DOI:
10.1039/C2RA20937C
(Paper)
RSC Adv., 2012,
2, 10505-10511
Effect of bismuth substitution on crystal chemistry, photocatalysis and conductivity in Sr3V2O8: a new structural type in palmierite class†
Received
15th May 2012
, Accepted 31st July 2012
First published on 6th September 2012
Abstract
The effect of Bi substitution has been investigated in the palmierite type system Sr3V2O8, wherein, a new solid solution Sr3−xBi2x/3V2O8 (0 ≤ x ≤ 0.4) has been established. The structure of the composition x = 0.20 (Sr2.8Bi0.13V2O8) was solved using single-crystal X-ray diffraction (XRD) technique. The compound crystallizes in the trigonal crystal system R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m with a palmierite structural type with a = 5.6067(4) Å, c = 20.0099(13) Å, V = 580.64(5) Å3 and Z = 3. The substituent Bi occupies a new unique 18h site resulting in a new series in palmierite class of compounds. The new solid solution has been characterized by synchrotron X-ray powder diffraction, solid-state UV-Vis diffuse-reflectance spectra, scanning electron microscopy and ionic conductivities and photocatalytic activities has been investigated. The solid solution compounds exhibited photocatalytic activity towards various classes of anionic dyes under UV radiation.
m with a palmierite structural type with a = 5.6067(4) Å, c = 20.0099(13) Å, V = 580.64(5) Å3 and Z = 3. The substituent Bi occupies a new unique 18h site resulting in a new series in palmierite class of compounds. The new solid solution has been characterized by synchrotron X-ray powder diffraction, solid-state UV-Vis diffuse-reflectance spectra, scanning electron microscopy and ionic conductivities and photocatalytic activities has been investigated. The solid solution compounds exhibited photocatalytic activity towards various classes of anionic dyes under UV radiation.
Introduction
Alkaline-earth metal phosphate, vanadates, arsenates and chromates with the general formula A3M2O8 (A = Sr, Ba; M = P, V, As, Cr) have been extensively investigated for their interesting optical, transport and electrical properties and for potential application in materials science, for example, as luminophors, as host materials for solid-state lasers, and as luminescent lamp coatings in television tubes.1–8 Strontium orthovanadate (Sr3V2O8) belongs to the palmierite (space group Rm) type of compounds9 and consists of [VO4]3− tetrahedra and two different kinds of Sr sites (Scheme 1). While one Sr atom occupies the m Wyckoff site, the other one sits at the 3m position, with coordination numbers of 6 and 10, respectively. It is observed that compounds of the type A2Ln2/3(VO4)2 belong to the palmierite class of compounds consisting of double vanadate units in the binary system A3V2O8–LnVO4, where A = Ca, Sr, Ba; Ln = La, Nd.10,11 In these compounds, Ln3+ and the A atom are statistically distributed in 6c (m) and 3a (3m) positions. The fact that the A atom sites are differently occupied, suggests that substitution at the A site would lead to solid solutions of varying compositions, leading to the possibility of forming hitherto unknown functional materials. For example, doping of Mn5+ and Eu3+ in Sr3V2O8 has been observed to enhance the near IR-laser action and rare-earth activated luminescence, respectively.3,12
 |
| | Scheme 1 General structure of palmierite showing polyhedral linkages. | |
Sr3V2O8 undergoes a pressure-induced first-order phase transition at around 150 kbar.6In situ high-pressure Raman studies reveal that the compound is transformed to the spinelloid type of structure by the mechanism in which the alternate V5+ ions pass through the triangular faces of their original distorted tetrahedral sites.6 Recently, a series of compounds with the formula Sr3−3xLa2x(V1−yPyO4)2 have been synthesized, and their cationic and electrical conductivities have been measured.13,14 The incorporation of Bi3+ in Sr sites in Sr3V2O8 has been described by Zhuravlev et al.15 The powder diffraction analysis has assigned 3a Wyckoff site to Bi atom (occupancy 0.667) and 6c site to Sr atom with full occupancy. However, it has been shown by our recent work using an accurate single-crystal data set that substitution of Bi3+ at the 3a Wyckoff site is not allowed in Pb3V2O8.16
Indeed, Bi3+ occupies a different Wyckoff site 18h in this solid solution and thus stabilizing the high-temperature γ form of Pb3V2O8. It is obvious that the position and occupancy of Bi3+ in relation to the A atom significantly influences the properties exhibited by these systems. Thus, a careful single-crystal study becomes a prerequisite to unequivocally establish the Bi site location and occupancy in substituted Sr3V2O8, to evaluate the structure–property correlation. In this study, Sr3−xBi2x/3V2O8 solid solutions have been synthesized and the limits of the solid solution and precise Bi atomic position have been determined through accurate single-crystal diffraction study, followed by the evaluation of photocatalytic and conductivity properties.
