Dendrimers with tetraphenylsilane core and 9-phenylcarbazole-based dendrons: synthesis, photophysics, and electrochemical behavior†
Jia
You
,
Guiyang
Li
and
Zhonggang
Wang
*
State Key Laboratory of Fine Chemicals, Department of Polymer Science and Materials, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: zgwang@dlut.edu.cn
First published on 30th August 2012
Abstract
Three generations of novel dendrimers (G1, G2 and G3) with a tetraphenylsilane core and 9-phenylcarbazole-based dendrons have been successfully synthesized. These dendrimers possess excellent thermal stability with 5% weight-loss temperatures in the range of 574 to 622 °C. Compared to the lower generation of dendrimer, G3 exhibits a high glass transition at 271 °C, indicating that the high content of 9-phenylcarbazole results in a dendrimer with significantly enhanced rigidity. The contorted dendrons effectively inhibit the aggregation of conjugative chromophore groups, leading to a fluorescence quantum yield of up to 99.1%. Moreover, all the dendrimers have high energy band gaps, a characteristic which is especially desirable for host materials in the fabrication of organic light-emitting devices.
Introduction
Dendrimers are monodispersed macromolecules which possess highly branched structures that can be precisely tailored with well-defined compositions and a high degree of order. The unique structures and properties provide an innovative opportunity in macromolecular chemistry and industrial applications in organic optoelectronic devices,1,2 drug delivery,3–6 multivalent bioconjugates and multivalent diagnostics for magnetic resonance imaging,7,8 efficient light-harvesting antennae,9,10 homogeneous catalysis11,12 and templates for the growth of single-wall carbon nanotubes.13As organic light-emitting materials, the properties of a dendrimer are tightly related to the generation number, chemical structure of the dendrons and stereo-configuration of the core. In previous reports, 1,3,5 or 1,2,4,5-substituted benzene have been utilized as cores to construct dendrimers.14,15 However, the planar 2D structure is prone to yielding π–π stacking between the cores and dendrons, leading to the occurrence of energy wastage for charge transfer and nonradiative decay. In this regard, a rigid compound with a tetrahedral stereo-configuration is favorable as a core monomer. The functional groups on the vertices direct four identical dendrons to stretch out to form a dendritic 3D architecture, which can prevent the effective packing of dendrimer molecules. On the other hand, in order to optimize the performance it is desirable that the dendrons of dendrimers are bulky, twisted and stiff so that the intercalation of dendrons is reduced or eliminated to a larger extent in order to suppress the aggregation and/or excimer formation.
Hence, we synthesized a series of novel dendrimers by introducing a 9-phenylcarbazole-based dendron and tetraphenylsilane core. The noteworthy features of a tetraphenylsilane core are its robust tetrahedral configuration and high PL efficiency due to the quantum confinement effects arising from the three-dimensional hindered structures.16–19 Carbazole and its derivatives have been widely employed as functional building blocks in the preparation of organic photoconductors, nonlinear optical and organic light emitting materials due to their electron-donating capability.20–23 Therefore, the combination of carbazole and tetraphenylsilane is expected to offer the dendrimer high luminescence efficiency. Moreover, owing to the interruption of the π-conjugated system by the Si atom and the varied distortion angle of the dendrons with the generational growth, we also suppose that the as-designed dendrimers would present an efficient hole-transport ability, a high triplet energy level and tunable energy gaps.
