Construction of five- and six-membered heterocycles on both Cp rings of the ferrocene moiety of α-oxoketene-S,S-acetal and β-oxodithioester via heteroaromatic annulation†
Girijesh Kumar
Verma
,
Rajiv Kumar
Verma
and
Maya Shankar
Singh
*
Department of Chemistry (Centre of Advanced Study), Faculty of Science, Banaras Hindu University, Varanasi-221005, India. E-mail: mssinghbhu@yahoo.co.in; Fax: +91-542-2368127; Tel: +91-542-6702502
First published on 30th October 2012
Abstract
1,1′-Bis(1,1-dimethylsulfanyl-3-oxo-1-propene)ferrocene and 1,1′-Bis(methyl-3-hydroxy-prop-2-ene-dithioate)ferrocene have been shown to be useful three-carbon synthons for the efficient synthesis of hitherto unreported and synthetically demanding Fc-heterocycles. Five-membered (pyrazole, isoxazole, and thiophene) and six-membered (pyrimidine, coumarin, and quinoline) heterocycles have been constructed on both Cp rings of the ferrocene matrix via regioselective heteroaromatic annulation.
Introduction
Since the accidental discovery of ferrocene1 in 1951 (an isostere of benzene), the fascinating metallocene has experienced a renaissance, and captured the attention of synthetic, materials, as well as medicinal chemists. Ferrocene appended scaffolds are important synthetic targets for both the pharmaceutical industry and academic laboratories, owing to their rich chemistry and numerous applications in organic synthesis,2,3 asymmetric catalysis,4 development of new materials,5 non-linear optics,6 and in bio-organometallic chemistry.7 In particular, ferrocene grafted compounds play a key role as scaffolds for ligands in asymmetric synthesis,8 and display a wide range of biological activities, such as anticancer9 (ferrocifen I, ferrocenyl acridine II), antimalarial10 (ferrochloroquine III), anti-proliferative,11 and inhibitors of enzymes12 (Chart 1). Incorporation of a ferrocene moiety into the skeleton of an organic compound often leads to enhanced activity or unexpected biological properties,13 which is due to their different membrane permeation properties and anomalous metabolism.14 There are many examples in the literature where a phenyl or alkyl group has been replaced by a ferrocene moiety as a drug design strategy. | ||
| Chart 1 Some important ferrocene appended scaffolds. | ||
Apart from the above properties, ferrocene based compounds have also been widely utilized as organic conductors15 (ferrocene-π-extended-dithiafulvalenes IV), redox-active coordination polymers16 (V), and as ligands in metal-catalyzed cyclopropanation and Diels–Alder reactions17 (VI) (Chart 1). Furthermore, several mono- and bis-urea substituted ferrocene receptors are synthesized and utilized for anion recognition and sensing.18 Recently, a ferrocene-functionalized polydiacetylene liposome has been developed,19 which has the propensity to respond colorimetrically and electrochemically upon binding to a cyclodextrin oligomer. Hence, there has been renewed interest in the synthesis of ferrocene based heterocycles with potential applications.
Functionalized α-oxoketene-S,S-acetals, due to their highly polarized push (MeS)–pull (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) interaction on the CC bond, have been proved to be versatile three-carbon building blocks20 for the construction of various heterocyclic systems, such as pyrazole,21a,b isoxazole,21c thiophene,21d,e pyrimidine,22a–c coumarin,22d,e and quinoline22f,g derivatives. Due to their broad array of applications in biology, materials science, and chemistry, there has been an ever increasing demand for ferrocene appended heterocycles. Recently, few synthetic approaches toward ferrocene-based heterocycles have been developed.23 The scarcity of resourceful synthetic methods for ferrocene-heterocycles synthesis, and the significant limitations of existing procedures prompted us to develop an efficient and more viable route for the synthesis of ferrocene grafted heterocycles.
O) interaction on the CC bond, have been proved to be versatile three-carbon building blocks20 for the construction of various heterocyclic systems, such as pyrazole,21a,b isoxazole,21c thiophene,21d,e pyrimidine,22a–c coumarin,22d,e and quinoline22f,g derivatives. Due to their broad array of applications in biology, materials science, and chemistry, there has been an ever increasing demand for ferrocene appended heterocycles. Recently, few synthetic approaches toward ferrocene-based heterocycles have been developed.23 The scarcity of resourceful synthetic methods for ferrocene-heterocycles synthesis, and the significant limitations of existing procedures prompted us to develop an efficient and more viable route for the synthesis of ferrocene grafted heterocycles.
To the best of our knowledge, there is no report available for the synthesis of ferrocene derivatives with 5- and 6-membered heterocycles installed on both Cp rings of a ferrocene matrix, utilizing a ferrocene based α-oxoketene-S,S-acetal and β-oxodithioester. As part of our ongoing studies on the chemistry of functionalized α-oxoketene acetals24 and β-oxodithioesters,25 we wish to report herein a highly efficient regioselective heteroannulation of ferrocene based α-oxoketene-S,S-acetal and β-oxodithioester to furnish hitherto unreported ferrocene-heterocycles containing five-/six-membered and fused heterocycles on both Cp rings.