Dye pollutants from the textile industry are an important source of environmental contamination in the form of wastewater. Photocatalysis is widely used for the cleaning of wastewater and extensive research is being carried out to overcome the problem of remediation of these wastes by developing new photocatalysts.17–19 Several factors such as band gap energies, and features associated with cation vacancy, oxygen deficiency and surface area influence the performance of materials used in photocatalytic degradation. Sr3−xBi2x/3V2O8, with its variability in site occupancies and structural features, may serve as a good candidate for use as a photodegradation material.
Results and discussion
X-Ray powder diffraction
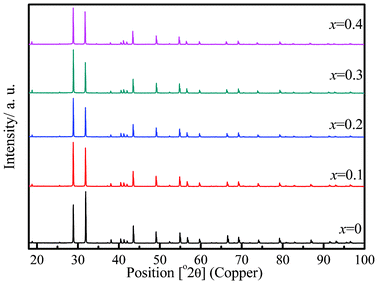
Phase purity of the solid solution (Sr3−xBi2x/3V2O8, x ≤ 0.4) members were confirmed by PXRD. Fig. 1 shows the powder X-ray diffractograms of various members of the solid solution. To establish the solid solution limit, Sr3−xBi2x/3V2O8, x = 0.45 has been synthesized and a comparison of the powder pattern with the solid-solution members x = 0.45 and x = 0.4 (see Fig. S1, ESI†) clearly shows the presence of impurity phases Bi2O3 and BiVO4 at x = 0.45, suggesting that the solid solution limit is x ≅ 0.4. Fig. 2 describes the changes in cell parameters obtained after profile refinement. While the length of the a axis increases, the length of the c axis decreases with the composition, indicating an opposite trend in the a and c axes with an increase in the Bi content in the series Sr3−xBi2x/3V2O8, 0 ≤ x ≤ 0.4. These observations are similar to those observed for the palmirite type structure Pb3V2O8.16
 |
| | Fig. 2 Variation in the cell parameters with the composition. | |
Single-crystal structure
Single-crystal XRD data on the composition x = 0.20 of the solid solution were indexed in a trigonal crystal system, space group Rm, with a = 5.6067(4) Å, c = 20.0099(13) Å, V = 580.64(5) Å3 and Z = 3 (Table 1). Initially, the positions of heavy atoms, Sr (Sr1 and Sr2) and V were obtained by Patterson methods using SHELXL97,20 and were found to be in similar positions as those found in the known structure of Sr3V2O8.21 The positions were refined using all observed reflections during the refinement cycles. All oxygen atoms were located from the subsequent difference Fourier synthesis. In order to account for the presence of Bi in the crystal structure, occupancies of Sr1 and Sr2 were allowed to refine keeping their respective displacement parameters constant. While no change in the occupancy value was observed for Sr1 (Wyckoff site 3a), the occupancy of Sr2 (Wyckoff site 6c) decreased considerably. The Bi atom was hence assigned to the 6c site along with the Sr2 atom and the occupancies were refined keeping the thermal parameters fixed. The resulting R factor was 0.0478 and the corresponding residual electron density map shows fairly significant residual density Δρ , ranging from +2.34 to −1.62 e Å−3. Fig. 3a shows the residual density map where the maximum is located at the 18h site. At this stage, it was thought worthwhile to consider the structure of the Bi-substituted Pb3V2O8 as a starting model since the parent structures Pb3V2O8 and Sr3V2O8 are isostructural. It is observed that upon substitution of Bi, the symmetry of Pb2 in Pb3V2O8 is lowered from the 6c to 18h site.16 Bi occupies the identical Wyckoff site (18h) as that of the atom Pb2. However, refinements on the Bi-substituted Sr3V2O8 based on this model become unsteady, suggesting that both Bi and Sr atoms cannot be present simultaneously at the 18h site unlike in the case of Bi-substituted Pb3V2O8. It may be argued that since Bi and Pb are neighbors in the periodic table, the assignment of the same site for Bi and Pb2 is appropriate in Bi substituted Pb3V2O8. Thus, it was decided that in further refinements to assign the Sr2 atom to the 6c site with lower occupancy and the Bi atom (lower occupancy) to the 18h site. However, there are no significant differences between the coordinates of Bi2 before and after the refinements of anisotropic displacement parameters. Refinements carried out with these features resulted in a R factor 0.0286 with Δρmin, max = 0.86, −0.96 e Å−3. This featureless residual map (Fig. 3b) thus provides proof for the formation of a new variant in the Palmierite class of compounds. The atomic coordinates and the thermal parameters are listed in Table 2. Fig. 4 shows the packing diagram depicting the nature of coordination in each unit cell and the extended network as viewed down the c-axis. Fig. 5 shows the extended features of the coordination environment for the metal atoms. High-resolution powder diffraction data collected at the Elettra synchrotron source for the composition x = 0.2 were modeled based on these new variant coordinates and subsequent Rietveld refinements gave a reasonable fit substantiating the observation (see Fig. S2, ESI†).