Experimental
Materials and instrumentation
Dibromobenzene, carbazole, tetraki(triphenylphosphine)palladium, n-BuLi and triisopropyl borate were purchased from Shanghai Regent Corporation Ltd and used without further purification. THF was freshly distilled from sodium–benzophenone prior to use. Other commercial reagents were of analytical grade and used without further purification unless otherwise specified. All chromatographic separations were carried out on silica gel (400 mesh).1H NMR spectra were record using a Varian INOVA at 400 MHz, with CDCl3 as the solvent and tetramethysilane as the internal standard. Gel permeation chromatography (GPC) analysis was conducted on a Water Associates Model PL-GPC-220 apparatus at room temperature with tetrahydrofuran (THF) as the eluent, at a flow rate of 1 mL min−1, calibrated with polystyrene standards. Molecular masses were determined by a MALDI micro MX laser desorption–ionization time-of-flight mass spectrometer (MALDI-TOF MS). Samples were analyzed using a THF solution of the sample (3 μL) mixed with 3 μL of a dithranol matrix (25 mg mL−l in THF) before being loaded onto a metal sample plate. Thermal gravimetric analyses (TGA) were performed on a NETZSCH TG209C in a nitrogen atmosphere at a heating rate of 10 °C min−1. Differential scanning calorimetries (DSC) were recorded on a NETZSCH DSC204 with a heating rate of 10 °C min−1 in a nitrogen atmosphere. Absorption and photoluminescence (PL) spectra were probed on a PTI-700 spectrophotometer and a HP854 luminescence spectrometer, respectively. Cyclic voltammetry (CV) curves were carried out on a BSA 100B electrochemical analyzer with a conventional three-electrode configuration consisting of a glassy carbon working electrode, a platinum auxiliary electrode and a non-aqueous Ag/AgNO3 reference electrode in a 0.1 M anhydrous acetonitrile solution of tetra-n-butylammonium hexafluorophosphate (nBu4NPF6) under nitrogen at room temperature. The dendrimer film deposited on the glassy carbon was used as the working electrode, while the counter electrode was a platinum wire and a Ag/Ag+ electrode was used as the reference electrode, which was calibrated using the ferrocene/ferrocenium (Fc/Fc+) redox couple (4.8 eV below the vacuum level).
The synthesis procedures for the G1–G3 dendrons G1–B(OH)2, G2–B(OH)2 and G3–B(OH)2 are described in the ESI.†
Synthesis of G1, G2, G3
The synthesis procedure of the G1 dendrimer is described as follows: G1–B(OH)2 (0.35 g, 12 mmol), tetrakis(4-bromophenyl)silane (0.13 g, 0.2 mmol) and tetrakis(triphenylphosphine) palladium (0.0233 g, 0.02 mmol) were added to an air-free two-phase system composed of toluene (30 mL) and a 2 M K2CO3 aqueous solution (10 mL). The resulting mixture was intensively stirred under an argon atmosphere at 80 °C for 24 h. The organic layer was separated and the aqueous phase was extracted with diethyl ether. The organic layers were combined and washed with brine and dried over anhydrous MgSO4. The solvent was evaporated and the residue went through a silica-gel column to obtain a 0.22 g white solid in an 85% yield. G2 and G3 were synthesized in similar procedures with yields of 69% and 43%, respectively.G1: Elemental analysis: calculated for C96H64N4Si, C 88.58%, H 4.96%, N 4.30%; found, C 88.54%, H 4.99%, N 4.35%. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.17 (d, 8H, J = 8.0 Hz; type a Ph–H), 7.92–7.88 (m, 16H; type g Ph–H + type f Ph–H), 7.83 (d, 8H, J = 8.0 Hz; type h Ph–H), 7.69 (d, 8H, J = 8.0 Hz; type e Ph–H), 7.51 (d, 8H, J = 7.6 Hz; type d Ph–H), 7.44 (t, 8H, J = 7.6 Hz; type c Ph–H), 7.31 (t, 8H, J = 7.6 Hz; type b Ph–H), MALDI-TOF ms (m/z): Calcd for C96H64N4Si 1301.65, found 1301.38 [M+].