Results and discussion
The utility of α-oxoketene acetals20–22 and β-oxodithioesters25,26 as versatile intermediates in organic synthesis has been well recognized. The desired ferrocene appended β-oxodithioester 2 and α-oxoketene dithioacetal 3 were not commercially sourced, and prepared in good yields according to reported procedures.27 The methodology involved reaction of the 1,1′-diacetylferrocene 1 with S,S-dimethyltrithiocarbonate in the presence of NaH in a mixture of N,N-dimethylformamide (DMF) and benzene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) at reflux temperature to yield 1,1′-bis(methyl-3-hydroxy-prop-2-ene-dithioate)ferrocene 2 in 65% yield. The β-oxodithioester 2 was further treated with methyl iodide, 50% solution of NaOH and tetrabutyl ammonium bromide (TBAB) in benzene to furnish 1,1′-bis(1,1-dimethylsulfanyl-3-oxo-1-propene)ferrocene 3 in 83% yield (Scheme 1).
:10) at reflux temperature to yield 1,1′-bis(methyl-3-hydroxy-prop-2-ene-dithioate)ferrocene 2 in 65% yield. The β-oxodithioester 2 was further treated with methyl iodide, 50% solution of NaOH and tetrabutyl ammonium bromide (TBAB) in benzene to furnish 1,1′-bis(1,1-dimethylsulfanyl-3-oxo-1-propene)ferrocene 3 in 83% yield (Scheme 1).
 | ||
| Scheme 1 The synthesis of ferrocene-based β-oxodithioester 2 and α-oxoketene dithioacetal 3. | ||
Substituted pyrazoles are important synthetic targets for the synthesis of several commercial drugs and functional materials. Recent studies have shown that incorporation of a ferrocenyl group into such structures may improve their biological activities or create new medicinal properties.28 Earlier studies have shown that it is possible to tune the reactivity of these ambident electrophiles toward unsymmetrical heterobinucleophiles by variation of reaction conditions to afford substituted five-/six-membered heterocycles in a highly regiocontrolled fashion.21–26
Our first attempt was to synthesize five-membered heterocyclic rings over both the Cp rings of ferrocene template via β-oxodithioester 2 and α-oxoketene dithioacetal 3 (Schemes 2–4). Thus, when β-oxodithioester 2 was treated with phenylhydrazine in refluxing ethanol, the corresponding 1,1′-bis(3-methylsulfanyl-1-phenyl-pyrazol-5-yl)ferrocene 4 was obtained in moderate yield (45%). Similarly, treatment of α-oxoketene dithioacetal 3 with hydrazine hydrate in refluxing ethanol furnished 1,1′-bis(3-methylsulfanyl-pyrazol-5-yl)ferrocene 5 in 73% yield. Further, when 3 was treated with phenylhydrazine in the presence of potassium tert-butoxide in refluxing tert-butanol provided 1,1′-bis(5-methylsulfanyl-1-phenyl-pyrazol-3-yl)ferrocene 6 in 63% yield. Only one regioisomeric pyrazole 6 was observed and no trace of the corresponding 1,1′-bis(3-methylsulfanyl-1-phenyl-pyrazol-5-yl)ferrocene 4 could be isolated from the reaction mixture, making the protocol highly regioselective (Scheme 2).

 | ||
| Scheme 3 The synthesis of ferrocene appended isoxazoles. | ||
 | ||
| Scheme 4 The synthesis of ferrocene appended thiophenes. | ||
The isoxazole ring is widely present in numerous natural products and synthetic pharmaceuticals. With a view to synthesize isoxazole rings over the ferrocene platform, the ketene dithioacetal 3 was next subjected to treatment with hydroxylamine hydrochloride in the presence of barium hydroxide and ethanol under reflux to give the corresponding 1,1′-bis(5-methylsulfanyl-isoxazol-3-yl)ferrocene 7 in 50% yield. Our attempt to prepare 1,1′-bis(5-ethoxy-isoxazol-3-yl)ferrocene 8 by the treatment of ketene dithioacetal 3 with hydroxylamine hydrochloride in the presence of sodium ethoxide under refluxing ethanol yielded a complex inseparable reaction mixture (Scheme 3).
