Advanced bimetallic In–Cu/Ag/Au nanostructures via microemulsion-based reaction†
Christian
Kind
a,
Radian
Popescu
b,
Reinhard
Schneider
b,
Erich
Müller
b,
Dagmar
Gerthsen
*b and
Claus
Feldmann
*a
aProf. Dr C. Feldmann, Dipl.-Chem. C. Kind, Institut für Anorganische Chemie, Karlsruhe Institute of Technology (KIT), Engesserstrasse 15, D-76131 Karlsruhe, Germany. E-mail: claus.feldmann@kit.edu; Fax: (+)49 721 608 44892; Tel: (+)49 721 608 42855
bProf. Dr D. Gerthsen, Dr R. Popescu, Dr Habil. R. Schneider, Dr E. Müller, Laboratorium für Elektronenmikroskopie, Karlsruhe Institute of Technology (KIT), Engesserstrasse 7, D-76131 Karlsruhe, Germany. E-mail: dagmar.gerthsen@kit.edu; Fax: (+)49 721 608 43721; Tel: (+)49 721 608 43200
First published on 3rd August 2012
Abstract
Bimetallic nanomaterials and nanostructures constituted of the coinage metals (Cu, Ag, Au) and indium with elaborate compositions and structures are realized via a microemulsion-based approach. In detail, this comprises Cu11In9@CuIn@In core@shell-A@shell-B nanoparticles, In-Ag Janushead-like nanoparticles, Ag0 hollow spheres, Ag3In@In core@shell nanoparticles, Au@AuIn2@In core@shell-A@shell-B nanoparticles and AuIn2 nanoparticles. To obtain these advanced architectures, two approaches are applied: (1) In0 nanoparticles—pre-synthesized in a microemulsion—were reacted in a follow-up reaction with CuCl2·2H2O, AgNO3 or KAuCl4; (2) simultaneous co-reduction of InCl3·4H2O together with CuCl2·2H2O, AgNO3 or KAuCl4 in a microemulsion. Characterization of the resulting advanced structures and compositions requires elaborate electron microscopy techniques, combined with energy dispersive X-ray spectroscopy for chemical analyses of single nanoparticles as well as X-ray powder diffraction and optical spectroscopy. The versatility of the experimental approach toward complex nanoparticle architectures is related to a precise control and fine-tuning of the experimental conditions. The resulting tool kit of In–Cu/In–Ag/In–Au-based bimetallic and intermetallic nanomaterials and, in general, of nanostructured metal architectures with such variability and complexity have not yet been described.
1. Introduction
Bimetallic and intermetallic nanomaterials with elaborate composition, structure and morphology are of general interest to fundamental science and of rapidly increasing relevance to technical application.1 In general, this is related to the unusual properties of nanoscaled metals, which can differ significantly from the bulk (e.g. due to small size, large surface, quantum-size effects).2 Thus, the chemical reactivity of nanoscaled metals can be dramatically increased, too. While a high reactivity can be disadvantageous (i.e. fast oxidation/hydrolysis, low temperature stability, rapid agglomeration/aggregation), reactive nanoparticles can be highly beneficial with regard to chemical synthesis, too.3 In particular, the high reactivity of nanoscaled metals can be used to initiate chemical reactions at ambient conditions and under kinetic control, which may allow the realization of new compositions, structures and morphologies that are far beyond the thermodynamically most stable phases, as they are typically given by the relevant phase diagrams.In particular, nanoscaled less-noble metals can represent attractive candidates for use as reactive precursors, as they could serve as pure element sources for various chemical conversions to bimetallic/intermetallic compounds. This is even more interesting since a transfer of standard approaches known from bulk metals (e.g. ball milling,4 melting techniques5) to the nanoscale is not straightforward. Dedicated strategies for the realization of nanoscaled bimetals and intermetallics, on the other hand, are currently limited.6 Shape- and composition-controlled synthesis of multi-metal nanoparticles has been largely limited to those metals that have similar electrochemical potential and reactivity.6,7 The systems Fe–Pt,8 Sn–Pt/Pb–Pt,9 or Ni–Au10 represent selected examples of metals exhibiting a significantly different electrochemical potential. Intermetallic nanoparticles containing In0—to the best of our knowledge—have only been reported for InPt3.11
Most intriguingly, Schaak et al. have prepared Sn0 nanoparticles via a polyol-mediated synthesis and reacted these as a sacrificial template (i.e. reduction of a second metal under partial oxidation of the pre-synthesized Sn0) with Co2+, Ni2+ or Pd2+, to gain intermetallic compounds.12 The authors also obtained AuCu and AuCu3 nanoparticles from thermal annealing of bimetallic Au0/Cu0 aggregates.13 AuCu and AuCu3 have been alternatively prepared via seed-based diffusion of reactive copper species into pre-synthesised Au0 nanocrystals.14 Recently, Armbrüster et al. synthesized intermetallic Ga–Pd nanoparticles based on pre-synthesized Pd0 nanoparticles.15 Reacting pre-synthesized metal nanoparticles can also result in core@shell structures, such as Ag@Au/Au@Ag,16 Pd@Au@FePt,17 or Au@Pb@Pt.18 The latter, more elaborate structures are however limited to a few examples. Note that most of the above reactions require an inert-gas atmosphere (N2, Ar) and elevated temperatures (>100 °C). In general, the synthetic strategies summarized above are most often limited to a single composition, structure and/or morphology.
To explore the potential of reactive, less-noble metals for conversion reactions and the formation of nanoscaled intermetallics, we have selected indium (In0) nanoparticles as a precursor. With its electrochemical potential (E0(bulk In0) = −0.34 V)19 and its further increased reactivity as a nanoparticle, In0 may serve as a promising precursor for various conversion reactions, without being too reactive to control these reactions. As a case study, we have reacted indium with the coinage metal salts (i.e. CuCl2·2H2O, AgNO3, KAuCl4) based on simple microemulsion techniques.20 The underlying mild, ambient conditions (i.e. water as the solvent; temperature ≤50 °C) enabled us to fine-tune the relevant experimental conditions far beyond the thermodynamically most stable phases. This strategy—namely follow-up and co-reduction reactions—allows us to precisely adjust the composition, structure and morphology of the resulting bimetallic and intermetallic nanomaterials, including advanced core–shell nanostructures, nanoscale hollow spheres and bimetallic Janushead-like nanoparticles. A synthetic strategy, as well as the resulting advanced architectures, are firstly reported and lead to a tool kit of bimetallic and intermetallic nanoparticles that may also be relevant when aiming at new materials for use in electronics,21 magnetism,8 optics22 and catalysis.23
2. Materials and methods
2.1 Pre-synthesized In0 nanoparticles
2.2 Follow-up approach
2.3 Co-reduction approach
2.4 Energy-dispersive X-ray spectroscopy (EDXS) and analysis of concentration profiles
Although advanced electron microscopy (including STEM, HAADF-STEM, HRTEM, SAED) is essential to characterize the as-prepared bimetallic nanostructures described here, the underlying equipment and techniques can be nevertheless denoted as standard techniques—and are therefore summarized in the ESI.† EDXS profile analysis, as used in the following to elucidate the complex composition, structure and morphology of the as-prepared elaborate nanoparticle architectures, is however new and far beyond standard characterization. Therefore, the underlying procedure is explained here in detail.EDXS line profiles were recorded with an FEI Titan3 80–300 microscope by applying a drift-correction routine via cross correlation of several images, which yields a local precision better than 1.0 nm. The drift-corrected EDXS line profiles were taken with a probe diameter of 0.5 nm and a distance of about 1.0 nm between two measuring points. With respect to a quantification of the EDX spectra from core–shell nanoparticles, one has to keep in mind that only compositions are obtained that are averaged along the electron-beam direction. In concrete, the whole volume along the electron trajectory contributes to the detected X-ray signal. To evaluate the composition of different shells of a core–shell nanoparticle, a procedure was developed which is outlined in the following.
