions. Moreover, as shown in the low-angle XRD pattern (Fig. 2b), the heterostructure shows a strong reflection peak of 1.46° at 2θ. This peak can be attributed to the d100 plane of the P6mm space group with a d-spacing of 60.4 Å,24 indicating that the ordered mesoporous structure of the lyotropic liquid crystalline (LLC) template was still retained in α-Co(OH)2 after the electrodeposition. The pore-to-pore distance for this mesoporous array given by d100/cos30 is determined to be 69.7 Å.
 | ||
| Fig. 2 (a and b) Wide-angle and low-angle XRD patterns of the α-Co(OH)2/ITO NWs heterostructure, respectively. (c) Room temperature Raman spectrum of the α-Co(OH)2/ITO NWs heterostructure. | ||
Fig. 2c demonstrates the Raman spectrum of the mesoporous α-Co(OH)2/ITO NWs heterostructure. ITO is known to possess a cubic structure that belongs to the I3a, T7h space groups. The spectrum shows vibrational modes at 110, 135, 308, 366, 495 and 631 cm−1, which is certainly the signature of the ITO cubic structure. The highest Raman shift at 631 cm−1 is largely due to the superposition of the In–O vibrational modes with a frequency 629 cm−1 and the Sn–O vibrational modes with a frequency 633 cm−1.35 The Raman spectrum also proves the presence of Co(OH)2 in the as-prepared heterostructure: three intense bands were detected at 456, 522 and 613 cm−1, which are attributed to the OCoO bending mode, CoO (Ag) symmetric stretching mode, and CoO (F2g) mode, respectively.36
Fig. 3a illustrates a typical cross-sectional FESEM image of the as-grown ITO NWs on the Ti substrate. ITO NWs with typical lengths of 50 μm in direct contact with the substrate surface were observed. Moreover, the nanowires were well separated as shown in a high-magnification FESEM image (Fig. 3b). A Au catalyst is of great import to promote such a vapor–liquid–solid (VLS) growth mechanism37,38 in an oxygen deficient environment. Fig. 3c presents an enlarged image of the tip of a typical nanowire. It is worth noting that the growth of nanowire was initiated by the hexagonal (111) crystal face, and the grown nanowire has a single crystal structure,39 in good agreement with the results obtained from the X-ray analysis presented above. Additionally, no catalyst droplets were observed at the tip of the ITO nanowire. Detailed crystal structures of the ITO NWs were investigated by TEM (Fig. 3d and 3e). The ITO nanowire has a smooth surface, as shown in a low magnification TEM image (Fig. 3d) and is a single crystalline growing along the [111] direction with an interlayer spacing of 2.9 Å, which is consistent with the result obtained from the wide-angle XRD characterization.
 | ||
| Fig. 3 FESEM images (cross-sectional view (a), top view (b) and high magnification view (c)) of ITO nanowires on the Ti substrate. (d) Low magnification TEM image of an individual ITO nanowire. (e) High-resolution TEM image of an ITO nanowire, showing the single crystalline structure. | ||
Fig. 4 shows the morphology, structural analysis and chemical composition of the mesoporous Co(OH)2/ITO NWs heterostructure. A uniform Co(OH)2 shell with a thickness of 200–300 nm was successfully deposited on the ITO NWs, as shown in FESEM images (Fig. 4a and 4b) and low magnification TEM image (Fig. 4c). The as-deposited Co(OH)2 exhibits densely interlaced nanosheets with a high active surface area. The cobalt hydroxide possesses a layered and mesoporous structure, as revealed by the TEM images at higher magnification (Fig. 4d and 4e). The bright dots represent pores formed by removing the surfactant template. The cylindrical pores typically have diameters of 2–3 nm with an interpore distance of about 7 nm, calculated from the low-angle XRD result. This mesoporous structure is expected to have a high surface area which is in turn beneficial to improve electrochemical properties. Chemical compositions of the heterostructure were analyzed using EDX attached to the FESEM system.
 | ||
| Fig. 4 (a and b) FESEM images of mesoporous α-Co(OH)2 coated along the ITO nanowires. (c and d) Low magnification TEM images of the α-Co(OH)2/ITO NWs heterostructure, showing a uniform distribution and layered structure of Co(OH)2. (e) High magnification TEM image of the Co(OH)2 coating, indicating a mesoporous structure. (f) EDX spectrum of the entire α-Co(OH)2/ITO NWs heterostructure. | ||
The major existences of In, Co and O are shown in the EDX spectrum recorded from the entire mesoporous Co(OH)2/ITO NWs heterostructure in Fig. 4f. From the results obtained from the physical characterizations, it was proven that mesoporous α-Co(OH)2 on uniform ITO NWs was successfully synthesized. Mesoporous α-Co(OH)2, which possesses a layered structure, acts as an electroactive material in the as-prepared composite electrode. On the other hand, ITO NWs play the role of “electron highways”, facilitating charge transport, which may have great potential in achieving good electrochemical properties.