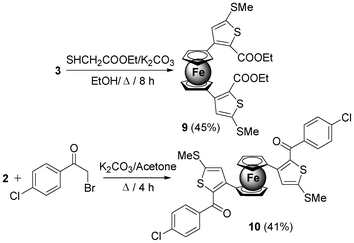
Redox-active molecules containing π-conjugated thiophene fragments have attracted increasing interest in materials science from the viewpoint of the fabrication of organic semi-conducting materials.29 To further add diversity to the ferrocene platform, ketene dithioacetal 3 was subjected to cyclocondensation with ethylthioglycolate in the presence of potassium carbonate in ethanol to afford 1,1′-bis(5-methylsulfanyl-2-carboethoxy-thiophen-3-yl) ferrocene 9 in moderate yield (45%). Similarly, addition of 2-bromo-1-(4-chlorophenyl)ethanone to a stirred solution of β-oxodithioester 2 and potassium carbonate in acetone produced 1,1′-bis(2-(4-chlorobenzoyl)-5-methylsulfanyl-thiophen-3-yl) ferrocene 10 in 41% yield (Scheme 4).
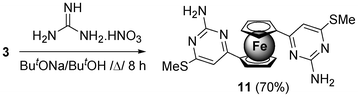
The successful use of 1,2-binucleophiles to construct five-membered heterocycles over the ferrocene platform prompted us to use 1,3-heterobinucleophiles with a view to construct six-membered heterocycles on the ferrocene matrix. Substituted pyrimidines not only represent an important class of six-membered heterocycles prevalent in bioactive natural products and pharmaceuticals, but also represent useful intermediates in organic synthesis. Thus, heterocyclization of 3 with guanidine nitrate in presence of sodium tert-butoxide in tert-butanol furnished 1,1′-bis(2-amino-4-methylsulfanyl-pyrimidin-6-yl) ferrocene 11 in 70% yield (Scheme 5). Compound 11 was found to be insoluble in common NMR solvents, such as CDCl3, CD2Cl2, CD3COCD3, and DMSO-d6, so the structure was assigned with the help of IR and LC-HRMS study. The versatility of heteroaromatic annulation was further demonstrated by the synthesis of ferrocene grafted coumarin 12, as shown in Scheme 6. Thus, (4 + 2) cycloaddition of ketene dithioacetal 3 with 5-bromo-2-hydroxybenzaldehyde in presence of InCl3 at 80 °C under solvent-free conditions produced 1,1′-bis(6-bromochromene-3-carbonyl)ferrocene 12 in 70% yield.
 | ||
| Scheme 5 The synthesis of a ferrocene appended pyrimidine. | ||

In order to get more insight into this type of annulation chemistry, in an another strategy, we further extended (4 + 2) cyclocondensation studies of ketene dithioacetal 3 for the synthesis of ferrocene appended quinoline derivatives to add further points of diversity. Thus, the annulation of 3 with o-aminobenzophenone/o-aminoacetophenone produced 1,1′-bis(4-phenyl/methyl-2-methylsulfanyl-quinoline-3-carbonyl)ferrocenes 13 in good yields (Scheme 7).
 | ||
| Scheme 7 The synthesis of ferrocene appended quinolines. | ||
In another alternative pathway, 3 was added to lithiated 3,4-dimethoxyaniline (prepared by drop wise addition of n-BuLi to a solution of 3,4-dimethoxyaniline in dry THF at room temperature under an argon atmosphere) at room temperature to produce the respective N,S-acetal 1,1′-bis(1-(3,4-dimethoxyamino)-1-methylsulfanyl-3-oxo-1-propene)ferrocene 14 in 35% yield. The N,S-acetal 14 thus obtained was treated with polyphosphoric acid (PPA) at 90 °C for 6 h to afford 1,1′-bis(6,7-dimethoxy-2-methylsulfanyl-quinoline-4-yl)ferrocene 15 in 70% yield (Scheme 7). The structural determinations of all the new compounds were done by IR, 1H and 13C NMR, and HRMS studies.
In summary, we have developed an efficient and regioselective protocol for the synthesis of ferrocene grafted five- and six-membered heterocycles via heteroaromatic annulation of β-oxodithioester and α-oxoketene acetal. Here, we have efficiently installed the five-/six-membered heterocycles over the ferrocene matrix. The methodology has been further elaborated to the corresponding N,S-acetal, leading to ferrocene appended quinoline, providing a further point of diversity in the newly synthesized heterocyclic frameworks. In view of the broad range of biological activities displayed by ferrocene grafted heterocycles, these newly synthesized compounds will prove useful for preliminary pharmacological screening. Our further efforts to improve the yields of 5-membered heterocycles and optimization of more annulation reactions on 1,1′-bis(1,1-dimethylsulfanyl-3-oxo-1-propene)ferrocene are in progress.