The analysis is based on the concentration profiles of different chemical elements within a nanoparticle determined from EDX spectra measured along a line scan that passes through its centre. The distance of 1 nm between two successive measured EDX spectra in the concentration profile suggests a division of the nanoparticle into sub-shells (SSn with n = 1,2,3…) with an equal thickness of 1 nm. Fig. 1 shows the 2-dimensional cross-section of an ideal spherical nanoparticle of radius R and the first two sub-shells represented by different colors. Here, the z-axis is parallel to the electron-beam direction, the x-axis represents the line-scan direction, and O is the nanoparticle centre. If cAi is the concentration of the chemical element i measured at point A, one can consider that it also represents the average concentration of element i within the first sub-shell cAi = ciSS1. Then, the concentration cBi measured at the next point B can be described as the weighted concentration of element i within the first and the second sub-shell. The weights w1 and w2 are proportional to the corresponding thickness of each sub-shell, measured along the vertical line that passes through B parallel to the z-axis (Fig. 1), i.e. to the sub-shell thicknesses passed by the electron beam. Thus, the average concentration of element i in the second sub-shell ciSS2 can be calculated using the ciSS1 value and the following relation:
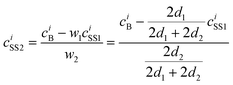 | (1) |
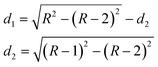 | (2) |
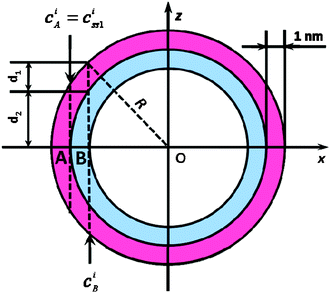 | ||
| Fig. 1 Cross-section of an ideal spherical nanoparticle (R: radius; z-axis parallel to electron-beam; x-axis corresponds to line-scan direction; with two sub-shells SS1 and SS2, both 1 nm in diameter, indicated by red/blue color. | ||
Similarly, one can calculate the average concentrations of all chemical elements in different sub-shells that are contained in the nanoparticle. As a result, the average composition of the nanoparticle shell/core can be calculated as the normalized concentration of all chemical elements found within different sub-shells that form the core/shell of the nanoparticle.
3. Results and discussion
3.1 Strategy of synthesis: follow-up and co-reduction approach
All the following bimetallic nanoparticles in the systems In–Cu, In–Ag and In–Au are realized based on microemulsion techniques. To obtain the resulting elaborate architectures, two slightly different approaches were applied (Table 1, Fig. 2):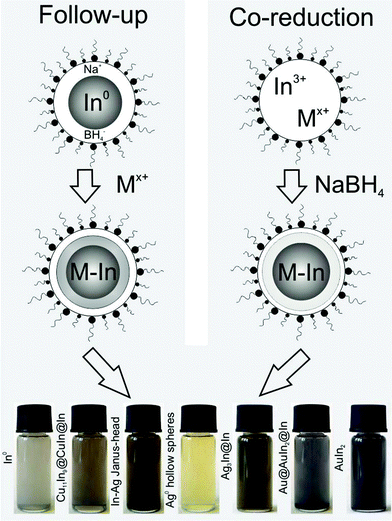 | ||
| Fig. 2 Scheme displaying the follow-up and co-reduction approach for the microemulsion-based reaction of In0 and the coinage metals as well as photos of the resulting nanoparticles: (left to right) In0 precursor; Cu11In9@CuIn@In; Ag hollow spheres; Ag3In@In; Au@AuIn2@In; AuIn2 (all suspensions in ethanol). | ||
| Type of Precursor | Type of approach | Molar ratio In![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M:BH4− :M:BH4− |
Resulting composition | Sizea/nm | |
|---|---|---|---|---|---|
| In | M | ||||
| a Average particle diameter according to BF-STEM analysis. | |||||
| InCl3·4H2O | — | — | 1:0:1 |
In precursor | 12(2) |
| In0-NP | CuCl2 | follow-up | 1:1:5 |
Cu11In9@CuIn@In core@shell | 14(6) |
| InCl3·4H2O | CuCl2 | co-reduction | 1:1:5 |
Cu11In9@CuIn@In core@shell | 6(2) |
| In0-NP | AgNO3 | follow-up | 1:1:5 |
Janushead-like In–Ag | 13(3) |
| In0-NP | AgNO3 | follow-up | 1:4:5 |
Ag hollow spheres | 11(2) |
| InCl3·4H2O | AgNO3 | co-reduction | 1:1:5 |
Ag3In@In core@shell | 9(3) |
| In0-NP | KAuCl4 | follow-up | 1:1:5 |
Au@AuIn2@In core@shell | 14(5) |
| InCl3·4H2O | KAuCl4 | co-reduction | 1:0.5:5 |
AuIn2 | 4(2) |
1. In0 precursor nanoparticles—pre-synthesized in a microemulsion—were reacted in a follow-up reaction with the coinage metal salts (i.e. CuCl2·2H2O, AgNO3, KAuCl4).
2. Co-reduction of InCl3·4H2O simultaneously with the coinage metal salt (i.e. CuCl2·2H2O, AgNO3, KAuCl4) in a microemulsion.
In the follow-up approach, In0 nanoparticles were first prepared by reduction of InCl3·4H2O with NaBH4 in a microemulsion (see section 3.2). Subsequently, these pre-synthesized “precursor In0 nanoparticles” were reacted with the much more noble coinage metals (i.e. CuCl2·2H2O, AgNO3, KAuCl4) to obtain bimetallic In–Cu, In–Ag and In–Au nanoparticles. The relevant coinage metal salt was added to the micellar system subsequent to the completed nucleation of In0. This point in time is qualitatively indicated by an ebbing of the hydrogen gas formation and the occurrence of a stable grey color of the suspension. Now, the coinage metal salt solution attains micelles that already contain In0 nanoparticles as well as excess NaBH4. Therefore, the coinage metal cations were instantaneously reduced to the elemental metal as well. Due to the spatially limited volume of the micelles, the In0 nanoparticles serve as a seed for the nucleation of the coinage metal. Depending on the interdiffusion of metals and the thermodynamics of alloy formation, nanoscaled intermetallic compounds or non-alloyed bimetallic nanoparticles—including core–shell and Janushead-like architectures—are obtained.
For this follow-up approach, the procedure of adding the coinage metal precursor to the micellar system turned out to be a key factor in controlling the reaction. If the coinage metal precursor was added too early, reduction and nucleation of indium might not be complete, resulting in quite a high excess of non-consumed NaBH4. As a consequence, non-controlled simultaneous reduction and nucleation of indium, as well as of the coinage metal, occurred and led to nanoparticles with inhomogeneous size and composition. If—by contrast—the coinage metal precursor was added too late, all NaBH4 was consumed by the aqueous medium due to hydrogen formation. This resulted in a very slow and incomplete reduction of the coinage metal. In such a case, reduction of the noble metal is driven by slow re-oxidation of the initial In0 nanoparticles—leading to formation of the elemental coinage metal only. Altogether, the course of the reaction has to be adjusted with regard to each relevant coinage metal. In detail, bimetallic nanoparticles containing copper and silver require a fast sequential injection of the coinage metal precursor as soon as the nucleation of indium is finished. This guarantees an appropriate amount of NaBH4 to remain for the reduction of the coinage metal. In contrast, a certain delay between the nucleation of In0 and the injection of the gold precursor is required to gain bimetallic indium–gold phases. Otherwise, the reduction proceeded too fast and initiated a separate nucleation of nanosized gold. Since the pure coinage metals and the bimetallic systems exhibit different surface-plasmon resonance absorptions, the course of the reaction can be qualitatively followed and visualized via the particular color (Fig. 2) and quantified by optical spectroscopy (Fig. S2, ESI†).
For the co-reduction approach, InCl3·4H2O and the relevant coinage metal salt (i.e. CuCl2·2H2O, AgNO3, KAuCl4) were both introduced in the microemulsion and simultaneously reduced by subsequent addition of excess NaBH4. Note that the co-reductions shown here were carried out at comparably low temperatures (e.g. 20–50 °C).