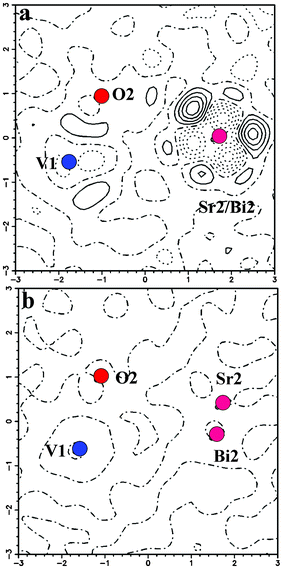 |
| | Fig. 3 An xy section of the difference Fourier map around Sr2 atom. Contour lines are drawn in the interval of 0.5 e Å−3. | |
 |
| | Fig. 4 Packing diagram of the compound Sr3−xBi2x/3V2O8, x = 0.2. | |
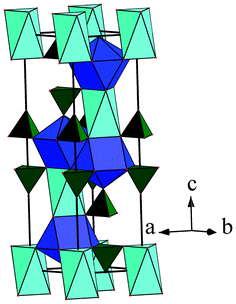 |
| | Fig. 5 Coordination environment around different metal atoms. | |
Table 1 Crystallographic refined parameters obtained from single-crystal data
| Empirical formula |
Sr2.8Bi0.13V2O8 |
|
M
r
|
503.08 |
| Crystal habit, colour |
Block, pale yellow |
| Crystal size/mm |
0.15 × 0.12 × 0.09 |
|
T/K |
120(2) |
| Radiation |
Mo-Kα |
| Monochromator |
Graphite |
|
λ/Å |
0.71069 |
| Crystal system |
Trigonal |
| Space group |
Rm |
|
a/Å |
5.6067(4) |
|
c/Å |
20.0099(13) |
|
V/Å3 |
580.64(5) |
|
Z
|
3 |
|
D
c/g cm−3 |
4.601 |
|
F(000) |
682 |
| Scan mode |
ω scan |
|
θ
max/° |
29.10 |
|
hkl
min, max
|
−7 to 7, −7 to 7, −27 to 26 |
| No of reflns measured |
2772 |
| No of unique reflns |
213 |
| No of parameters |
25 |
| Absorption correction |
Numerical from crystal shape |
|
μ/mm−1 |
26.137 |
| Refinement |
F
2
|
|
R (all), R (obs) |
0.0345, 0.0285 |
| wR2 (all), wR2 (obs) |
0.0558, 0.0551 |
| GoF |
1.208 |
| Δρmax, min/e Å−3 |
0.890, −0.960 |
| Atom (Wyckoff position) |
x
|
y
|
z
|
U
eq/Å2 |
Occupancy (site multiplicity) |
| Sr1 (3a) |
0 |
0 |
0 |
0.0154(4) |
1 (12) |
| Sr2 (6c) |
0 |
0 |
0.20132(6) |
0.0072(3) |
0.9 (6) |
| Bi2 (18h) |
0.513(5) |
0.257(8) |
0.1335(11) |
0.060(7) |
0.022 (2) |
| V1 (6c) |
1/3 |
2/3 |
0.07265(8) |
0.0079(4) |
1 (6) |
| O1 (6c) |
1/3 |
2/3 |
−0.0113(4) |
0.0229(17) |
1 (6) |
| O2 (18h) |
0.1682(5) |
0.3364(10) |
0.1009(2) |
0.0231(14) |
1 (2) |
In VO4 tetrahedra there are two kinds of V–O distances making the tetrahedra distorted. The apical V–O1 [O atom shared by both Sr2, Bi2 and V] bond length (1.680(8) Å) is shorter that the other three bonds (V–O2 = 1.700(5) Å). These bond lengths are comparable with those reported for the analogous compound Pb3−xBi2x/3V2O8.16 The atom Sr1 forms an octahedron with six equal bond distances of 2.597(5) Å while Sr2 forms a SrO10 polyhedron with axial Sr2–O1 distance of 2.415(8) Å and hexagonal set Sr2–O2 at distances of 2.872(1) Å respectively. The other three Sr2–O2 bond distances are 2.589(5) Å. Similarly Bi2 also forms BiO10 polyhedra and the axial bond length of Bi2–O1 (2.557(10) Å) is longer than the corresponding Bi2–O1 (2.420(14) Å) reported for Pb3−xBi2x/3V2O8. The three similar Bi2–O2 bond lengths (2.434(18) Å) are shorter than Pb2/Bi2–O2 bond (2.668(8) Å) whereas the six similar bond lengths are 2.972(47) Å, longer than the bonds in Pb3−xBi2x/3V2O8.16
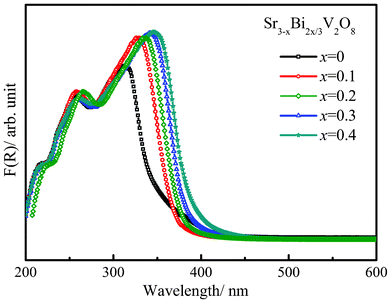