Experimental
General
Commercially available reagents were purchased from Merck, Aldrich, and Fluka, and used as received without further purification. β-Oxodithioester 2 and α-oxoketene acetal 3 were prepared according to the protocol described in the literature26 and displayed in Scheme 1. Solvents were dried before use by standard procedures. 1H and 13C NMR spectra were recorded on a JEOL AL 300 FT-NMR spectrometer operating at 300 and 75 MHz, respectively. Chemical shifts are given as δ values with reference to tetramethylsilane (TMS) as the internal standard. J values are given in Hz. The IR spectra were recorded on a Varian 3100 FT-IR spectrophotometer. Mass spectra were recorded on a micro TOF-Q II 10330, WATERS-Q-Tof Premier-HAB213 and Q-Tof micro (YA-105) instruments. All the reactions were monitored by TLC using precoated sheets of silica gel G/UV-254 of 0.25 mm thickness (Merck 60 F254) using UV light (254 nm/365 nm) for visualization. Melting points were determined with a Büchi B-540 melting point apparatus and are uncorrected.Synthesis of starting materials and final compounds
:1) as eluent to give 2 as a shiny violet solid (1.1 g, 65%). mp 155–156 °C. IR (KBr): 3437, 1668, 1568, 1455, 1233, 1095 cm−1. 1H NMR (300 MHz, CDCl3): δ 14.76 (s, 2H, 2 × OH, D2O exchangeable), 6.41 (s, 2H, 2 × CH), 4.79 (s, 4H, HCp), 4.54(s, 4H, HCp), 2.60 (s, 6H, 2 × SCH3). 13C NMR (75 MHz, CDCl3): δ 213.6, 171.5, 107.9, 79.2, 73.3, 69.4, 16.8.
:1) as eluent to give 3 as red solid (0.9 g, 83%). mp 186–187 °C. IR (KBr): 1608, 1494, 1374, 1250, 1100 cm−1. 1H NMR (300 MHz, CDCl3): δ 6.18 (s, 2H, 2 × CH), 4.74 (s, 4H, HCp), 4.46 (s, 4H, HCp), 2.55 (s, 6H, 2 × SCH3), 2.50 (s, 6H, 2 × SCH3). 13C NMR (75 MHz, CDCl3): δ 188.0, 162.9, 110.7, 83.0, 72.7, 70.7, 17.3, 15.0. LC-HRMS [ESI, (M + H)+]: C20H22FeO2S4, Calcd: 478.9930, Found 478.9925.
:1) as eluent to give 4 as an orange colored solid (0.253 g, 45%). mp 160–161 °C. IR (KBr): 3059, 2923, 1595, 1499, 1356, 1195 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.38–7.27 (m, 10H, ArH), 6.26 (s, 2H, ArH), 4.09 (s, 4H, HCp), 3.97 (s, 4H, HCp), 2.58 (s, 6H, 2 × SCH3). 13C NMR (75 MHz, CDCl3): δ 147.8, 142.0, 139.8, 128.9, 128.1, 128.0, 126.0, 106.2, 75.4, 70.4, 15.7. HRMS [ESI, (M + H)+]: C30H26FeN4S2, Calcd: 563.1026, Found: 563.1157.
:1) as eluent to give 6 as an orange solid (0.349 g, 62%). mp 175–176 °C. IR (KBr): 2922, 2854, 1658, 1595, 1498, 1420, 1364, 1030 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.62 (d, J = 7.5 Hz, 4H, ArH), 7.47–7.35 (m, 6H, ArH), 6.26 (s, 2H, ArH), 4.62 (s, 4H, HCp), 4.26 (s, 4H, HCp), 2.31 (s, 6H, 2 × SCH3). 13C NMR (75 MHz, CDCl3): δ 139.6, 138.0, 128.8, 127.4, 124.5, 105.9, 79.0, 69.8, 68.1, 17.9. HRMS [ESI, (M + H)+]: C30H26FeN4S2, Calcd: 563.1026, Found: 563.1023.
:1) as eluent to yield 9 as a red solid (0.264 g, 45%). mp 179–180 °C. IR (KBr): 2924, 2858, 1700, 1507, 1420, 1284, 1090 cm−1. 1H NMR (300 MHz, CDCl3): δ 6.85 (s, 2H, ArH), 4.77 (s, 4H, HCp), 4.29–4.21 [m, 8H, (4H, HCp and 2 × CH2)], 2.56 (s, 6H, 2 × SCH3), 1.33 (t, J = 6.9 Hz, 6H, 2 × CH3). 13C NMR (75 MHz, CDCl3): δ 161.3, 145.0, 143.5, 130.3, 124.4, 80.2, 71.9, 70.2, 60.7, 19.3, 14.2. HRMS [ESI, (M + Na)+]: C26H26FeO4S4, Calcd: 608.9961, Found: 608.9960.