3.2 In0 precursor nanoparticles
Microemulsion techniques are well known for the synthesis of various inorganic nanomaterials.24 Although this already comprises several metals (e.g. Bi, Co, Ni, Pd, Pt, Cu, Ag, Au),25 nanoscale indium—to the best of our knowledge—has not yet been prepared via microemulsion techniques. Here, InCl3·4H2O was introduced into a standard w/o-(water-in-oil) microemulsion. Subsequent to addition of excess NaBH4 as the reducing agent, nucleation of In0 is indicated by a deep grey color occurring within a few seconds. The course of the reaction can be clearly monitored by this color change and the simultaneous gas formation due to hydrogen release from NaBH4. Finally, the microemulsion was destabilized by the addition of diethylene glycol (DEG).20b At the same time, the nanoparticles accumulate in the DEG bottom phase. After centrifugation and careful washing, the In0 precursor nanoparticles were resuspended in ethanol to form a grayish dispersion, which is stable against reoxidation and agglomeration for months (Fig. 2). Previous studies have evidenced that these suspensions can be stored in air without the need for any inert gas atmosphere and without formation of In(OH)3 or In2O3.26Size and shape of the In0 precursor nanoparticles are elucidated by electron microscopy (Fig. 3). Low-energy BF-STEM and TEM overview images show a manifold of spherical and nearly monodisperse particles, 12(2) nm in diameter (Fig. 3a,b,h). According to HRTEM images, the In0 nanoparticles show small crystalline areas with an average lattice fringe distance of 276(10) pm (Fig. 3c), which corresponds well with tetragonal bulk-In0 (d101: 272 pm). The small dimensions of the crystalline areas points to an amorphous or quasi-molten state of the nanoparticles due to high-energy electron bombardment. Taking the low melting point of bulk-In0 (156 °C) into account, such behavior is expected and has been frequently observed for nanostructured indium samples.26,27
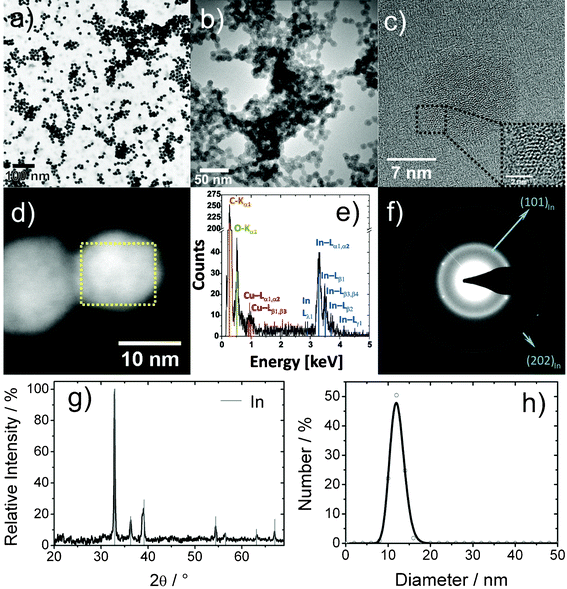 | ||
| Fig. 3 Analysis of In0 precursor nanoparticles: (a) low-energy BF-STEM overview image; ( b) TEM overview image; (c) typical HRTEM image; (d) HAADF-STEM image; (e) area-EDX spectrum (of dashed box in 3d); (f) Debye-SAED pattern; (g) XRD pattern; (h) size distribution according to statistical evaluation of STEM images. | ||
SAED patterns show an intense diffraction ring corresponding to a distance of 272(3) pm (Fig. 3f), which is again consistent with bulk-In0 (d101: 272 pm). The broadening of the ring and the absence of further diffraction rings can be ascribed to structural disorder. Some discrete (101) Bragg reflections are nevertheless visible and evidence the crystallinity of at least some nanoparticles. Since structural disorder might be an artifact of high-energy electron illumination, X-ray diffraction was involved to acquire a statistically relevant number of nanoparticles as present in a powder sample (Fig. 3g). The lattice parameters of the nanoparticles derived from XRD line positions (a = 325.5(5), c = 493.4(7) pm) are in good agreement with tetragonal bulk-In0 (a = 324.8, c = 494.8 pm). This demonstrates the crystallinity and purity of the pre-synthesized In0 precursor nanoparticles.
Considering the electrochemical potential of bulk-In0 (E0: −0.34 V),19 fast re-oxidation—especially of high-surface nanoparticles—could have been expected. However, the In0 precursor nanoparticles turned out to be stable even in the absence of inert conditions. With regard to this finding, the surface capping of the purified nanoparticles was analyzed by FT-IR spectroscopy (Fig. S1, ESI†). Besides a broad vibration at 3450–3250 cm−1 (ν(OH)) with a shoulder at 2950–2850 cm−1 (ν(CH)), several vibrations in the fingerprint area (1300–800 cm−1) indicate the presence of DEG as a capping agent. Hence, the unexpected stability of the In0 nanoparticles is ascribed to effective protection of its surface by DEG. This view is consistent with the role of DEG as reducing agent and colloidal stabilizer in the polyol-mediated synthesis of, for instance, Co0, Ni0 or Bi0 nanoparticles.28 The overall good stability of pre-synthesized In0 is also beneficial in view of a controlled reaction to intermetallic compounds.
The surface conditioning of the In0 precursor nanoparticles was further investigated by optical spectroscopy (Fig. S2, ESI†). Here, the surface-plasmon resonance absorption of indium (240–280 nm)26,29 is overlapped by the strong and broad absorption of DEG in the UV range, and hence not clearly visible. While a characteristic plasmon-resonance absorption of In0 is, in fact, not observed, UV-Vis spectroscopy is nevertheless a useful tool to analyze the plasmon-resonance absorption of the coinage metals when next performing the follow-up and co-reduction approach.
Finally, EDX spectra from single In0 precursor nanoparticles were taken at a primary electron energy of 300 keV. Due to the sensitivity of the In0 nanoparticles to the high-energy electron beam, EDXS line scans could not be performed. However, EDX spectra were obtained during scanning of a rectangular area within a single particle for about 120 s (Fig. 3d). The resulting EDX spectrum contains the characteristic L-lines of indium as well as of carbon (Kα1), oxygen (Kα1) and the L-lines of copper (Fig. 3e). While considering DEG as the surface capping, the presence of carbon and oxygen is not a surprise. Note that the copper signal, as well as a certain amount of carbon and oxygen, also stem from the carbon-film copper grid used as the sample holder. In summary, the presence and composition of DEG-capped In0 precursor nanoparticles is reliably evidenced based on the above discussed analytical tools.
3.3 The In–Cu system
:Cu ratio of 1:1, as introduced in the synthesis. The absence of Cu0 is also validated by comparing UV-Vis spectra of the sample to reference spectra of Cu0 nanoparticles (Fig. S2, ESI†).32 The observed strong absorption at 230 nm originates from oleylamine as a surface capping. Based on the above findings, now, the question arises as to whether Cu11In9, CuIn and In0 are obtained as separate, individual nanoparticles or whether a certain gradient or core–shell structure emerges for each single particle.