Both cyclic voltammetry (CV) and chronopotential charge–discharge methods were used to investigate capacitor behavior of the as-prepared electrodes. All electrochemical measurements were conducted in a 1 M KOH aqueous solution. Fig. 5a and 5b represent the CV response of α-Co(OH)2/Ti and α-Co(OH)2/ITO NWs electrodes with the same passed charges and loading masses within potential ranges from −0.2 to 0.65 V. It is interesting to note that one pair of redox peaks were observed in CV curves at all scan rates, which is distinct from the shape of CV curves of EDLCs (close to ideal rectangular shape), likely due to the electron transfer process in pseudocapacitive α-Co(OH)2/Ti and α-Co(OH)2/ITO NWs. The surface faradaic reaction can be given as follows:40
| Co(OH)2 + OH− ↔ CoOOH + H2O + e− | (3) |
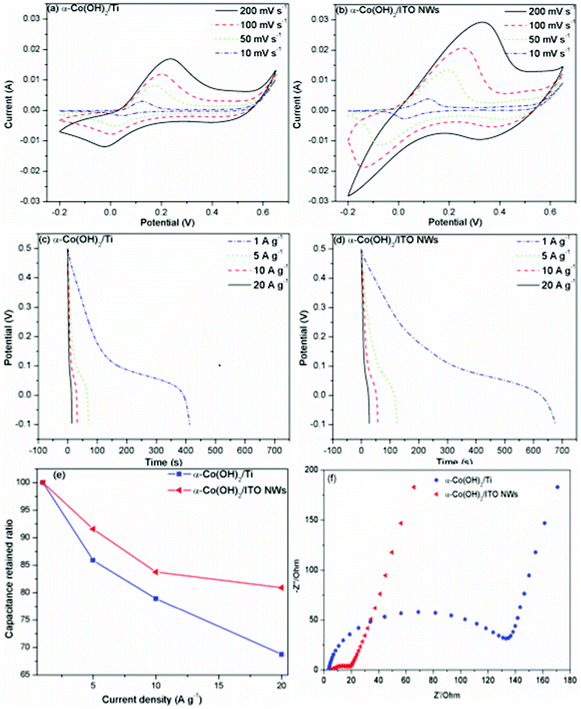 | ||
| Fig. 5 Electrochemical performance. (a and b) CV plots at different scan rates. (c and d) Discharge curves at different current densities in the potential range of −0.1 V to 0.5 V. (e) Capacitance retained ratio versus current density. (f) Complex-plane impedance spectra of the α-Co(OH)2/Ti and α-Co(OH)2/ITO NWs electrodes. | ||
It can be observed that the shapes of CV curves change with scan rates and those changes can be attributed to peak current (Ip) and peak potential separation (ΔEp).29 In the case of α-Co(OH)2/Ti, the anodic peak current increases from 0.00298 A to 0.00813 A, 0.01188 A and 0.01692 A and peak potential separation increases from 0.081 V to 0.156 V, 0.201 V and 0.261 V as the potential scan rate increases from 10 mV s−1 to 50 mV s−1, 100 mV s−1 and 200 mV s−1. Similar trends are observed in the CV curves of α-Co(OH)2/ITO NWs at scan rates equal to 10 mV s−1 to 50 mV s−1 and 100 mV s−1. However, no cathodic peak was observed at a scan rate of 200 mV s−1 due to the quasi-reversible characteristic of the faradaic reaction (eqn (3)) and uncompensated potential drop (IR drop). The anodic peak (above 0.1 V) is due to the oxidation of Co(OH)2 to produce Co(OOH) and the cathodic peak (below 0.1 V) is due to the reverse process. As compared to the CV curves of α-Co(OH)2/Ti, α-Co(OH)2/ITO NWs generate a much larger cathodic and anodic area at all scan rates, indicating its enhanced capacitive behavior.