:1) as eluent to yield 10 as a red solid (0.294 g, 41%). mp 194–195 °C. IR (KBr): 3094, 2922, 2852, 1670, 1607, 1409, 1284, 1086 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.54 (d, J = 8.4 Hz, 4H, ArH), 7.21 (d, J = 8.4 Hz, 4H, ArH), 6.88 (s, 2H, ArH), 4.20 (s, 4H, HCp), 4.03 (s, 4H, HCp), 2.60 (s, 6H, 2 × SCH3). 13C NMR (75 MHz, CDCl3): δ 187.2, 145.7, 143.9, 138.5, 136.5, 134.5, 131.0, 129.5, 128.2, 81.4, 71.0, 70.3, 19.4. HRMS [ESI, (M + H)+]: C34H24Cl2FeO2S4, Calcd: 718.9464, Found: 718.9474.
General procedure for the synthesis of 13
A mixture of 2-aminoaryl/alkyl ketone (2.0 mmol) and α-oxoketene dithioacetal 3 (1.0 mmol) was heated at 80 °C in the presence of InCl3 (20 mol %, 0.2 mmol, 44 mg) for the stipulated period of time (1.5–2.0 h) until the completion of the reaction (monitored by TLC). The mixture was treated with water (20 mL) and extracted with EtOAc (2 × 20 mL). The combined organic extract was washed with water (2 × 20 mL) followed by brine (15 mL) and dried over anhydrous Na2SO4. The solvent was evaporated under vacuum. The crude product thus obtained was purified by column chromatography on silica gel (ethyl acetate/n-hexane, 1:19).
PPA (0.500 g) and N,S-acetal 14 (0.240 g, 0.35 mmol) was mixed well and heated at 90 °C for 6 h. After the completion of reaction (monitored by TLC), the reaction mixture was carefully neutralized by saturated aqueous solution of NaHCO3 (400 mL). The reaction mixture was extracted with EtOAc (2 × 25 mL), washed with water (3 × 20 mL) followed by brine (20 mL), and dried over anhydrous Na2SO4. The crude product was purified over neutral alumina using 20% EtOAc in n-hexane to yield the red colored compound 15 (0.457 g, 70%). mp 197–198 °C. IR (KBr): 2957, 2924, 2853, 1579, 1485, 1464, 1248, 1103, 1006 cm−1. 1H NMR (300 MHz, DMSO-d6): δ 8.31 (s, 2H, ArH), 7.56 (s, 2H, ArH), 7.19 (s, 1H, ArH), 7.15 (s, 1H, ArH), 4.97 (s, 4H, HCp), 4.54 (s, 4H, HCp), 3.90 (s, 6H, 2 × OMe), 3.88 (s, 6H, 2 × OCH3), 2.54 (s, 6H, 2 × SCH3). 13C NMR (75 MHz, CDCl3): δ 156.6, 151.9, 148.2, 146.0, 142.5, 119.5, 119.1, 107.8, 103.9, 84.6, 71.7, 71.3, 56.0, 14.1. HRMS [ESI, (M + H)+]: C34H32FeN2O4S2, Calcd: 653.1231, Found: 653.1238.
Acknowledgements
We gratefully acknowledge the generous financial support from the Council of Scientific and Industrial Research (CSIR) and the Department of Science and Technology (DST), New Delhi. The authors (G.K.V. & R.K.V.) are thankful to UGC and CSIR, New Delhi for senior research fellowships.References
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039 CrossRef CAS.
- (a) Ferrocenes: Homogenous Catalysis. Organic Synthesis, Materials Science, ed. A. Togni and T. Hayashi, VCH, Weinheim, Germany, 1995 Search PubMed; (b) Metallocenes, ed. A. Togni and R. L. Halterman, VCH, Weinheim, Germany, 1998 Search PubMed; (c) Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Stepnicka, John Wiley & Sons, Inc., New York, 2008, p 39 Search PubMed.
- (a) V. S. Sudhir, N. B. R. Baig and S. Chandrasekharan, Eur. J. Org. Chem., 2009, 5365 CrossRef CAS; (b) M. A. Sierra, M. Rodrıguez-Fernandez, L. Casarrubios, M. Gomez-Gallego, C. P. Allen and M. J. Mancheno, Dalton Trans., 2009, 39, 8399 RSC; (c) P. James, J. Neudorfl, M. Eissmann, P. Jesse, A. Prokop and H. -G. Schmalz, Org. Lett., 2006, 8, 2763 CrossRef CAS; (d) J. Tauchman and P. Stepnicka, Inorg. Chem. Commun., 2010, 13, 149 CrossRef CAS; (e) A. Garcia, B. Insuasty, M. A. Herranz, R. Martinez-Alvarez and N. Martin, Org. Lett., 2009, 11, 5398 CrossRef CAS; (f) V. J. Chebny, D. Dhar, S. V. Lindeman and R. Rathore, Org. Lett., 2006, 8, 5041 CrossRef CAS.