![Analysis of Cu11In9@CuIn@In nanoparticles (follow-up approach): (a) low-energy BF-STEM overview image; (b) typical HRTEM image; (c) experimental diffractogram of inner particle region (of dashed area in 4b) and calculated diffraction pattern of monoclinic bulk-Cu11In9 ([001] zone axis, white circles); (d) HAADF-STEM image; (e) EDXS line scan (of yellow line in 4d); (f) 3D reconstruction of nanoparticle from composition profile; (g) indexed SAED pattern of nanoparticle ensemble; (h) XRD pattern; (i) size distribution according to statistical evaluation of STEM images.](/image/article/2012/RA/c2ra21659k/c2ra21659k-f4.gif) | ||
| Fig. 4 Analysis of Cu11In9@CuIn@In nanoparticles (follow-up approach): (a) low-energy BF-STEM overview image; (b) typical HRTEM image; (c) experimental diffractogram of inner particle region (of dashed area in 4b) and calculated diffraction pattern of monoclinic bulk-Cu11In9 ([001] zone axis, white circles); (d) HAADF-STEM image; (e) EDXS line scan (of yellow line in 4d); (f) 3D reconstruction of nanoparticle from composition profile; (g) indexed SAED pattern of nanoparticle ensemble; (h) XRD pattern; (i) size distribution according to statistical evaluation of STEM images. | ||
BF-STEM images allow verification of the size and morphology of the obtained nanoparticles (Fig. 4a). With a mean size of 14(6) nm, particle diameter and size distribution are slightly broadened as compared to the initial In0 nanoparticles (Fig. 4i). HRTEM images of a single In–Cu nanoparticle show the crystalline, single phase Cu11In9 as the central core (Fig. 4b). This is also evidenced by good agreement between the experimental diffractogram and the calculated diffraction pattern of monoclinic bulk-Cu11In9 oriented along the [001] zone axis (Fig. 4c). In contrast, no lattice fringes were observed for the outer nanoparticle shell, which might be due to its small thickness, as well as due to an amorphous or quasi-molten state of the shell (see sections 3.2, 3.4 and 3.5). This view is supported by a distance of 275 pm, deduced from the diffuse diffraction ring in Fig. 4c that can be correlated to bulk-In0 (d101: 272 pm). SAED analysis of single nanoparticles, however, confirms the results stemming from XRD (Fig. 4g). All observed Bragg peaks are again in accordance with Cu11In9, CuIn and In0.
HAADF-STEM images and EDXS line profiles measured across a single In–Cu nanoparticle with a diameter of 39 nm further elucidate the nanoparticle architecture (Fig. 4d). Quantification of In and Cu concentrations from EDX spectra was performed using the Cu–K and In–L series (Fig. 4e). Thus, a ∼2 nm thin outer shell of In0 can be deduced from the concentration profile. A more detailed composition analysis (see section 2.4) indicates the presence of a second shell, 3–4 nm in diameter, with an average composition Cu0.53(9)In0.47(9) that corresponds approximately to the CuIn phase. These two shells surround a nanoparticle core with a diameter of 27–29 nm. This particle core exhibits the composition Cu0.64(15)In0.36(15), which can be assigned to Cu11In9, as already suggested by the diffractogram (Fig. 4c).
Based on the analysis of single nanoparticles (HRTEM, HAADF-STEM, EDXS composition profiles) and based on the analysis of a statistically relevant number of particles in a powder sample (STEM, XRD, SAED), the complex composition and structure of Cu11In9@CuIn@In core@shell-A@shell-B nanoparticles is reliably evidenced. The formation of this advanced architecture can be ascribed to a diffusion of copper into the initially prepared In0 precursor nanoparticles. While the synthesis of Cu11In9@CuIn@In was performed at low temperature (50 °C), an equilibration of the system to form CuIn as a single phase is obviously suppressed. In contrast, Cu11In9 was formed as the most stable intermetallic compound under these conditions. This is even more surprising since Cu11In9 as the bulk phase is typically formed at temperatures above 150 °C.33 After a certain reduction of the copper concentration, CuIn becomes the more stable compound and is formed next. After the reduction of all the copper—with an overall In:Cu ratio of 1:1—excess In0 remains. While In0 represents the most mobile and the least stable compound, it forms the outer shell of the nanoparticle architecture. The obtained elaborate composition and structure is first observed and can be highly relevant to catalysis as well as a precursor for CIS solar cells.34
Similar to the Cu11In9@CuIn@In core@shell-A@shell-B nanoparticles that were yielded via the follow-up approach (Fig. 4), the composition, structure and morphology of the here obtained particle architecture were validated based on advanced electron microscopy (BF-STEM, HAADF-STEM, HRTEM), detailed analysis of X-ray and electron diffraction data and concentration profiles measured via EDXS line scans (Fig. S3, ESI†). Again, a complex Cu11In9@CuIn@In core@shell-A@shell-B structure was validated. While being similar to the results of the follow-up approach, these data are discussed in detail in the ESI.†
3.4 The system In–Ag
![In–Ag Janushead-like nanoparticles (follow-up approach): (a) BF-STEM overview image; (b) typical HRTEM image; (c and d) experimental diffractograms of left-hand/right-hand particle side (of dashed area in 5b) as well as calculated diffraction pattern of cubic bulk-Ag0 ([110] zone axis, white circles) and of tetragonal bulk-In0 ([102] zone axis, white circles); (e) HAADF-STEM image; (f) spot EDX spectra (of blue/orange spots in 5e); (g) indexed SAED pattern of nanoparticle ensemble; (h) XRD pattern; (i) size distribution according to statistical evaluation of STEM images.](/image/article/2012/RA/c2ra21659k/c2ra21659k-f5.gif) | ||
| Fig. 5 In–Ag Janushead-like nanoparticles (follow-up approach): (a) BF-STEM overview image; (b) typical HRTEM image; (c and d) experimental diffractograms of left-hand/right-hand particle side (of dashed area in 5b) as well as calculated diffraction pattern of cubic bulk-Ag0 ([110] zone axis, white circles) and of tetragonal bulk-In0 ([102] zone axis, white circles); (e) HAADF-STEM image; (f) spot EDX spectra (of blue/orange spots in 5e); (g) indexed SAED pattern of nanoparticle ensemble; (h) XRD pattern; (i) size distribution according to statistical evaluation of STEM images. | ||
The composition of the two separate regions was further analyzed by EDXS of a single particle with a probe size of 1 nm at the positions indicated by the orange and blue dot in the HAADF-STEM image (Fig. 5e). Here, the EDX spectra taken in the Ag-rich region (right-hand side, orange dot) indeed show Ag–L lines with high intensity, whereas In–L lines only had low intensity (Fig. 5f). Quantification yielded 92(2) at% Ag and 8(1) at% In for the particle side showing a bright contrast (Fig. 5e). EDX spectra recorded in the In-rich side (left-hand side, blue dot), in contrast, show Ag–L lines with low intensity, and instead the In–L series with high intensity (Fig. 5f), resulting in 4(1) at% Ag and 96(2) at% In (Fig. 5e). This again evidences a “Janushead-like” architecture with silver and indium as opposite “particle faces”. The bimetallic structure of the In–Ag nanoparticles is further confirmed by XRD and SAED. Thus, XRD patterns indicate the presence of Ag0 (a = 405.9(5) pm) and In0 (a = 325.3(1), c = 493.7(1) pm) with calculated lattice parameters that are well in agreement with the reference data for cubic bulk-Ag0 and tetragonal bulk-In0 (Fig. 5h). Similarly, the Bragg reflections in SAED patterns can be attributed to the presence of Ag0 and In0 (Fig. 5g).
Qualitatively, the formation of the Janushead-like In–Ag nanoparticles can already be followed by the naked eye in the course of the reaction. Hence, the pristine grey suspension of In0 precursor nanoparticles turned to a characteristic dark-brown subsequent to the addition of AgNO3. Quantification via UV-Vis spectroscopy evidences the characteristic surface-plasmon resonance of silver (Fig. S2, ESI†). Thus, the absorption maximum appears at 389 nm, which is in good agreement with literature data referring to values of around 400 nm for nanoscaled silver of similar size.35 A certain blue-shift of the plasmon resonance can be attributed to the electronic structure of Ag0, which is influenced here by intimate contact with In0.36 While the less-noble indium will naturally transfer electrons to silver, such blue-shift is to be expected for bimetallic Janushead-like architecture.23c In addition, the oleylamine capping causes a strong absorption at around 230 nm that overlaps the surface-plasmon resonance of In0 (see section 3.2).
In summary, a Janushead-like architecture with In0 and Ag0 as opposite “faces” is obtained. The asymmetric structure of the nanoparticles can be explained by formation of a first silver seed crystal on the surface of the In0 precursor nanoparticles, right at the beginning of the reduction of Ag+. While the reduction of AgNO3 proceeds, the growth of this first Ag0 seed crystal is obviously favored, as compared to the formation of additional seeds on the indium surface. This finding is in accordance with the fact that alloying of pre-formed indium and silver normally requires elevated temperatures (e.g. furnace, arc-melting techniques, microwave synthesis).37 As a consequence of the proceeding growth of the once formed silver seed, bimetallic Janushead-like nanoparticles are finally obtained. When comparing the diameter of the initial In0 (12 nm) and the diameter of In0 remaining in the bimetallic In–Ag nanoparticles (5 nm), at least a partial dissolution of In0 becomes obvious. This finding can be attributed to the nanosized bimetal, which also represents an electrochemical local cell. Altogether, heterostructured bimetallic nanoparticles with diameters <50 nm are rare and limited to, for instance, Au–Pt heterodimers38 or Au–Ag heterometallic nanorods and icosahedra.39 A Janushead-like In–Ag architecture is first described.