G2: Elemental analysis: calculated for C240H152N12Si, C 89.19%, H 4.74%, N 5.20%; found, C 89.24%, H 4.71%, N 5.15%. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.59 (s, 8H; type i Ph–H), 8.17 (d, 16H, J = 8.0 Hz; type a Ph–H), 8.03–7.97 (m, 32H; type m Ph–H + type f Ph–H + type k Ph–H), 7.91 (d, 8H, J = 6.8 Hz; type j Ph–H), 7.86–7.83 (m, 16H; type g Ph–H + type h Ph–H), 7.71–7.68 (m, 24H; type e Ph–H + type l Ph–H), 7.52 (d, 16H, J = 8.0 Hz; type d Ph–H), 7.45 (d, 16H, J = 7.4 Hz; type c Ph–H), 7.32 (t, 16H, J = 7.6 Hz; type b Ph–H). MALDI-TOF ms (m/z): Calcd for C240H152N12Si 3231.94, found 3231.09 [M+].
G3: Elemental analysis: calculated for C528H328N28Si, C 89.41%, H 4.66%, N 5.53%; found, C 89.46%, H 4.71%, N 5.49%. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.64 (s, 8H; type p Ph–H), 8.58 (s, 16H; type i Ph–H), 8.17 (d, 32H, J = 7.6 Hz; type a Ph–H), 8.07 (d, 16H, J = 7.2 Hz; type g Ph–H), 8.00–7.96 (m, 40H; type f Ph–H + type q Ph–H), 7.92–7.76 (m, 56H; type o Ph–H + type l Ph–H + type k Ph–H + type j Ph–H + type n Ph–H), 7.69–7.62 (m, 64H; type e Ph–H + type h Ph–H + type m Ph–H + type r Ph–H), 7.50 (d, 32H, J = 7.6 Hz; type d Ph–H), 7.42 (t, 32H, J = 7.6 Hz; type c Ph–H), 7.29 (t, 32H, J = 7.6 Hz; type b Ph–H). MALDI-TOF ms (m/z): Calcd for C528H328N28Si 7092.53, found 7096.24 [M+].
Results and discussion
Synthesis and characterization
The synthesis routes and chemical structures of the dendrimers G1, G2 and G3 are presented in Scheme 1 and 2, which were synthesized by Suzuki cross-coupling from the core monomer tetrakis(4-bromophenyl)silane and corresponding dendron GnB(OH)2 (n = 1, 2, 3). The reaction was carried out using a toluene–water system for 48 h in the presence of tetrakis(triphenylphosphine)palladium, K2CO3 and tricaprylylmethylammonium chloride (Aliquat 336). It is possible to yield small amounts of side products containing only one, two or three dendrons during the Suzuki coupling between tetrakis(4-bromophenyl)silane and Gn–B(OH)2. By taking advantage of the differences in polarity and molecular weights, the target compounds were separated from the side products by using a silica gel column.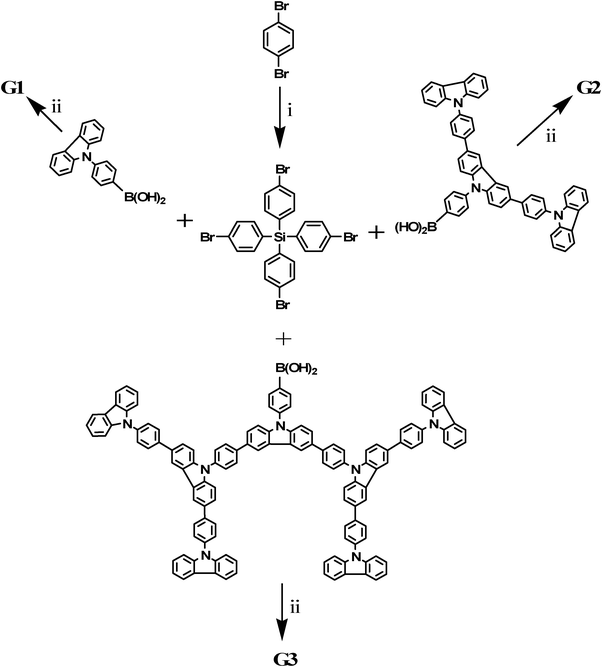 | ||