- (a) Q. Li, C. -H. Ding, X. -L. Hou and L. -X. Dai, Org. Lett., 2010, 12, 1080 CrossRef CAS; (b) S. L. Marquard, D. C. Rosenfeld and J. F. Hartwig, Angew. Chem., Int. Ed., 2010, 49, 793 CrossRef CAS; (c) T. J. Colacot, Chem. Rev., 2003, 103, 3101 CrossRef CAS; (d) L. -X. Dai, T. Tu, S. -L. You, W. -P. Deng and X. -L. Hou, Acc. Chem. Res., 2003, 36, 659 CrossRef CAS; (e) R. G. Arrayas, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674 CrossRef; (f) R. C. J. Atkinson, V. C. Gibson and N. J. Long, Chem. Soc. Rev., 2004, 33, 313 RSC.
- (a) W. Wang, H. Sun and A. E. Kaifer, Org. Lett., 2007, 9, 2657 CrossRef CAS; (b) T. Moriuchi and T. Hirao, Chem. Soc. Rev., 2004, 33, 294 RSC; (c) M. Even, B. Heinrich, D. Guillon, D. M. Guldi, M. Prato and R. Deschenaux, Chem.–Eur. J., 2001, 7, 2595 CrossRef CAS; (d) Z. Li, J. F. Godsell, J. P. O'Byrne, N. Petkov, M. A. Morris, S. Roy and J. D. Holmes, J. Am. Chem. Soc., 2010, 132, 12540 CrossRef CAS; (e) J. Yoshino, R. Shimizu, N. Hayashi and H. Higuchi, Bull. Chem. Soc. Jpn., 2011, 84, 110 CrossRef CAS; (f) B. Fabre, Acc. Chem. Res., 2010, 43, 1509 CrossRef CAS; (g) M. Ortiz, E. M. Wajse, A. Fragoso and C. K. O'Sullivan, Chem. Commun., 2012, 48, 1045 RSC; (h) D. Astruc, New J. Chem., 2011, 35, 764 RSC.
- (a) S. Barlow and S. R. Marder, in Functional Organic Materials, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VCHWeinheim, 2007, 393 Search PubMed; (b) S.-I. Kato, M. T. R. Beels, P. La Porta, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht, I. Biaggio and F. Diederich, Angew. Chem., Int. Ed., 2010, 49, 6207 CrossRef CAS.
- (a) D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931 CrossRef CAS; (b) H. -B. Kraatz, J. Inorg. Organomet. Polym. Mater., 2005, 15, 83 CrossRef CAS; (c) S. Chowdhury, G. Schatte and H. -B. Kraatz, Angew. Chem., Int. Ed., 2006, 45, 6882 CrossRef CAS; (d) C. Drexler, M. Milne, E. Morgan, M. Jennings and H. -B. Kraatz, Dalton Trans., 2009, 4370 RSC; (e) D. Hamels, P. M. Dansette, E. A. Hillard, S. Top, A. Vessieres, P. Herson, G. Jaouen and D. Mansuy, Angew. Chem., Int. Ed., 2009, 48, 9124 CrossRef CAS; (f) U. Schatzschneider and N. Metzler-Nolte, Angew. Chem., Int. Ed., 2006, 45, 1504 CrossRef CAS; (g) W. Balemans, N. Lounis, R. Gilissen, J. Guillemont, K. Simmen, K. Andries and A. Koul, Nature, 2010, 463, E3 CrossRef CAS; (h) S. I. Kirin, H. -B. Kraatz and N. Metzler-Nolte, Chem. Soc. Rev., 2006, 35, 348 RSC; (i) S. Matric, M. Labib, P. O. Shipman and H. -B. Kraatz, Dalton Trans., 2011, 40, 7264 RSC; (j) M. C. Semencic, K. Heinze, C. Foerster and V. Rapic, Eur. J. Inorg. Chem., 2010, 1089 CrossRef CAS.
- (a) F. Fache, E. Schulz, M. L. Tommasino and M. Lemaire, Chem. Rev., 2000, 100, 2159 CrossRef CAS; (b) H. -U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103 CrossRef CAS; (c) J. Rajput, A. T. Hutton, J. R. Moss, H. Su and C. Imrie, J. Organomet. Chem., 2006, 691, 4573 CrossRef CAS; (d) D. Vinci, N. Mateus, X. Wu, F. Hancock, A. Steiner and J. Xiao, Org. Lett., 2006, 8, 215 CrossRef CAS; (e) D. Vinci, N. Martins, O. Saidi, J. Bacsa, A. Brigas and J. Xiao, Can. J. Chem., 2009, 87, 171 CrossRef CAS; (f) N. Martins, N. Mateus, D. Vinci, O. Saidi, A. Brigas, J. Bacsa and J. Xiao, Org. Biomol. Chem., 2012, 10, 4036 RSC.