Room-temperature UV-visible spectra were collected in the diffuse-reflectance mode for various compositions in the series Sr3−xBi2x/3V2O8. The optical absorption was represented by the Kubelka–Munk function (F(R) = (1 − R)2/2R) calculated from the diffuse reflectance spectra at constant scattering (Fig. 6). The steep shape of the spectra signified that absorption was due to the band-gap transition. The experimental band gap for Sr3V2O8 was calculated to be 3.45 eV. It is of interest to note that upon substitution, the band gap of the material decreases and reaches a value 3.15 eV for the composition x = 0.4. This can be attributed to the fact that the Bi 6p orbital makes a significant contribution to the conduction band.22 Fig. S3 (ESI†) is an SEM micrograph showing particle sizes in the μm range and single-crystal images for the composition Sr3−xBi2x/3V2O8 (x = 0.2).
Photocatalysis
The photocatalytic behavior of Sr3V2O8 was studied under UV radiation by degrading anionic and cationic dyes. Dye solutions were stirred in the presence of the catalyst for 1 h prior to the UV radiation exposure to monitor the changes in dye concentration due to adsorption. No significant adsorption (<5%) was observed. Experiments were also carried out in the absence of catalyst (photolysis) and less than 5% degradation was observed.
The inset to Fig. 7 shows the degradation profile for cationic dyes MB and RB and anionic dye OG in the presence of Sr3V2O8. After 1 h of UV radiation, less than 10% degradation was observed for cationic dyes MB and RB while 30% degradation was observed for the anionic dye OG. It was therefore concluded that the catalyst may be active for only anionic dyes. Further to study the effect of Bi substitution, OG was degraded under UV light using all compositions of Bi-substituted Sr3V2O8 (Fig. 7). It is observed that upon increased substitution the photocatalytic activity decreases. Further, photocatalytic dye degradation experiments were carried out using unsubstituted Sr3V2O8. Fig. 8 shows the degradation profile for different anionic dyes under UV light in the presence of Sr3V2O8. Complete degradation was observed in case of IC within 30 min of UV exposure while EY was completely degraded in 60 min. For RBBR and AB, degradation was 60 and 35%, respectively, while degradation was around 30% for both OG and AG. Therefore, from the degradation profile we can conclude that the degradation rate follows the trend IC > EY > RBBR > AB > AG ≅ OG. This observation is consistent with the order observed with TiO2.23 The powder pattern (Fig. S4, ESI†) of the compound remains identical even after several degradation experiments, suggesting that the material is stable under the degradation conditions. The degradation of EY was compared with commercial catalysts Degussa P-25 and the activity was found to be similar (Fig. 8).