Fig. 5c and 5d show the chronopotential discharge curves of α-Co(OH)2/Ti and α-Co(OH)2/ITO NWs electrodes at different discharge current densities in the potential window of −0.1 to 0.5 V. Their specific capacitance can be calculated from the discharge curve using following equation:
 | (4) |
Electrochemical impedance spectroscopy was carried out in the frequency range from 30 kHz to 0.1 Hz, with the amplitude of the perturbation signal equal to 50 mV in order to verify the conductivity enhancement, as presented in Fig. 5f. The measurement was performed on two types of electrodes (α-Co(OH)2/Ti and α-Co(OH)2/ITO NWs electrodes) with the same mass of active material. The semicircular loops at high frequencies are attributed to charge-transfer resistances of the electrodes and the straight segments at lower frequencies represent capacitive behaviors of the electrodes. The diameter of the semicircular loop for the α-Co(OH)2/ITO NWs electrode is determined to be 11.74 Ω, which is much smaller than that of the α-Co(OH)2/Ti electrode (129.63 Ω), indicating that the mesoporous nanowire structure can significantly improve electron transfer and reduce interfacial resistance during the reaction.
When particles of Co(OH)2 are used for the electrode, it is required that particles of carbon black and binder are uniformly distributed on the electrode, the thickness of composite is thin and uniform, and the contact between the composite and the substrate is good, to achieve uniform current distribution on the electrode, which leads to the full capacity of the active material in the system. However, it is often difficult to satisfy all the requirements. To overcome such problems, an Co(OH)2 nanowire array electrode was introduced using a template method.24 However, some drop of capacitance of the nanowire array was reported due to the collapse of the end parts of the nanowires during the synthesis procedure. Thus, this proposed heterostructure for α-Co(OH)2/ITO NWs electrode is quite promising because it is a binder-free structure. Moreover, the proposed structure provides an increased accessibility of electrolyte to the electrode and prevents the aggregation of nanowires as presented in Fig. 1. Such advantages lead not only to reduce charge transfer resistance in the electrode, but also provide the shortest electron transfer pathway through the nanowires. Nevertheless, further work is required to optimize the formation of the α-Co(OH)2/ITO NWs electrode, which will lead to more enhanced electrochemical properties.
The study of electrochemical stability of the α-Co(OH)2/ITO NWs electrode was performed in 1 M aqueous KOH electrolyte by charge–discharge cycling. At a current density of 10 A g−1, the electrode retained 90.9% of its initial specific capacitance after 500 cycles as shown in Fig. 6, indicating that the as-prepared electrode has long-term electrochemical stability in alkaline solution.
 | ||
| Fig. 6 Cycle life data for the α-Co(OH)2/ITO NWs electrode at a discharge current density of 10 A g−1 in the potential range of −0.1 to 0.5 V. | ||
Conclusions
In conclusion, we demonstrated a new method of combining CVD and potentiostatic electrodeposition methods to synthesize a α-Co(OH)2/ITO NWs heterostructure, which possesses an enhanced supercapacitive behavior and an improved high discharge rate capability. It has been shown that uniform α-Co(OH)2 along the ITO NWs can be obtained by electrodeposition at −0.75 V. In addition, the α-Co(OH)2 has a mesoporous and layered structure with a high surface area, which results in excellent electrochemical properties. Both CV and chronopotential charge–discharge curves showed that the supercapacitive behavior of the α-Co(OH)2/ITO NWs electrode was superior to that of the α-Co(OH)2/Ti electrode at all scan rates and current densities, due to its one-dimensional-like mesoporous structure, which has a high surface area and improved electron transport from the indirect connection of Co(OH)2 to the Ti current collector through ITO NWs. The unique design of the nanoscale heterostructure which combines the advantages of highly active Co(OH)2 and the excellent conductivity of ITO NWs can be considered as a new technique to fabricate porous electrodes for ECs and even for Li-ion battery applications.Acknowledgements
This work is supported by a Startup Grant of the Nanyang Technological University and Academic Research Fund (RG21/09) of the Ministry of Education in Singapore.References
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- A. Rudge, I. Raistrick, S. Gottesfeld and J. P. Ferraris, Electrochim. Acta, 1994, 39, 273–287 CrossRef CAS.
- A. Rudge, J. Davey, I. Raistrick, S. Gottesfeld and J. P. Ferraris, J. Power Sources, 1994, 47, 89–107 CrossRef CAS.
- A. A. F. Grupioni, E. Arashiro and T. A. F. Lassali, Electrochim. Acta, 2002, 48, 407–418 CrossRef CAS.
- D. Zhao, W. Zhou and H. Li, Chem. Mater., 2007, 19, 3882–3891 CrossRef CAS.
- J. P. Zheng, P. J. Cygan and T. R. Jow, J. Electrochem. Soc., 1995, 142, 2699–2703 CrossRef CAS.
- V. Gupta, T. Kusahara, H. Toyama, S. Gupta and N. Miura, Electrochem. Commun., 2007, 9, 2315–2319 CrossRef CAS.
- Z. Liu, R. Ma, M. Osada, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2005, 127, 13869–13874 CrossRef CAS.
- B. E. Conway, J. Electrochem. Soc., 1991, 138, 1539–1548 CrossRef CAS.