- (a) S. Top, A. Vessieres, C. Cabestaing, I. Laios, G. Leclercq, C. Provot and G. Jaouen, J. Organomet. Chem., 2001, 637–639, 500 CrossRef CAS; (b) P. N. Kelly, A. Pretre, S. Devoy, I. O'Rielly, R. Devery, A. Goel, J. F. Gallagher, A. J. Lough and P. T. M. Kenny, J. Organomet. Chem., 2007, 692, 1327 CrossRef CAS; (c) C. -W. Ong, J. -Y. Jeng, S. -S. Juang and C. -F. Chen, Bioorg. Med. Chem. Lett., 1992, 2, 929 CrossRef CAS; (d) Y. N. Vashisht Gopal, D. Jayaraju and A. K. Kondapi, Arch. Biochem. Biophys., 2000, 376, 229 CrossRef CAS.
- (a) X. Wu, P. Wilairat and M. -L. Go, Bioorg. Med. Chem. Lett., 2002, 12, 2299 CrossRef CAS; (b) C. Biot, L. Delhaes, C. M. N'Diaye, L. A. Maciejewski, D. Camus, D. Dive and J. S. Brocard, Bioorg. Med. Chem., 1999, 7, 2843 CrossRef CAS; (c) C. Biot, G. Glorian, L. A. Maciejewski, J. S. Brocard, O. Domarle, G. Blampain, P. Millet, A. J. Georges, H. Abessolo and D. Dive, J. Med. Chem., 1997, 40, 3715 CrossRef CAS; (d) O. Domarle, G. Blampain, H. Agnaniet, T. Nzadiyabi, J. Lebibi, J. Brocard, L. Maciejewski, C. Biot, A. J. Georges and P. Millet, Antimicrob. Agents Chemother., 1998, 42, 540 CAS.
- (a) J. C. Swarts, D. M. Swarts, D. M. Maree, E. W. Neuse, C. La Madeleine and J. E. Van Lier, Anticancer Res., 2001, 21, 2033 CAS; (b) M. T. Johnson, E. Kreft, D.D. N'Da, E. W. Neuse and C. E. J. van Rensburg, J. Inorg. Organomet. Polym., 2003, 13, 255 CrossRef CAS.
- (a) R. P. Hanzlik, P. Soine and W. H. Soine, J. Med. Chem., 1979, 22, 424 CrossRef CAS; (b) J. Spencer, J. Amin, R. Boddiboyena, G. Packham, B. E. Cavell, S. S. Syed Alwi, R. M. Paranal, T. D. Heightman, M. Wang, B. Marsden, P. Coxhead, M. Guille, G. J. Tizzard, S. J. Coles and J. E. Bradner, Med. Chem. Commun., 2012, 3, 61 RSC.
- H. Runqiu and W. Qingmin, J. Organomet. Chem., 2001, 637–639, 94 CrossRef.
- (a) L. Chen, Q. Wang, R. Huang, C. Mao, J. Shang and H. Song, Appl. Organomet. Chem., 2005, 19, 45 CrossRef CAS; (b) L. Chen, J. X. Chen, L. Sun and Q. Xie, Appl. Organomet. Chem., 2005, 19, 1038 CrossRef CAS.
- (a) A. A. O. Sarhan, O. F. Mohammed and T. Izumi, Beilstein J. Org. Chem., 2009, 5(6) CAS; (b) A. El-W. Sarhan, T. Kijima and T. Izumi, J. Organomet. Chem., 2003, 682, 49 CrossRef; (c) A. El-W. Sarhan and T. Izumi, Synth. Met., 2004, 140, 95 CrossRef.
- R. Horikoshi, T. Mochida and H. Moriyama, Inorg. Chem., 2002, 41, 3017 CrossRef CAS.
- Z. Qiao, X. Y. Shi, Q. H. Bian, S. C. Hou and M. Wang, Chin. Chem. Lett., 2004, 15, 1015 CAS.
- M. D. Pratt and P. D. Beer, Polyhedron, 2003, 22, 649 CrossRef CAS.
- C. E. Maclean, C. L. Parkinson, P. J. Davis, M. J. Davis and G. Cooke, Tetrahedron Lett., 2012, 53, 2948 CrossRef CAS.
- (a) R. K. Dieter, Tetrahedron, 1986, 42, 3029 CrossRef CAS; (b) H. Junjappa, H. Ila and C. V. Asokan, Tetrahedron, 1990, 46, 5423 CrossRef CAS; (c) H. Ila, H. Junjappa and P. K. Mohanta, Progress in Heterocyclic Chemistry, 2001, vol. 13, ch.1 Search PubMed.
- (a) S. Peruncheralathan, T. A. Khan, H. Ila and H. Junjappa, J. Org. Chem., 2005, 70, 10030 CrossRef CAS; (b) S. M. S. Chauhan and H. Junjappa, Synthesis, 1975, 798 CrossRef CAS; (c) M. L. Purkayastha, H. Ila and H. Junjappa, Synthesis, 1989, 20 CrossRef CAS; (d) G. Sommen, A. Comel and G. Kirsch, Synthesis, 2003, 735 CAS; (e) V. J. Ram, A. Goel, P. K. Shukla and A. Kapil, Bioorg. Med. Chem. Lett., 1997, 7, 3101 CrossRef CAS.