 |
| | Fig. 7 Degradation profile of OG in presence of different compositions of Sr3V2O8 (inset degradation profile for cationic dyes MB and RB and anionic dye OG in the presence of Sr3V2O8). | |
 |
| | Fig. 8 Degradation profiles of different anionic dyes in the presence of Sr3V2O8. The open diamonds represent the degradation profile EY in presence of Degussa P 25. | |
The structure of the dye plays an important role in its degradation rate. The rate of degradation of IC is higher than that of the other dyes and is attributed to the very reactive central –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– bond.23 It is found that intermolecular hydrogen bonds in water disrupt the planarity and the central bond is readily cleaved leading to rapid degradation. The degradation of AG is higher than OG because of the role of the sulfate ions. The sulfonyl group attached to the benzene ring in AG makes it more reactive than when it is attached to naphthalene, as in the case of OG.23 Similarly, the rate of OG and AB can be directly compared because the former is a monoazo dye while the later is a diazo dye. AB has two azo groups functions, namely an amino and a nitro group, which can be decolorized faster with the evolution of ammonium ion, nitrates and nitrogen compared to OG, which has only one azo group. The higher rate of degradation of the diazo relative to the monoazo dye is consistent with previous studies.23,24 The mechanism of degradation of the anthraquinonic dye, RBBR, suggests that it degrades faster than azo dyes.25 An important reaction during photocatalysis is the photo-Kolbe decarboxylation reaction. This involves the reaction of the carboxyl radical with a valance band hole. As the carboxyl ion is present in EY the reaction is dominant and leads to a higher degradation of EY.
C– bond.23 It is found that intermolecular hydrogen bonds in water disrupt the planarity and the central bond is readily cleaved leading to rapid degradation. The degradation of AG is higher than OG because of the role of the sulfate ions. The sulfonyl group attached to the benzene ring in AG makes it more reactive than when it is attached to naphthalene, as in the case of OG.23 Similarly, the rate of OG and AB can be directly compared because the former is a monoazo dye while the later is a diazo dye. AB has two azo groups functions, namely an amino and a nitro group, which can be decolorized faster with the evolution of ammonium ion, nitrates and nitrogen compared to OG, which has only one azo group. The higher rate of degradation of the diazo relative to the monoazo dye is consistent with previous studies.23,24 The mechanism of degradation of the anthraquinonic dye, RBBR, suggests that it degrades faster than azo dyes.25 An important reaction during photocatalysis is the photo-Kolbe decarboxylation reaction. This involves the reaction of the carboxyl radical with a valance band hole. As the carboxyl ion is present in EY the reaction is dominant and leads to a higher degradation of EY.
Ionic conductivity
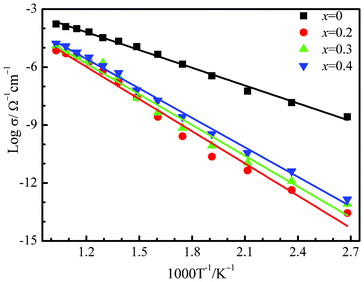
The measured conductivity vs. temperature of Sr3V2O8 is shown in Fig. 9 in the form of an Arrhenius plot (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) σ vs. 1000/T). The value of the bulk ionic conductivity was calculated from the intercept of the single semicircular arcs obtained in the complex impedance plots of Re(Z′)–Im (Z′′) plots (Z = impedance). The conductivity values showed a linear relationship with the temperature. The bulk ionic conductivity of Sr3V2O8 reaches a value of 10−3 Ω−1 cm−1 at 750 °C while the substituted compounds showed a decrease in the ionic conductivity value in the range 10−4–10−5 Ω−1 cm−1.The activation energy calculated from the Arrhenius plot was found to be 0.61 eV whereas the activation energy for all the substituted compounds were found to be around 1 eV (x = 0.2, Ea = 1.10 eV; x = 0.3, Ea = 1.05 eV; x = 0.4, Ea = 1.00 eV). Sr ion conductivity is also observed in similar materials Sr3−3xLa2x□x(VO4)226 La1−xSrxCo1−yFeyO3−δ,27 and La1−xSrxFeO3−δ.28 It has been earlier reported that the conduction mechanism is primarily the result of the formation of vacancies and interstitial Sr ions with an increase in the temperature in a solid solution, Sr2.4La0.4(V1−yPyO4)2, with the Palmierite structural type.14 A similar mechanism can be invoked in case of the Bi-substituted Sr3V2O8. It is interesting to note that the substituent Bi3+ (1.17 Å) is smaller in size than Sr2+ (1.36 Å, 10 coordination) but heavier than strontium. However, the presence of the substituted heavy Bi3+ ions causes a reduction in the number of Sr2+ ions and subsequently the vacancies, resulting in the observed decrease in conductivity. There is no clear trend observed in case of differently substituted Bi compounds as their conductivity values lie within the error limits.