| Scheme 1 Synthesis routes to the dendrimers (G1, G2, G3) by a convergent approach. Reagents and conditions: (i) n-BuLi, SiCl4, ether, −50 °C; (ii) Pd(PPh3)4, K2CO3, toluene, 50 °C. | ||
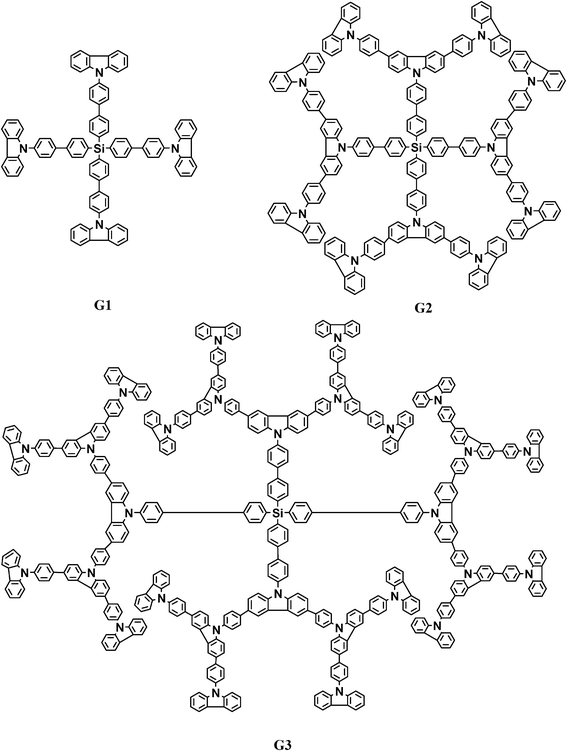 | ||
| Scheme 2 Chemical structures of the 1st–3rd generation of dendrimers (G1, G2, G3). | ||
We used 1H NMR, gel permeation chromatography (GPC), MALDI-TOF mass spectroscopy and elemental analyses to identify the chemical structures and molecular weights of the dendrimers. In the 1H NMR spectra (Fig. 5S–7S, ESI†), all the resonance peaks are well assigned to the corresponding protons in the dendrons and dendrimers. The GPC measurements give number-average molecular weights for G1, G2 and G3 of 753, 2524 and 6922, with polydispersities of 1.036, 1.024 and 1.004, respectively (Fig. 1). It is noted that the molecular weight obtained by GPC can only be a rough estimate due to the big difference in the hydrodynamic radius between the dendrimer and the polystyrene standard. In this regard, MALDI-TOF mass spectrometry is more effective as definitive evidence of the molecular weight and chemical structure. As illustrated in Fig. 2, all the MALDI-TOF mass spectra display only a single intense signal, whereas no signals corresponding to unreacted dendrons or the tetraphenylsilane core can be detected. The calculated and experimentally measured m/z values are in good agreement within the range of accuracy of the instrument. For example, in the MALDI-TOF mass spectrum of G3, the sharp molecular ion peak appears at 7096.24, while the calculated value of the molecular ion according to the molecular formula of G3 (C528H328N28Si) is 7092.53. The other two dendrimers (G1 and G2) also show correct molecular ion peaks which are consistent with the calculated molecular weights.