According to BF-STEM and TEM overview images the nanoparticles exhibit an average diameter of 11(2) nm, which is very similar to the initial In0 nanoparticles (12 nm) (Fig. 6a,b,h). The nanoparticles exhibit a ring-type contrast, indicating the presence of an inner cavity (Fig. 6a,b). HRTEM images evidence the formation of nanoscale hollow spheres with an outer diameter of 18 nm and a wall thickness of 7 nm (Fig. 6c). The crystallinity and composition of the sphere wall are confirmed as elemental silver with lattice distances of 237(14) pm (cubic bulk-Ag0 d111: 236 pm).41 Some hollow spheres, however, seem to be not completely empty, but to exhibit a certain inner structure. Note furthermore that the XRD pattern indicates the presence of In0 in addition to Ag0 (Fig. 6g). Here, the calculated lattice parameters with a = 408.9(9) pm, and a = 326.9(9), c = 491.9(14) pm are in agreement with cubic bulk-Ag0 and tetragonal bulk-In0. Moreover, all Bragg reflections in SAED patterns can be also attributed to In0 and Ag0 (Fig. 6f).
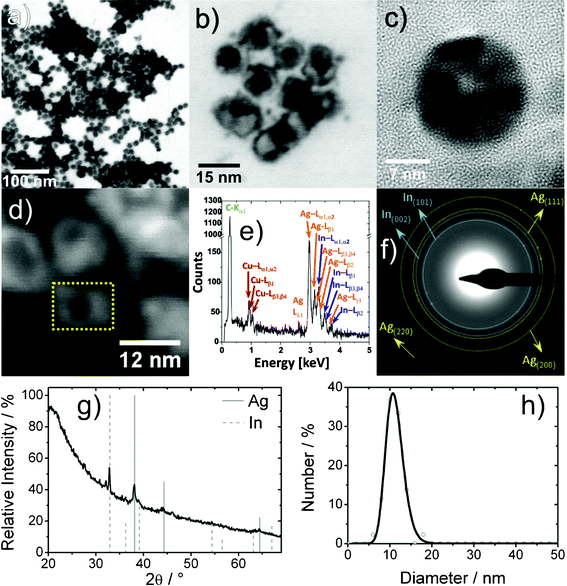 | ||
| Fig. 6 Ag hollow spheres (follow-up approach): (a) low-energy BF-STEM overview image; (b) BF-STEM detail image; (c) HRTEM image; (d) HAADF-STEM image; (e) area EDX spectrum (of dashed area in 6d); (f) indexed SAED pattern of nanoparticle ensemble; (g) XRD pattern; (h) size distribution according to statistical evaluation of STEM images. | ||
To verify the chemical composition, EDX spectra were recorded from single Ag0 hollow spheres at 30 keV primary electron energy by scanning a selected rectangular area for 120 s (Fig. 6d). Such an EDX spectrum reveals the characteristic L-lines of silver and indium as well as the lines of carbon (Kα1) and copper (L-lines) from the supporting film and the grid (Fig. 6e). Based on the observed Ag–L and In–L series, the chemical composition can be quantified to 68(7) at% Ag and 33(3) at% In. Obviously, non-consumed indium remains inside the silver shell, at least for some hollow spheres. The limited diffusibility of In3+ through the growing silver shell can be regarded as a kinetic reason for this residual In0.
Size and shape are well known to influence the surface-plasmon resonance, and thereby the optical properties of metal nanoparticles.42 In this regard, “hollowing” of silver or gold nanospheres typically leads to a red-shifted absorption maximum of the surface-plasmon resonance.43 This effect is observed for Ag0 hollow spheres also (Fig. S2, ESI†). UV-Vis spectra show the absorption maximum at 423 nm, as compared to 400 nm for massive Ag0 nanoparticles of similar size.44 Besides the surface-plasmon resonance, a strong absorption is observed at around 210 nm, which can be attributed to surface-bound DEG and polyvinylpyrrolidone (PVP) on the silver hollow spheres. These capping agents are also responsible for the comparatively low intensity of the plasmon-resonance absorption. Note finally that the color of the suspension of the Ag0 hollow spheres is significantly different from the In0 precursor nanoparticles, the In–Ag Janushead-like nanoparticles and the Ag3In@In core@shell nanoparticles (Fig. 2, Fig. S2, ESI†). This again evidences the different architecture of the nanoparticles obtained in the In–Ag system.
:Ag ratio of 1:1 results in the formation of Ag3In@In core–shell nanoparticles (Table 1, Fig. 2). Again, uniform and spherical particles are obtained with a mean size of 9(3) nm (Fig. 7a,k). While the follow-up approach did not lead to any intermetallic phase, the co-reduction of In3+ and Ag+ gives access to Ag3In. XRD patterns confirm the presence of a hexagonal (h) Ag3In as well as a cubic (c) Ag3In phase (Fig. 7j). The resulting lattice parameters of a = 301.3(4), c = 465.2(6) pm (bulk h-Ag3In: a = 299.0, c = 480.5 pm)45 and a = 409.9(4) pm (bulk c-Ag3In: a = 414(9) pm) are well in agreement with the literature data. Note that Bragg peaks of In0 are not observed.
![h-/c-Ag3In@In nanoparticles (co-reduction approach): (a) low-energy BF-STEM overview image; (b) HRTEM image of h-Ag3In@In nanoparticle; (c) experimental diffractogram of the inner particle region (of dashed area in 7b) and calculated indexed diffraction pattern of bulk h-Ag3In ([100] zone axis, white circles); (d) indexed SAED pattern of nanoparticle ensemble; (e) HRTEM image of c-Ag3In@In nanoparticle; (f) experimental diffractogram of the inner particle region (of dashed area in 7e) and calculated indexed diffraction pattern of bulk c-Ag3In ([110] zone axis, white circles); (g) typical HAADF-STEM image; (h) EDXS line scan (of yellow line in 7g); (i) 3D reconstruction of nanoparticle from composition profile; (j) XRD pattern; (k) size distribution according to statistical evaluation of STEM images.](/image/article/2012/RA/c2ra21659k/c2ra21659k-f7.gif) | ||
| Fig. 7 h-/c-Ag3In@In nanoparticles (co-reduction approach): (a) low-energy BF-STEM overview image; (b) HRTEM image of h-Ag3In@In nanoparticle; (c) experimental diffractogram of the inner particle region (of dashed area in 7b) and calculated indexed diffraction pattern of bulk h-Ag3In ([100] zone axis, white circles); (d) indexed SAED pattern of nanoparticle ensemble; (e) HRTEM image of c-Ag3In@In nanoparticle; (f) experimental diffractogram of the inner particle region (of dashed area in 7e) and calculated indexed diffraction pattern of bulk c-Ag3In ([110] zone axis, white circles); (g) typical HAADF-STEM image; (h) EDXS line scan (of yellow line in 7g); (i) 3D reconstruction of nanoparticle from composition profile; (j) XRD pattern; (k) size distribution according to statistical evaluation of STEM images. | ||
The HRTEM and HAADF images show evidence of a core–shell structure for the obtained In–Ag nanoparticles (Fig. 7b,e,g). Here, the particle core represents either a h-Ag3In monocrystal oriented along the [100] zone axis (Fig. 7c) or a c-Ag3In monocrystal with a [110] zone axis (Fig. 7f).45 In contrast, the particle shell does not show any lattice fringes. This again points to an amorphous or quasi-molten shell similar to the In0 precursor nanoparticles (see section 3.2) as well as to Cu11In9@CuIn@In (see section 3.3) and Au@AuIn2@In (see section 3.5) nanoparticles. This view is confirmed by electron diffraction patterns (Fig. 7d). Here, all Bragg reflections can be rationalized based on the presence of h-/c-Ag3In as well as of In0.