σ vs. 1000/T). The value of the bulk ionic conductivity was calculated from the intercept of the single semicircular arcs obtained in the complex impedance plots of Re(Z′)–Im (Z′′) plots (Z = impedance). The conductivity values showed a linear relationship with the temperature. The bulk ionic conductivity of Sr3V2O8 reaches a value of 10−3 Ω−1 cm−1 at 750 °C while the substituted compounds showed a decrease in the ionic conductivity value in the range 10−4–10−5 Ω−1 cm−1.The activation energy calculated from the Arrhenius plot was found to be 0.61 eV whereas the activation energy for all the substituted compounds were found to be around 1 eV (x = 0.2, Ea = 1.10 eV; x = 0.3, Ea = 1.05 eV; x = 0.4, Ea = 1.00 eV). Sr ion conductivity is also observed in similar materials Sr3−3xLa2x□x(VO4)226 La1−xSrxCo1−yFeyO3−δ,27 and La1−xSrxFeO3−δ.28 It has been earlier reported that the conduction mechanism is primarily the result of the formation of vacancies and interstitial Sr ions with an increase in the temperature in a solid solution, Sr2.4La0.4(V1−yPyO4)2, with the Palmierite structural type.14 A similar mechanism can be invoked in case of the Bi-substituted Sr3V2O8. It is interesting to note that the substituent Bi3+ (1.17 Å) is smaller in size than Sr2+ (1.36 Å, 10 coordination) but heavier than strontium. However, the presence of the substituted heavy Bi3+ ions causes a reduction in the number of Sr2+ ions and subsequently the vacancies, resulting in the observed decrease in conductivity. There is no clear trend observed in case of differently substituted Bi compounds as their conductivity values lie within the error limits.
Experimental
Materials
Bi2O3 (Aldrich, 99.9%) was dried at 350 °C for 6 h before use while V2O5 and SrCO3 (from S. D. Fine-Chem Ltd., India, 99%) were used as received. Methylene blue (MB), Rhodamine B (RB), Remazol brilliant blue R (RBBR), Orange G (OG), Eosin Y (EY), Amido black 10B (AB) (all from S. D. Fine Chem Ltd, India), Alizarin Green (AG), Indigo carmine (IC) (all from Rolex Labs, India) were used as supplied without further purification. Water was distilled twice and filtered through a Millipore membrane before use.
Preparation and crystal growth
Polycrystalline samples of Sr3−xBi2x/3V2O8 (x = 0, 0.1, 0.2, 0.3 and 0.4) were synthesized by a solid-state route using the starting materials SrCO3, Bi2O3 and V2O5 in stoichiometric ratios. The starting reactants were initially ground well in an agate mortar and pestle and were subjected to heat treatment at 650 °C for 6 h to decompose the carbonates. The resultant mixture was ground well and subjected to heat treatment for 6 h at 750 °C, at 900 °C for 24 h and at 950 °C for 24 h with two intermediate regrindings. All the syntheses were carried out under an oxygen atmosphere. The unsubstituted Sr3V2O8 was colorless, while Bi-substituted products were light yellow. The progress of the reaction was monitored by powder X-ray diffraction that confirmed the formation of a single phase in case of Sr3−xBi2x/3V2O8. Single-crystals of the composition x = 0.2 were obtained by melting a polycrystalline sample at 1100 °C for 1 h in a platinum crucible followed by slow cooling at 5 °C h−1 to 900 °C and then furnace cooling to room temperature.
Powder XRD
Powder XRD data were collected using a Philips X-pert Pro diffractometer with Cu-Kα radiation over the 2θ range 5–100°, with a step width of 0.0167°. Le Bail profile fitting followed by Rietveld analysis were carried out using the XRD data by the JANA200029 suite of programs. The background was estimated by a Legendre polynomial function consisting of 15 coefficients, and the peak shapes were described by a pseudo-Voigt function, varying five profile coefficients. A scale factor, a zero error factor, and a shape function were refined.
High-resolution Powder XRD data were collected at the Powder Diffraction Beam line (MCX) equipped with fast scintillator detector (Bede) at synchrotron Elettra, Trieste, Italy. The wavelength used for the experiment was 1.0 Å, calibrated against silicon (NIST-SRM660a) standards. The data was collected with a step size of 0.02° in the range 5–60°. The samples were loaded in a capillary and sonicated to pack the capillary uniformly. The capillary was rotated during data accumulation.