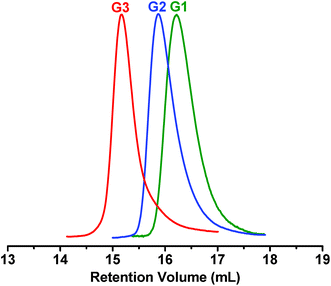 | ||
| Fig. 1 GPC chromatograms of G1–G3. | ||
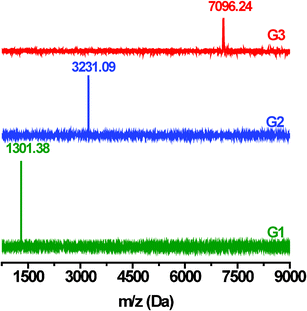 | ||
| Fig. 2 MALDI-TOF ms spectra of dendrimers G1–G3. | ||
Computer modelling of the dendrimers was carried out to illustrate the evolution of the topologies with the generational growth and elucidate the effects on the crystalline behavior, photophysical and electrochemical properties. Molecular models were obtained from the Materials Studio software and energy minimized via the conjugate-gradient method. The algorithm was set to Polak–Ribiere and the convergence and line search were equal to 0.1 and 0.5, respectively. After successive geometry and molecular mechanics optimizing, the energy-minimized structures are relaxed through dynamic calculations at 298 K for 200 ps, based on the COMPASS force field, in order to obtain the molecular models (Fig. 3). It can be seen that the torsional angle between the benzene ring and carbazole ring as well as the bending angle of the C–N–C bond gives rise to the twisted dendron. In particular, for the 3rd generation dendrimer G3, the tetrahedral robust core and the bulky, rigid and contorted dendrons result in the dendrimers very extended topological structure.
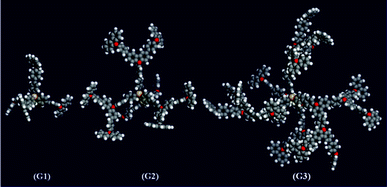 | ||
| Fig. 3 Computer simulations for the dendrimers G1–G3. | ||
Thermal properties
The thermal properties of these dendrimers were evaluated by thermogravimetric analysis (TGA) (Fig. 4) and differential scanning calorimetry (DSC) (Fig. 5) under a nitrogen atmosphere. All the three dendrimers exhibit excellent thermal stability with an onset of decomposition over 480 °C and 5% weight-loss temperatures for G1, G2 and G3 as high as 574, 622 and 594 °C, respectively. The first heating DSC curve of G1 shows a melting peak at 241 °C but this peak disappears in the second heating run. Moreover, no melting peaks for G2 and G3 were detected in both the first and second heating runs, indicating that the introduction of the rigid tetraphenylsilane moiety and bulky, twisted dendrons can considerably reduce the crystalline ability. The molecular weight of G1 is too low to display an obvious glass transition. With the increase of the dendron size in the second heating curves, G2 and G3 show glass transitions (Tg) at 122 and 271 °C, respectively. Compared to G2, the significantly increased Tg value for G3 indicates the greatly restricted motion ability of the dendrons.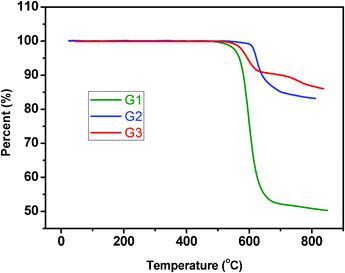 | ||
| Fig. 4 TGA traces of the dendrimers measured at a heating rate of 10 °C min−1. | ||
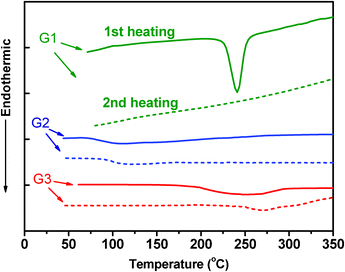 | ||
| Fig. 5 DSC curves of the dendrimers at a heating rate of 10 °C min−1. | ||
Photophysical properties of the dendrimer films
The UV-vis absorption and PL emission spectra of the dendrimer films were recorded (Fig. 6). The films were spin-coated from a THF solution on quartz substrates. It can be seen that the absorption spectra of these dendrimers show two major absorption bands. The high energy absorption peak appearing at 298 nm and the low energy absorption centred at around 330 nm are attributed to the n–π and π–π transitions of the 9-phenylcarbazole dendron, respectively.26 Notably, with the generational growth of the dendrimers, the second absorption bands of G2 and G3 become