Silver and indium concentration profiles were measured via EDXS line scans across a single nanoparticle with a diameter of 31 nm (Fig. 7g). Quantification was performed by using the Ag–L and In–L series and confirms the presence of an In-rich shell, about 5 nm in diameter (Fig. 7h,i). The outer 1–2 nm of this shell is composed of pure In0, whereas the remaining 3–4 nm of the shell has an average composition of In0.87(5)Ag0.13(5) (see section 2.4). However, the absence of any well-defined In–Ag phases around this composition suggests that the whole nanoparticle shell is indium-rich with an average composition of In0.92(4)Ag0.08(4) (Fig. 7h,i). In addition, the particle core has a diameter of 21–23 nm and a chemical composition of Ag0.71(8)In0.29(8), which can be assigned to Ag3In, as already suggested by the diffractograms (Fig. 7c,f). As expected, no compositional difference can be detected in view of an inner core of h- and c-Ag3In.
In summary, all applied analytical tools (BF-STEM, HRTEM, SAED, HAADF-STEM, EDXS) suggest that a h-/c-Ag3In@In core@shell structure is obtained. The absence of diffraction lines from In0 in the XRD pattern can again be attributed to an amorphous or quasi-molten outer particle shell (see sections 3.2, 3.4 and 3.5). The dark yellowish brown color of the h-/c-Ag3In@In suspension was finally quantified by UV-Vis measurements (Fig. S2, ESI†). Besides the strong absorption of oleylamine as a capping (around 230 nm), a comparatively weak absorption is observed with a maximum at 363 nm. Taking a silver-rich intermetallic compound such as Ag3In into account, a plasmon-resonance absorption in the region of elemental silver (around 400 nm) is within expectations, and well-known for silver-rich Au–Ag alloys.7a,42a The obvious blue shift can be explained by the electronic influence of the less-noble indium (see section 3.4). To our knowledge Ag3In has not yet been prepared as a nanomaterial; comparison of the optical properties to a nanoscaled reference material is therefore not possible.
3.5 The system In–Au
![Au@AuIn2@In nanoparticles (follow-up approach): (a) low-energy BF-STEM overview image; (b) typical HRTEM image; (c) experimental diffractogram of inner particle region (of dashed area in 8b) and calculated indexed diffraction pattern of cubic bulk-Au0 ([110] zone axis, white circles); (d) HAADF-STEM image; (e) EDXS line scan (of yellow line in 8d); (f) 3D reconstruction of nanoparticle from composition profile; (g) indexed SAED pattern of nanoparticle ensemble; (h) XRD pattern; (i) size distribution according to statistical evaluation of STEM images.](/image/article/2012/RA/c2ra21659k/c2ra21659k-f8.gif) | ||
| Fig. 8 Au@AuIn2@In nanoparticles (follow-up approach): (a) low-energy BF-STEM overview image; (b) typical HRTEM image; (c) experimental diffractogram of inner particle region (of dashed area in 8b) and calculated indexed diffraction pattern of cubic bulk-Au0 ([110] zone axis, white circles); (d) HAADF-STEM image; (e) EDXS line scan (of yellow line in 8d); (f) 3D reconstruction of nanoparticle from composition profile; (g) indexed SAED pattern of nanoparticle ensemble; (h) XRD pattern; (i) size distribution according to statistical evaluation of STEM images. | ||
The architecture of the In–Au nanoparticles is further confirmed by SAED and XRD. All Bragg reflections observed in SAED patterns can be attributed to AuIn2, Au0 and In0 (Fig. 8g). While HRTEM and SAED evidence the structure and composition of single particles, the XRD patterns confirm the presence of AuIn2 and Au0 for a statistically relevant number of particles in a powder sample (Fig. 8h). The calculated lattice parameters of a = 408.6(1) pm (cubic Au) and a = 650.1(2) pm (cubic AuIn2) are in good agreement with the bulk references.46,47 Note that Bragg reflections of In0 were not observed.
Typical HAADF images of the In–Au nanoparticles support their core–shell structure with a clear contrast between its shell and core (Fig. 8d). Concentration profiles from EDXS line scans across a single nanoparticle, about 24 nm in diameter, are quantified based on Au–L and In–L series (Fig. 8e). Accordingly, an outer shell of 1–2 nm is composed of In0, followed by a second shell of 2–3 nm with a composition Au0.27(4)In0.73(5) (see section 2.4). The latter corresponds well with AuIn2. Finally, the particle core with a diameter of 14–18 nm is composed of Au0, as already suggested by the diffractogram. In summary, the above considerations and analytical tools confirm an Au@AuIn2@In core@shell-A@shell-B architecture as the result of the follow-up approach (Fig. 8f).
Optically, the Au@AuIn2@In nanoparticles exhibit a weak absorption around 539 nm (Fig. S2, ESI†) that refers to the characteristic surface-plasmon resonance of Au0 nanoparticles (typically positioned at around 520 nm). While considering Au0 as the inner core of the nanoparticles, the weak absorption might point to a small number of particles with a comparably thin shell. An appreciable number of “naked” Au0 nanoparticles in the sample would, in contrast, lead to a much more pronounced characteristic absorption, as was indeed found in reference experiments.
The formation of the Au@AuIn2@In particle architecture can be ascribed to a diffusion of as-reduced Au0 atoms into the initial In0 nanoparticles. A similar observation was described previously by Mori et al., based on indium and gold nanoparticles synthesized via evaporation techniques inside of a TEM.48 While pure gold is obviously the most stable phase under the conditions of the follow-up approach, Au0 represents the inner particle core. As the concentration of gold decreases over the course of the reaction, the intermetallic phase AuIn2 is formed next as a shell. With regard to the introduced In:Au ratio of 1:1, residual In0 remains as a thin outer shell on the nanoparticles. As discussed for all other nanostructures presented here, a precise adjustment of the experimental conditions turned out to be crucial in controlling the formation of one or the other phase composition.
BF-STEM images show spherical nanoparticles with a mean diameter of 4(2) nm (Fig. 9a,h). XRD patterns evidence the formation of the intermetallic AuIn2 phase (Fig. 9g). From the diffraction line positions, a lattice parameter of a = 648.9(10) pm was calculated that is in agreement with cubic bulk-AuIn2.47 Note that no Bragg reflections of In0 or Au0 were observed. The structure of the In–Au nanoparticles is further validated by the HRTEM images (Fig. 9b). Herein, the whole particle turned out to be crystalline AuIn2, as demonstrated by the excellent agreement between its diffractogram and the calculated diffraction pattern of cubic bulk-AuIn2 oriented along the [111] zone axis (Fig. 9c). SAED further confirms the purity of AuIn2 (Fig. 9f). HAADF images of the nanoparticles do not suggest the presence of any outer shell here (Fig. 9d). EDX spectra were recorded in a rectangular area of a single AuIn2 nanoparticle (Fig. 9e) and reveal the characteristic lines of gold (M-series) and indium (L-series). Moreover, the lines of carbon (Kα1) and copper (L-series) from the supporting film and grid are observed. As a result, the In and Au content were quantified to Au35(9)In65(6), which fits well with the intermetallic phase AuIn2.