Single-crystal XRD
Good quality crystals were chosen under a polarizing microscope for single-crystal X-ray diffraction studies. Single-crystal X-ray diffraction data were collected on Xcalibur™ Mova E diffractometer with a four-circle kappa goniometer and Eos CCD detector in a CrysAlis CCD, Oxford Diffraction apparatus employing graphite-monochromated Mo-Kα (λ = 0.7107 Å) radiation at 120(2) K with X-ray generator (50 kV and 0.8 mA). The cell refinement and data reduction were accomplished using CrysAlis RED.30 The diffraction intensities were corrected for Lorentz and polarization effects. The data were reduced using CrysAlisRED (special programs available with the diffractometer), the shape was determined with the video microscope attached to the diffractometer, and an analytical absorption correction (after Clark and Reid) was applied. The structures were solved by the direct method using SHELXL9720 using the program suite WINGX (version 1.70.01).31 The packing diagrams were generated using package Diamond, Version 2.1c.32
Characterization
Morphology and composition analysis of all samples of Sr3−xBi2x/3V2O8 were carried out with the help of FEI Quanta scanning electron microscope. UV-visible diffuse-reflectance spectra were recorded on a Perkin-Elmer Lambda 35 UV-visible spectrophotometer.
Photocatalytic experiments
The photochemical reactor used in this study was made of a jacketed quartz tube of 3.4 cm internal diameter, 4 cm outer diameter and 21 cm length and an outer Pyrex glass reactor of 5.7 cm internal diameter and 16 cm length. A high-pressure 125 W mercury vapor lamp (Philips, India) was placed inside the jacketed quartz tube after the removal of the outer shell. To avoid fluctuations in the input, the supply ballast and capacitor were connected in series with the lamp. Ice water was circulated through the annulus of the quartz tube to avoid heating of the solution. The lamp radiated predominantly at 365 nm corresponding to an energy of 3.4 eV and photon flux of 6.1 × 10−6 mol photons s−1.
Aqueous solutions of all dyes and organic compounds were taken in the reactor with a catalyst loading of 1 g L−1. 100 ml of the solution was taken in the outer reactor and continuously stirred to ensure that the suspension of the catalyst was uniform. The mixture was stirred for 12 h in the dark and the resulting concentration was taken as the initial concentration after adsorption. The reactions were carried out at ∼30 °C, which was maintained by circulating water in the annulus of the jacketed quartz reactor. No adjustment of pH was done during the experiment. The degradation experiment was carried out for 1 h during which samples were collected at regular time intervals. The collected samples were then filtered and centrifuged for about an hour to remove any catalyst particles before the analysis. All dyes were analyzed with a UV-visible spectrometer (Lambda 35 Perkin-Elmer). The calibration for MB, RB, RBBR, OG, EY, AB, AG and IC were based on the Beer–Lambert law at their maximum absorption wavelength, λmax of 664, 554, 592, 480, 517, 618, 640 and 661 nm, respectively.
The ac-impedance measurements were carried out using pelletized powder samples. Prior to measurement, the pellets were annealed at 900 °C for 6 h. Silver paste was applied on both sides of the pellets for better ohmic contact. The coated pellets were heated at 200 °C for 1 h to remove the organic component of the Ag-paste. The diameter and thickness of the pellet were approximately 10 mm and 1 mm, respectively. The annealed pellets were then placed between two steel electrodes of a homemade conductivity cell. The ac-impedance measurements were carried out (Novocontrol Alpha-A) in the frequency range 1 to 3 × 106 Hz (signal amplitude = 0.05 V) from 30 to 700 °C at intervals of 50 °C up to 750 °C. The temperature of samples was equilibrated for 30 min at each temperature point (±2 °C, programmable Thermolyne furnace) prior to the impedance measurements.
Conclusions
The effect of substitution of Bi atom (varying size, oxidation state and atomic number) in Sr3V2O8 is found to result in a new variant in palmeirite type compounds with varying properties. Accurate location of the substituent can only be achieved from single-crystal data using difference Fourier methods. Structural basis for the selective photodegradation in dyes reveals the importance of the location of the substituent in the solid solution.
Acknowledgements
We thank DST-FIST (level II) for funding the Oxford diffraction X-ray facility. D. S. thanks DST-India and Sincrotrone Trieste for financial support during the synchrotron data collection at Trieste, Italy. D. S. also thanks Dr Jasper Plaisier for scientific assistance during the synchrotron data collection. T. N. G. thanks DST-India for a J. C. Bose fellowship for funding.