wider, reflecting the complexity of the conjugative system for the higher generation dendrimers. As a matter of fact, the generational growth can bring about two opposing roles. The seriously twisted dendron for the higher generation dendrimers can interrupt the conjugation effect, leading to a blue-shift of the absorption band. On the other hand, the increased content of 9-phenylcarbazole is favorable for the conjugative length, resulting in a red-shift. Relative to G1, the wide UV adsorptions for the higher generation dendrimers are the result of the common action of the above two factors. The band gaps of G1, G2 and G3, as estimated from the onset position of its absorption in the solid state, are 3.22, 2.97 and 3.27 eV, respectively. Apparently, for G3, the latter factor plays a more important role.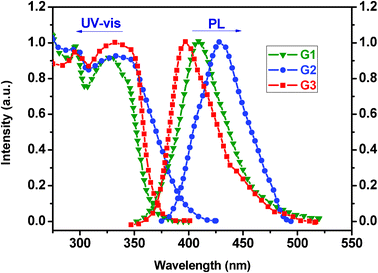 | ||
| Fig. 6 UV-vis and PL spectra of the dendrimer films. | ||
The emissions of G1, G2 and G3 are located at 409, 428 and 397 nm, respectively. Compared to G1, the red-shift of G2 means that the effective conjugation is elongated with the dendron size. However, the further generational increase makes the emission of G3 blue-shifted relative to G2 and the results agree well with those of the UV adsorptions. The full widths at half-maximum (FWHM) of these dendrimer PL emission bands are quite narrow at only around 45–54 nm (Table 1). In addition, using quinine sulfate (0.1N in H2SO4) as a reference, the measured fluorescence yields of G1–G3 are in the range of 98.9 to 99.1%, suggesting that the three-dimensional topologies of these dendrimers can effectively prevent molecular aggregation to yield significantly high PL quantum efficiencies.
| Dendrimers | UV Abs (nm) | PL [FWHM] (nm) | ΦPLa (%) | Band gapb (eV) |
|---|---|---|---|---|
| a Fluorescence quantum efficiency measured in THF using quinine sulfate dehydrate as a standard. b Band gaps were calculated from the onsets of the UV-visible absorption spectra of G1–G3 in solid films. | ||||
| G1 | 330, 298 | 409 [48] | 98.9 | 3.22 |
| G2 | 339, 298 | 428 [54] | 96.3 | 2.97 |
| G3 | 331, 298 | 397 [45] | 99.1 | 3.27 |
Electrochemical behavior of the dendrimers films
The electrochemical behavior of G1–G3 were examined by cyclic voltammetry (CV), using a standard three-electrode electrochemical cell in a 0.1 M anhydrous acetonitrile solution of tetra-n-butylammonium hexafluorophosphate (nBu4NPF6) under nitrogen at room temperature. As illustrated in Fig. 7, the dendrimers display two electrochemical oxidation peaks owing to the tetraphenylsilane core and carbazole dendron, respectively.25 The reduction curves shift to the left and exhibit a strong wave with the incremental generational increase. The reason can be attributed to the fact that the higher generation dendrimer has the more twisted structure, which interrupts its π-delocalization effect. With the generational growth, the strong electron-donating effect of carbazole leads to the decreasing trend in the onset oxidation potentials for G1, G2 and G3 of 0.70, 0.68 and 0.59 eV, respectively. The oxidation and reduction onset potential allows us to calculate the energy of the highest occupied molecular orbital (HOMO) and the electrochemical energy gap (Eg) according to the equations: HOMO = −Eonset(ox) − 4.8 eV and Eg = Eonset(ox) −Eonset(red). As a