![AuIn2 nanoparticles (co-reduction approach): (a) low-energy BF-STEM overview image; (b) typical HRTEM image; (c) experimental diffractogram of inner particle region (of dashed area in 9b) and calculated indexed diffraction pattern of cubic bulk-AuIn2 ([111] zone axis, white circles); (d) HAADF-STEM image; (e) area EDX spectrum (of dashed area in 9d); (f) indexed SAED pattern of nanoparticle ensemble; (g) XRD pattern; (h) size distribution according to statistical evaluation of STEM images.](/image/article/2012/RA/c2ra21659k/c2ra21659k-f9.gif) | ||
| Fig. 9 AuIn2 nanoparticles (co-reduction approach): (a) low-energy BF-STEM overview image; (b) typical HRTEM image; (c) experimental diffractogram of inner particle region (of dashed area in 9b) and calculated indexed diffraction pattern of cubic bulk-AuIn2 ([111] zone axis, white circles); (d) HAADF-STEM image; (e) area EDX spectrum (of dashed area in 9d); (f) indexed SAED pattern of nanoparticle ensemble; (g) XRD pattern; (h) size distribution according to statistical evaluation of STEM images. | ||
4. Conclusions
Follow-up approach (i.e. reaction of pre-synthesized In0 precursor nanoparticles with Cu2+, Ag+ or Au3+) and co-reduction approach (i.e. simultaneous reduction of In3+ together with Cu2+, Ag+ or Au3+) in a micellar system results in a wide range of different compositions, structures and morphologies. This includes Cu11In9@CuIn@In core@shell-A@shell-B nanoparticles, In–Ag Janushead-like nanoparticles, Ag hollow spheres, Ag3In@In core@shell nanoparticles, Au@AuIn2@In core@shell-A@shell-B nanoparticles and AuIn2 nanoparticles. Due to the spatially limited volume of the micelles, particle nucleation and growth are very much limited, and lead to nanoparticles in a narrow size range. The elaborate architecture of the nanoparticles is firstly described and requires advanced electron microscopic techniques for reliable characterization (e.g. BF-/HAADF-STEM at 300 keV and energies ≤30 keV, HRTEM, SAED, spot/line-scan/area EDXS). In addition to the analysis of selected individual nanoparticles, the results were validated based on the analysis of powder samples containing a statistically relevant number of nanoparticles (STEM, XRD, UV-Vis).The formation of the different particle architectures can be attributed, first of all, to the low reaction temperature (≤50 °C). As a consequence, the syntheses are performed under kinetic control, allowing a fine-tuning of the experimental conditions and the resulting nanostructures. Depending on the interdiffusion of the relevant metals and the thermodynamics of alloy formation, nanoscaled intermetallic compounds or non-alloyed bimetallic nanoparticles, including core–shell and Janushead-like structures, are obtained. In general, the follow-up approach favors the formation of core–shell structures, while the co-reduction approach favors the formation of intermetallic compounds. Whereas the composition and architecture of the obtained nanoparticles are significantly different, the very similar experimental conditions (type of microemulsion, concentration of reactants, temperature, etc.) guarantee a very similar size range 5–15 nm of all obtained nanostructures.
Based on the variety of compositions, structures and morphologies that can be obtained via the follow-up and co-reduction approach for the systems In–Cu, In–Ag and In–Au, many more compounds and elaborate architectures can be expected from extending the synthesis and its flexibility toward other metals. The resulting tool kit of new architectures of bimetallic and intermetallic nanostructures may also be relevant in view of new materials for catalysis, thin-film electronics or solar cells.
Acknowledgements
The authors are grateful to the Center for Functional Nanostructures (CFN) of the Deutsche Forschungsgemeinschaft (DFG) at the Karlsruhe Institute of Technology (KIT) for financial support (analytical tools and equipment). We also acknowledge the DFG for individual grant within the project FE 911/4-1.References
- C. J. Serpell, J. Cookson, D. Ozkaya and P. D. Beer, Nat. Chem., 2011, 3, 478 CAS.
- (a) C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS; (b) N. Toshima and T. Yonezawa, New J. Chem., 1998, 1179 RSC; (c) H. Goesmann and C. Feldmann, Angew. Chem., Int. Ed., 2010, 49, 1362 CrossRef CAS; (d) G. A. Ozin and A. C. Arsenault, Nanochemistry, RSC Publishing, Cambridge, 2005 Search PubMed.
- (a) U. Jeong, P. H. C. Camargo, Y. H. Lee and Y. Xia, J. Mater. Chem., 2006, 16, 3893 RSC; (b) Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711 CrossRef CAS; (c) Y. G. Sun and Y. N. Xia, Science, 2002, 298, 2176 CrossRef CAS; (d) Y. Vasquez, A. K. Sra and R. E. Schaak, J. Am. Chem. Soc., 2005, 127, 12504 CrossRef CAS; (e) J. M. Nedeljković, O. I. Mićić, S. P. Ahrenkiel, A. Miedaner and A. J. Nozik, J. Am. Chem. Soc., 2004, 126, 2632 CrossRef; (f) Z. Bai, L. Yang, J. Zhang, L. Li, J. Lv, C. Hu and J. Zhou, Catal. Commun., 2010, 11, 919 CrossRef CAS.
- C. Suryanarayana, E. Ivanov and V. V. Boldyrev, Mater. Sci. Eng. A, 2001, 151, 304 Search PubMed.
- E. Zintl, A. Harder and W. Haucke, Z. Phys. Chem., 1937, 35, 354 Search PubMed.
- (a) R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845 CrossRef CAS; (b) D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044 CrossRef CAS; (c) X. Liu and X. Liu, Angew. Chem., 2012, 124, 3367 CrossRef; (d) M. Boutonnet, S. Lögdberg and E. E. Svensson, Curr. Opin. Colloid Interface Sci., 2008, 13, 270 CrossRef CAS; (e) M. P. Pileni, Nat. Mater., 2003, 2, 145 CrossRef CAS.
- (a) S. Liu, G. Chen, P. N. Prasad and M. T. Swihart, Chem. Mater., 2011, 23, 4098 CAS; (b) Y. Liu, M. Chi, V. Mazumder, K. L. More, S. Soled, J. D. Henao and S. Sun, Chem. Mater., 2011, 23, 4199 CAS; (c) Y. Liu and A. R. Walker, Angew. Chem., Int. Ed., 2010, 49, 6781 CrossRef CAS; (d) M. L. Wu, D. H. Chen and T. C. Huang, Langmuir, 2001, 17, 3877 CrossRef CAS.
- (a) S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989 CrossRef CAS; (b) K. M. Hyie and I. I. Yaacob, J. Mater. Process. Technol., 2007, 191, 48 CrossRef CAS.
- (a) R. E. Cable and R. E. Schaak, J. Am. Chem. Soc., 2006, 128, 9588 CrossRef CAS; (b) Z. Liu, D. Reed, G. Kwon, M. Shamsuzzoha and D. E. Nikles, J. Phys. Chem. C, 2007, 111, 14223 CrossRef CAS; (c) Z. Liu, G. S. Jackson and B. W. Eichhorn, Angew. Chem., Int. Ed., 2010, 49, 3173 CrossRef CAS; (d) Y. Liu, D. Li, V. R. Stamenkovic, S. Soled, J. D. Henao and S. Sun, ACS Catal., 2011, 1, 1719 CrossRef CAS; (e) L. M. Magno, W. Sigle, P. van Aken, D. Angelescu and C. Stubenrauch, Phys. Chem. Chem. Phys., 2011, 13, 9134 RSC.
- (a) B. J. Auten, B. P. Hahn, G. Vijayaraghavan, K. J. Stevenson and B. D. Chandler, J. Phys. Chem. C, 2008, 112, 5365 CrossRef CAS; (b) S. Zhou, Z. Ma, H. Yin, Z. Wu, B. Eichhorn, S. H. Overbury and S. Dai, J. Phys. Chem. C, 2009, 113, 5758 CrossRef CAS.
- W. Wang, Q. Peng and Y. Li, Nano Res., 2010, 3, 574 CrossRef.
- N. H. Chou and R. E. Schaak, J. Am. Chem. Soc., 2007, 129, 7339 CrossRef CAS.
- A. K. Sra and R. E. Schaak, J. Am. Chem. Soc., 2004, 126, 6667 CrossRef CAS.
- (a) W. Chen, R. Yu, L. Li, A. Wang, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2010, 49, 2917 CrossRef CAS; (b) R. E. Schaak, A. K. Sra, B. M. Leonard, R. E. Cable, J. C. Bauer, Y.-F. Han, J. Means, W. Teizer, Y. Vasquez and E. S. Funck, J. Am. Chem. Soc., 2005, 127, 3506 CrossRef CAS.
- M. Armbrüster, G. Wowsnick, M. Friedrich, M. Heggen and R. Cardoso-Gil, J. Am. Chem. Soc., 2011, 133, 9112 CrossRef.