References
- V. D. Zhuravlev, A. A. Fotiev and B. V. Shul'gin, Izv. Akad. Nauk SSSR Neorg. Mater., 1979, 15, 2003–2006 CAS.
- V. D. Zhuravlev and A. A. Fotiev, Zh. Neorg. Khim., 1980, 25, 2560 CAS.
- L. Merkle, D. A. Pinto, H. Verdún and R. B. McIntosh, Appl. Phys. Lett., 1992, 61, 2386–2388 CrossRef CAS.
- B. Buijsse, J. Schmidt, I. Y. Chan and D. J. Singel, Phys. Rev. B: Condens. Matter, 1995, 51, 6215 CrossRef CAS.
- L. V. Kristallov and A. A. Fotiev, Zh. Neorg. Khimii, 1981, 26, 2718 CAS.
- A. Grzechnik and P. F. McMillan, J. Solid State Chem., 1997, 132, 156–162 CrossRef CAS.
- P. Roux and G. Bonel, Rev. Chim. Miner., 1985, 22, 767–775 CAS.
- P. Khatri, B. Behera and R. N. P. Choudhary, Phys. Status Solidi B, 2009, 246, 1118–1123 CrossRef CAS.
- A. Durif, Acta Crystallogr., 1959, 12, 420–421 CrossRef CAS.
- G. Le Flem and R. Olascuaga, Bull. Soc. Chem. Fr., 1968, 2769–2780 CAS.
- M. S. Skakle, A. M. Coats and J. Marr, J. Mater. Sci., 2000, 35, 3251–3256 CrossRef.
- H. Zhang, M. Lü, Z. Yang, Z. Xiu, G. Zhou, S. Wang, Y. Zhou and S. Wang, J. Alloys Compd., 2006, 426, 384–389 CrossRef CAS.
- I. Leonidov, O. Leonidova and V. Slepukhin, Inorg. Mater., 2000, 36, 72–75 CrossRef CAS.
- O. N. Leonidova and E. I. Leonidova, Solid State Ionics, 2008, 179, 188–191 CrossRef CAS.
- V. D. Zhuravlev, A. P. Tyutyunnik, Y. A. Velikodnyi, V. G. Zubkov, V. G. Bamburov, I. R. Shein and A. L. Ivanovskii, Dokl. Phys. Chem., 2007, 415, 186–189 CrossRef CAS.
- P. P. Sahoo, E. Gaudin, J. Darriet and T. N. G. Row, Inorg. Chem., 2010, 49, 5603–5610 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- U. Stafford, K. A. Gray and P. V. Kamat, Heterog. Chem. Rev., 1996, 3, 77–104 CrossRef CAS.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 64, 112–122 CrossRef.
- W. Carrillo-Cabrera and H. G. Von Schnering, Z. Kristallogr., 1993, 205, 271–276 CrossRef CAS.
- M. W. Stoltzfus, P. M. Woodward, R. Seshadri, J. H. Klepeis and B. Bursten, Inorg. Chem., 2007, 46, 3839–3850 CrossRef CAS.
- R. Vinu, S. U. Akki and G. Madras, J. Hazard. Mater., 2010, 176, 765–773 CrossRef CAS.
- C. u. G. Silva, W. Wang and J. L. s. Faria, J. Photochem. Photobiol., A, 2006, 181, 314–324 CrossRef CAS.
- A. S. Mahmoud, M. S. Brooks and A. E. Ghaly, Am. J. Appl. Sci., 2007, 4, 1054–1062 CrossRef CAS.
- O. Leonidova, I. Leonidov, G. Dontsov and A. Zhukovskaya, Phys. Solid State, 1998, 40, 200–203 CrossRef.
- Y. Teraoka, H. M. Zhang, K. Okamoto and N. Yamazoe, Mater. Res. Bull., 1988, 23, 51–58 CrossRef CAS.
- M. V. Patrakeev, J. A. Bahteeva, E. B. Mitberg, I. A. Leonidov, V. L. Kozhevnikov and K. R. Poeppelmeier, J. Solid State Chem., 2003, 172, 219–231 CrossRef CAS.
-
V. Petricek, M. Dusek and L. Palatinus, JANA2000, 2007 Search PubMed.
-
Oxford Diffraction, Oxford Diffraction Ltd., Abingdon, Oxfordshire, UK, 2009 Search PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
-
K. Brandenburg, Crystal Impact GbR, Bonn, Germany, 1999 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.