result, the HOMO levels of G1, G2 and G3 are −5.50, −5.48 and −5.39 eV, respectively, very close to the work function of the ITO/PEDOT electrode (−5.2 eV). Apparently, the HOMO levels of the dendrimers shift to higher energies with the increase in the generation. This characteristic can effectively reduce the energy barrier for the hole-injection between the hole-injecting layer and emitting layer. Furthermore, it is found that the incorporation of the tetrahedral core and bulky, twisted 9-phenylcarbazole-based dendrons give rise to the dendrimers wide Eg values because of the reduced coplanarity and interruption of the π-conjugation along the dendrons (Table 2). Moreover, the Eg values can be tuned in the range of 3.01 to 3.29 eV by the variation of the generation.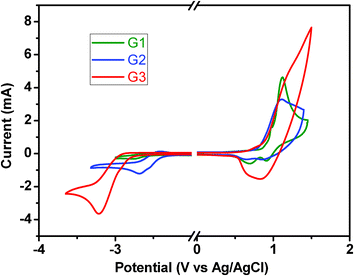 | ||
| Fig. 7 Cyclic voltammograms of the dendrimer thin films . | ||
| Dendrimer | E onset(ox) (V)a | E onset(red) (V)a | HOMO (eV)b | LUMO (eV)b | Eg (eV)c |
|---|---|---|---|---|---|
| a Onset oxidation and reduction potentials versus Ag/Ag+. b Estimated from the onset oxidation and reduction potential using HOMO = −Eonset(ox) − 4.8 eV and LUMO = −Eonset(red) − 4.8 eV. c Electrochemical band gaps determined using Eg = Eonset(ox) − Eonset(red). | |||||
| G1 | 0.70 | −2.53 | −5.50 | −2.27 | 3.23 |
| G2 | 0.68 | −2.33 | −5.48 | −2.47 | 3.01 |
| G3 | 0.59 | −2.70 | −5.39 | −2.10 | 3.29 |
Conclusion
In summary, we have synthesized a series of novel dendrimers (G1, G2 and G3) containing a tetrahedral tetraphenylsilane core and twisted 9-phenylcarbazole dendrons via a Suzuki coupling reaction through a convergent approach. The DSC and TGA results demonstrate that the 3rd generation dendrimer, G3, has a high glass transition temperature of 270 °C and a 5% weight-loss temperature of 594 °C. The UV absorption and fluorescence spectra show that the molecular aggregation has been significantly suppressed by the tetrahedral core and rigid, twisted dendrons, leading to the extremely high fluorescence quantum yields of 98.9–99.1%. In addition, with the generational growth, the high content of the carbazole unit greatly increases the hole-injection ability of the dendrimers because of the electron-donating effect of carbazole. In particular, the contorted dendron reduces its effective length of conjugation, leading to the high triplet energy band gaps of the dendrimers from 3.01 to 3.29 eV. The combination of these excellent properties increases the dendrimers application potential in the optoelectronic field as hole-transporting and/or host materials.References
- T. S. Qin, G. Zhou, H. Scheiber, R. E. Bauer, M. Baumgarten, C. E. Anson, E. J. W. List and K. Mullen, Angew. Chem., Int. Ed., 2008, 47, 8292–8296 CrossRef CAS.
- J. You, G. Y. Li, R. J. Wang, Q. P. Nie, Z. G. Wang and J. Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 17825–17830 RSC.
- E. R. Gillies and J. M. Frechet, J. Am. Chem. Soc., 2002, 124, 14137–14146 CrossRef CAS.
- S. Li, M. L. Szalai, R. M. Kevwitch and D. V. McGrath, J. Am. Chem. Soc., 2003, 125, 10516–10517 CrossRef CAS.
- F. M. H. de Groot, C. Albrecht, R. Koekkoek, P. H. Beusker and H. W. Scheeren, Angew. Chem., Int. Ed., 2003, 42, 4490–4494 CrossRef CAS.
- R. J. Amir, N. Pessah, M. Shamis and D. Shabat, Angew. Chem., Int. Ed., 2003, 42, 4494–4499 CrossRef CAS.