- (a) Q. Zhang, J. Xie, Y. Yu and J. Y. Lee, Nanoscale, 2010, 2, 1962 RSC; (b) Y. Okuno, K. Nishioka, A. Kiya, N. Nakashima, A. Ishibashi and Y. Niidome, Nanoscale, 2010, 2, 1489 RSC.
- V. Mazumder, M. Chi, K. L. More and S. Sun, Angew. Chem., Int. Ed., 2010, 49, 9368 CrossRef CAS.
- S. Patra and H. Yang, Bull. Korean Chem. Soc., 2009, 30, 1485 CrossRef CAS.
- S. Hakomori, J. Am. Chem. Soc., 1930, 52, 2372 CrossRef CAS.
- (a) M. P. Pileni, J. Phys. Chem., 1993, 97, 6961 CrossRef CAS; (b) D. H. M. Buchold and C. Feldmann, Adv. Funct. Mater., 2008, 18, 1002 CrossRef CAS.
- X. L. Wang, W. Q. Han, J. Chen and J. Graetz, ACS Appl. Mater. Interfaces, 2010, 2, 1548 CAS.
- M. G. Blaber, M. D. Arnold and M. J. Ford, J. Phys.: Condens. Matter, 2010, 22, 143201 CrossRef CAS.
- (a) A. Elattar, T. Takeshita, W. E. Wallace and R. S. Craig, Science, 1977, 196, 1093 CAS; (b) H. Abe, F. Matsumoto, L. R. Alden, S. C. Warren, H. D. Abruña and F. J. Di Salvo, J. Am. Chem. Soc., 2008, 130, 5452 CrossRef CAS; (c) T. Yonezawa and N. Toshima, J. Mol. Catal., 1993, 83, 167 CrossRef CAS.
- (a) M. P. Pileni, Cryst. Res. Technol., 1998, 7–8, 1155 CrossRef; (b) C. Destrée and J. B. Nagy, Adv. Colloid Interface Sci., 2006, 123–126, 353 CrossRef; (c) J. Eastoe, M. J. Hollamby and L. Hudson, Adv. Colloid Interface Sci., 2006, 128–130, 5 CrossRef CAS.
- (a) B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893 CrossRef CAS; (b) I. Capek, Adv. Colloid Interface Sci., 2004, 110, 49 CrossRef CAS.
- C. Kind and C. Feldmann, Chem. Mater., 2011, 23, 4982 CrossRef CAS.
- (a) G. L. Allen, R. A. Bayles, W. W. Gile and W. A. Jesser, Thin Solid Films, 1986, 144, 297 CrossRef CAS; (b) D. Xie, M. P. Wang, W. H. Qi and L. F. Cao, Mater. Chem. Phys., 2006, 96, 418 CrossRef CAS; (c) T. H. Lim, B. Ingham, K. H. Kamarudin, P. G. Etchegoin and R. D. Tilley, Cryst. Growth Des., 2010, 10, 3854 CrossRef CAS.
- (a) F. Fievet, J. P. Lagier and M. Figlarz, Mater. Res. Bull., 1989, 29 CAS; (b) A. Luz and C. Feldmann, J. Mater. Chem., 2009, 19, 8107 RSC.
- (a) P. Singh, S. Kumar, A. Katyal, R. Kalra and R. Chandra, Mater. Lett., 2008, 62, 4164 CrossRef CAS; (b) N. H. Chou, X. Ke, P. Schiffer and R. E. Schaak, J. Am. Chem. Soc., 2008, 130, 8140 CrossRef CAS; (c) P. K. Khanna, K.-W. Jun, K. B. Hong, J.-O. Baeg, R. C. Chikate and B. K. Das, Mater. Lett., 2005, 59, 1032 CrossRef CAS; (d) S. Cingarapu, Z. Yang, C. M. Sorensen and K. J. Klabunde, Inorg. Chem., 2011, 50, 5000 CrossRef CAS.
- (a) T. P. Rajasekharan and K. Schubert, Z. Metallkd., 1981, 72, 275 CAS; (b) V. Simić and Ž. Marinković, J. Less Common Met., 1980, 72, 133 CrossRef.
- M. E. Straumanis, P. B. Rao and W. J. James, Z. Metallkd., 1971, 62, 493 CAS.
- (a) P. K. Khanna, S. Gaikwad, P. V. Adhyapak, N. Singh and R. Marimuthu, Mater. Lett., 2007, 61, 4711 CrossRef CAS; (b) M. Singh, I. Sinha, M. Premkumar, A. K. Singh and R. K. Mandal, Colloids Surf., A, 2010, 359, 88 CrossRef CAS; (c) C. Kind, A. Weber and C. Feldmann, J. Mater. Chem., 2012, 22, 987 RSC.
- H. Okamoto, J. Phase Equilib., 2005, 26, 645 Search PubMed.
- C. Kind, C. Feldmann, A. Quintilla and E. Ahlswede, Chem. Mater., 2011, 23, 5269 CrossRef CAS.
- (a) C. Petit, P. Lixon and M. P. Pileni, J. Phys. Chem., 1993, 97, 12974 CrossRef CAS; (b) A. Slistan-Grijalva, R. Herrera-Urbina, J. F. Rivas-Silva, M. Ávalos-Borja, F. F. Castillón-Barraza and A. Posada-Amarillas, Phys. E., 2005, 27, 104 CrossRef CAS.
- P. Mulvaney, Langmuir, 1996, 12, 788 CrossRef CAS.
- J. W. Lekse, T. J. Stagger and J. A. Aitken, Chem. Mater., 2007, 19, 3601 CrossRef CAS.
- S. I. Lim, M. Varon, I. Ojea-Jiménez, J. Arbiol and V. Puntes, J. Mater. Chem., 2011, 21, 11518 RSC.
- M. R. Langille, J. Zhang and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 3543 CrossRef CAS.
- (a) Y. Sun and Y. Xia, Anal. Chem., 2002, 74, 5297 CrossRef CAS; (b) Y. Sun, B. Mayers and Y. Xia, Adv. Mater., 2003, 15, 641 CrossRef CAS; (c) X. Lu, H.-Y. Tuan, J. Chen, Z.-Y. Li, B. A. Korgel and Y. Xia, J. Am. Chem. Soc., 2007, 129, 1733 CrossRef CAS.
- E. A. Owen and E. L. Yates, J. Chem. Phys., 1935, 3, 605 CrossRef.
- (a) Y. Sun and Y. Xia, Analyst, 2003, 128, 686 RSC; (b) E. Hutter and J. H. Fendler, Adv. Mater., 2004, 16, 1685 CrossRef CAS.
- (a) H. P. Liang, L. J. Wan, C. L. Bai and L. Jiang, J. Phys. Chem. B, 2005, 109, 7795 CrossRef CAS; (b) M. Gellner, B. Küstner and S. Schlücker, Vib. Spectrosc., 2009, 50, 43 CrossRef CAS.
- C. Kind, R. Popescu, E. Müller, D. Gerthsen and C. Feldmann, Nanoscale, 2010, 2, 2223 RSC.
- (a) A. N. Campbell and W. F. Reynolds, Can. J. Chem., 1962, 40, 37 CrossRef CAS; (b) A. N. Campbell, R. Wagemann and R. B. Ferguson, Can. J. Chem., 1970, 48, 1703 CrossRef CAS.
- J. J. Couderc, G. Garique, L. Lafourcade, Q. T. Nguyen and Z. Metallkd, Z. Metallkd., 1959, 50, 708 Search PubMed.
- O. Kubaschewski and Z. Elektrochem, Angew. Phys. Chem., 1938, 44, 870 CAS.
- H. Yasuda and H. Mori, Mater. Sci. Forum, 1999, 307, 167 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information regarding the starting materials and the analytical methods are described. Moreover, the analysis of the surface capping of the In0 precursor nanoparticles, the complete investigation of the surface-plasmon resonance absorption of all obtained nanoparticles as well as the complete analysis of the Cu11In9@CuIn@In core@shell-A@shell-B nanoparticles gained via the co-reduction approach can be found here. See DOI: 10.1039/c2ra21659k |
| This journal is © The Royal Society of Chemistry 2012 |