- (a) S. B. Katiyar, I. Bansal, J. K. Saxena and P. M. S. Chauhan, Bioorg. Med. Chem. Lett., 2005, 15, 47 CrossRef CAS; (b) A. Mathews and C. V. Asokan, Tetrahedron, 2007, 63, 7845 CrossRef CAS; (c) S. M. S. Chauhan and H. Junjappa, Tetrahedron, 1976, 32, 1779 CrossRef CAS; (d) H. S. P. Rao and S. Sivakumar, J. Org. Chem., 2006, 71, 8715 CrossRef CAS; (e) H.-J. Yuan, M. Wang, Y.-J. Liu and Q. Liu, Adv. Synth. Catal., 2009, 351, 112 CrossRef CAS; (f) P. K. Mahata, C. Venkatesh, U. K. S. Kumar, H. Ila and H. Junjappa, J. Org. Chem., 2003, 68, 3966 CrossRef CAS; (g) C. Venkatesh, G. S. M. Sundaram, H. Ila and H. Junjappa, J. Org. Chem., 2006, 71, 1280 CrossRef CAS.
- (a) W. J. Lee, J. M. Chea and Y. Jahng, Bull. Korean Chem. Soc., 2009, 30, 3061 CrossRef CAS; (b) O. N. Chupakhin, I. A. Utepova, I. S. Kovalev, V. L. Rusinov and Z. A. Starikova, Eur. J. Org. Chem., 2007, 857 CrossRef CAS; (c) M. Zora and M. Gormen, J. Organomet. Chem., 2007, 692, 5026 CrossRef CAS.
- Recent papers: (a) R. K. Verma, H. Ila and M. S. Singh, Tetrahedron, 2010, 66, 7389 CrossRef CAS; (b) M. S. Singh, G. C. Nandi and S. Samai, Green Chem., 2012, 14, 447 RSC; (c) R. K. Verma, G. K. Verma, G. Shukla and M. S. Singh, RSC Adv., 2012, 2, 2413 RSC; (d) T. Chanda, R. K. Verma and M. S. Singh, Chem.–Asian J., 2012, 7, 778 CrossRef CAS.
- (a) G. C. Nandi, S. Samai and M. S. Singh, J. Org. Chem., 2010, 75, 7785 CrossRef CAS; (b) R. K. Verma, G. K. Verma, K. Raghuvanshi and M. S. Singh, Tetrahedron, 2011, 67, 584 CrossRef CAS; (c) S. Chowdhury, G. C. Nandi, S. Samai and M. S. Singh, Org. Lett., 2011, 13, 3762 CrossRef CAS; (d) G. C. Nandi, S. Samai and M. S. Singh, J. Org. Chem., 2011, 76, 8009 CrossRef CAS; (e) R. K. Verma, G. K. Verma, G. Shukla, A. Nagaraju and M. S. Singh, ACS Comb. Sci., 2012, 14, 224 CrossRef CAS; (f) G. C. Nandi, M. S. Singh, H. Ila and H. Junjappa, Eur. J. Org. Chem., 2012, 967 CrossRef CAS; (g) S. Samai, G. C. Nandi and M. S. Singh, Tetrahedron, 2012, 68, 1247 CrossRef CAS.
- (a) O. M. Singh, N. S. Devi, D. S. Thokchom and G. J. Sharma, Eur. J. Med. Chem., 2010, 45, 2250 CrossRef CAS; (b) O. M. Singh and N. S. Devi, J. Org. Chem., 2009, 74, 3141 CrossRef CAS; (c) R. Samuel, P. Chandran, S. Retnamma, K. A. Sasikala, N. K. Sreedevi, E. R. Anabha and C. V. Asokan, Tetrahedron, 2008, 64, 5944 CrossRef CAS.
- (a) G. Singh, S. S. Bhattacharjee, H. Ila and H. Junjappa, Synthesis, 1982, 693 CrossRef CAS; (b) B. N. Ahamed, M. Arunachalam and P. Ghosh, Inorg. Chem., 2010, 49, 4447 CrossRef CAS.
- (a) S. Top, A. Vessieres, G. Leclercq, J. Quivy, J. Tang, J. Vaissermann, M. Huche and G. Jaouen, Chem.–Eur. J., 2003, 9, 5223 CrossRef CAS; (b) G. Jaouen, S. Top, A. Vessieres, G. Leclercq and M. J. McGlinchey, Curr. Med. Chem., 2004, 11, 2505 CrossRef CAS.
- S. Ogawa, H. Kabayashi and H. Muraoka, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 1238 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: original 1H, 13C NMR and HRMS spectra of compounds. See DOI: 10.1039/c2ra21744a |
| This journal is © The Royal Society of Chemistry 2013 |