- H. Kobayashi, S. Kawamoto, R. A. Star, T. A. Waldmann, Y. Tagaya and M. W. Brechbiel, Cancer Res., 2003, 63, 271–276 CAS.
- D. A. Fulton, E. M. Elemento, S. Aime, L. Chaabane, M. Botta and D. Parker, Chem. Commun., 2006, 1064–1066 RSC.
- D. W. Brousmiche, J. M. Serin, J. M. J. Frechet, G. S. He, T. C. Lin, S. J. Chung and P. N. Prasad, J. Am. Chem. Soc., 2003, 125, 1448–1449 CrossRef CAS.
- T. H. Ghaddar, J. F. Wishart, D. W. Thompson, J. K. Whitesell and M. A. Fox, J. Am. Chem. Soc., 2002, 124, 8285–8289 CrossRef CAS.
- Y. Ribourdouille, G. D. Engel, M. Rchard-Plouet and L. H. Gade, Chem. Commun., 2003, 1228–1229 RSC.
- E. Delort, T. Darbre and J. L. Reymond, J. Am. Chem. Soc., 2004, 126, 15642–15643 CrossRef CAS.
- P. B. Amama, M. R. Maschmann, T. S. Fisher and T. D. Sands, J. Phys. Chem. B, 2006, 110, 10636–10644 CrossRef CAS.
- S. C. Lo, R. E. Harding, E. Brightman, P. L. Burn and I. D. W. Samuel, J. Mater. Chem., 2009, 19, 3213–3227 RSC.
- B. L. Rupert, W. J. Mitchell, A. J. Ferguson, M. E. Kose, W. L. Rance, G. Rumbles, D. S. Ginley, S. E. Shaheen and N. Kopidakis, J. Mater. Chem., 2009, 19, 5311–5324 RSC.
- X. Ren, J. Li, R. J. Holmes, P. I. Djurovich, S. R. Forrest and M. E. Thompson, Chem. Mater., 2004, 16, 4743–4747 CrossRef CAS.
- S. J. Yeh, M. F. Wu, C. T. Chen, Y. H. Song, Y. Chi, M. H. Ho, S. F. Hsu and C. H. Chen, Adv. Mater., 2005, 17, 285–289 CrossRef CAS.
- M. H. Tsai, H. W. Lin, H. C. Su, T. H. Ke, C. C. Wu, F. C. Fang, Y. L. Liao, K. T. Wong and C. I. Wu, Adv. Mater., 2006, 18, 1216–1220 CrossRef CAS.
- C. He, Y. Xiao, J. Huang, T. Lin, K. Y. Mya and X. Zhang, J. Am. Chem. Soc., 2004, 126, 7792–7793 CrossRef CAS.
- M. Tabata, T. Fukushima and Y. Sadahiro, Macromolecules, 2004, 37, 4342–4350 CrossRef CAS.
- N. Drolet, J. F. Morin, N. Leclerc, S. Wakim, Y Tao and M. Leclerc, Adv. Funct. Mater., 2005, 14, 1671–1682 CrossRef.
- V. Percec, M. Obata, J. G. Rudick, B. B. De, M. Glodde, T. K. Bera, S. N. Magonov, V. S. K. Balagurusamy and P. A. Heiney, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3509–3533 CrossRef CAS.
- Y. Q. Fu, M. H. Sun, Y. G. Wu, Z. S. Bo and D. G. Ma, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1349–1356 CrossRef CAS.
- J. H. Fournier, X. Wang and J. D. Wuest, Can. J. Chem., 2003, 81, 376–380 CrossRef CAS.
- A. Kulasi, H. Yi and A. Iraqi, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5957–5967 CrossRef CAS.
- Q. Zhang, J. Chen, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, J. Mater. Chem., 2004, 14, 895–900 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra21439c |
| This journal is © The Royal Society of Chemistry 2012 |