- C. C. Hu and W. C. Chen, Electrochim. Acta, 2004, 49, 3469–3477 CrossRef CAS.
- W. Xing, F. Li, Z. F. Yan and G. Q. Lu, J. Power Sources, 2004, 134, 324–330 CrossRef CAS.
- C. Lin, J. A. Ritter and B. N. Popov, J. Electrochem. Soc., 1998, 145, 4097–4103 CrossRef CAS.
- S. C. Pang, M. A. Anderson and T. W. Chapman, J. Electrochem. Soc., 2000, 147, 444–450 CrossRef CAS.
- L. Cao, L. B. Kong, Y. Y. Liang and H. L. Li, Chem. Commun., 2004, 1646–1647 RSC.
- L. Cao, F. Xu, Y. Y. Liang and H. L. Li, Adv. Mater., 2004, 16, 1853–1857 CrossRef CAS.
- T. Zhao, H. Jiang and J. Ma, J. Power Sources, 2011, 196, 860–864 CrossRef CAS.
- Z. Hu, L. Mo, X. Feng, J. Shi, Y. Wang and Y. Xie, Mater. Chem. Phys., 2009, 114, 53–57 CrossRef CAS.
- T. Y. Wei, C. H. Chen, K. H. Chang, S. Y. Lu and C. C. Hu, Chem. Mater., 2009, 21, 3228–3233 CrossRef CAS.
- S. F. E. Hosono, I. Honma, M. Ichihara and H. Zhou, J. Power Sources, 2006, 158, 779–783 CrossRef.
- S. Chen, J. Zhu and X. Wang, J. Phys. Chem. C, 2010, 114, 11829–11834 CAS.
- C. G. Liu, Y. S. Lee, Y. J. Kim, I. C. Song and J. H. Kim, Synth. Met., 2009, 159, 2009–2012 CrossRef CAS.
- T. Xue, X. Wang and J. M. Lee, J. Power Sources, 2012, 201, 382–386 CrossRef CAS.
- D. Lin, H. Wu, R. Zhang and W. Pan, Nanotechnology, 2007, 18, 465301 CrossRef.
- H. W. Kim, H. S. Kim, H. G. Na, J. C. Yang, R. Choi, J. K. Jeong, C. Lee and D. Y. Kim, J. Solid State Chem., 2010, 183, 2490–2495 CrossRef CAS.
- J.-M. Lee, Electrochim. Acta, 2006, 51, 3256–3260 CrossRef CAS.
- Y. Cao, J.-M. Lee and A. C. West, Plating and Surface Finishing, 2003, 90, 40–45 Search PubMed.
- C. Yan, H. Jiang, T. Zhao, C. Li, J. Ma and P. S. Lee, J. Mater. Chem., 2011, 21, 10482–10488 RSC.
- C. Yan and P. S. Lee, J. Phys. Chem. C, 2009, 113, 14135–14139 CAS.
- N. Singh, T. Zhang and P. S. Lee, Nanotechnology, 2009, 20, 195605–195611 CrossRef.
- W. J. Zhou, M. W. Xu, D. D. Zhao, C. L. Xu and H. L. Li, Microporous Mesoporous Mater., 2009, 117, 55–60 CrossRef CAS.
- M. S. Yarger, E. M. P. Steinmiller and K. S. Choi, Chem. Commun., 2007, 159–161 RSC.
- V. Gupta, S. Gupta and N. Miura, J. Power Sources, 2008, 175, 680–685 CrossRef CAS.
- O. M. Berengue, A. D. Rodrigues, C. J. Dalmaschio, A. J. C. Lanfredi, E. R. Leite and A. J. Chiquito, J. Phys. D: Appl. Phys., 2010, 43, 045401 CrossRef.
- Y. Jing, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2009, 114, 111–119 Search PubMed.
- H. S. Jang, D. H. Kim, H. R. Lee and S. Y. Lee, Mater. Lett., 2005, 59, 1526–1529 CrossRef CAS.
- M. K. Fung, Y. C. Sun, A. M. C. Ng, X. Y. Chen, K. K. Wong, A. B. Djurišić and W. K. Chan, Appl. Phys. A: Mater. Sci. Process., 2011, 104, 1075–1080 CrossRef CAS.
- C. J. Chen, W. L. Xu and M. Y. Chern, Adv. Mater., 2007, 19, 3012–3015 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc, 2001 Search PubMed.
- X. Ji, P. M. Hallam, S. M. Houssein, R. Kadara, L. Lang and C. E. Banks, RSC Adv., 2012, 2, 1508–1515 RSC.
| This journal is © The Royal Society of Chemistry 2012 |