DOI:
10.1039/C2RA21657D
(Communication)
RSC Adv., 2012,
2, 10821-10828
Carbazole oligomers revisited: new additions at the carbazole 1- and 8-positions†
Received
1st August 2012
, Accepted 14th September 2012
First published on 17th September 2012
Abstract
In recent years low molecular weight carbazole-based materials have attracted much interest as hole-transporting and host materials for organic light emitting diode (OLED) applications. Herein we report the tri- and tetra-bromination of carbazole and the subsequent synthesis of novel carbazole oligomers substituted at the 1- and 8-positions. The oligomer series range from mono-carbazole (Cz-1) to quinque-carbazole (Cz-5) and we have carried out a comparative study of their thermal, optical, electrochemical and optoelectronic properties. The glass transition temperature increases as the carbazole core is enlarged (0 °C to 54 °C) and all oligomers exhibit excellent thermal stability with high thermal decomposition temperatures in the range of 442 °C to 494 °C. From optical data and Raman spectroscopy we observe negligible changes in the effective conjugation length as the number of peripheral carbazole units increases. However, the oxidation potential decreases as the number of carbazole units increase, in which Cz-5 exhibits the highest HOMO level (−5.19 eV) compared to other carbazole oligomers in the series. All carbazole materials emit deep blue photoluminescence ranging from 416 to 432 nm in the solid-state. Finally, electrophosphorescence was demonstrated from solution processable guest-host organic light- emitting diodes (OLEDs) fabricated using Cz(2–5) as the host materials with device configuration ITO/PEDOT/Cz1–5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10% Ir(mppy)3/TPBi/LiF/Al. The device efficiency decreases from Cz-3 to Cz-5. The best OLED device that incorporates Cz-3 as the host exhibits a turn-on voltage of 2.8 V, maximum brightness of 2310 cd m−2, maximum power efficiency of 33.6 lm W−1, maximum current efficiency 35.5 cd A−1 and EQE of 10.6%.
:10% Ir(mppy)3/TPBi/LiF/Al. The device efficiency decreases from Cz-3 to Cz-5. The best OLED device that incorporates Cz-3 as the host exhibits a turn-on voltage of 2.8 V, maximum brightness of 2310 cd m−2, maximum power efficiency of 33.6 lm W−1, maximum current efficiency 35.5 cd A−1 and EQE of 10.6%.
Introduction
Historically, carbazole has shown great potential in applications, such as electrophotography,1 solar cells,2 organic light-emitting diodes (OLEDs)3 and as photorefractive materials.4 Its efficient use in organic semiconductor devices is primarily due to the formation of stable radical cations and its efficient hole-mobility properties. Indeed, the first polymeric electroluminescent device, fabricated by Partridge (1983), incorporated polyvinylcarbazole (PVK) as the active emitting layer.5 More recently, carbazole based materials have shown great potential as host materials in phosphorescent OLED devices, due to their high triplet energy levels and low oxidation potentials.6 Although carbazole-based small molecules and polymeric materials have both been applied, small molecules have improved chemical purity compared to the latter because they can be purified by chromatography, recrystallisation or train sublimation.
Oligomeric materials have proven to be good model systems for polymeric systems due to their well-defined structure, mono-dispersity and high purity.7 Oligocarbazoles linked at the 3,6- and 2,7- positions have been synthesized previously by Strohriegl and co-workers.8,9 They demonstrated that 2,7-oligocarbazoles are optically similar to their fluorene counterparts. However, 3,6-oligocarbazoles have limited conjugation and display an effective conjugation that is more appropriately defined as a “substituted 1,4-diaminodiphenyl”, or in other words meta-linked polyphenylenes. Interestingly, compared to their sister 2,7-oligofluorenes, the 2,7-oligocarbazoles do not exhibit a thermotropic liquid crystalline phase. With respect to carbazole-based semiconductor materials, nearly all of the previous literature has focused on the 2,7-positions, 3,6-positions and the N-position. In contrast to fluorene, carbazole is activated towards electrophiles at its 3,6-positions,8 and, therefore, alternative pathways are needed to synthesise 2,7-dibromocarbazole.9 The N-position can be easily functionalised by nucleophilic substitution using base and alkyl halides and Ullmann condensations.10 Relatively unexplored positions are the 1 and 8 positions (Fig. 1), which can also be functionalized by bromination, although very little research has been done about it. Although the syntheses of tri- and tetra-brominated carbazole have been reported using N-bromosuccinimide (NBS)-silica gel in methylene chloride,11 we were unable to reproduce these findings using carbazole. Due to the uncharted 1- and 8-positions and the absence of reliable literature procedures concerning selective tri- and tetra-bromination, it encouraged us to unequivocally synthesise 1,3,6-tribromocarbazole and 1,3,6,8-tetrabromocarbazole. Subsequent palladium-catalysed cross couplings with 9-octylcarbazole-3-boronic afforded a series of carbazole oligomeric materials (Cz-2 to Cz-5), and we have carried out a comparative study of their optical, electrochemical, thermal and optoelectronic properties.
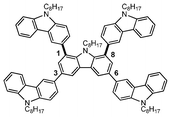 |
| | Fig. 1 Molecular structure of the X-shaped carbazole pentamer (Cz-5), formed by substitution at the carbazole 1-, 3-, 6- and 8-positions. | |
Results and discussion
Synthesis
The selective carbazole brominations are outlined in Scheme 1. Mono and di-brominated carbazole (Cz-1Br and Cz-2Br) were easily synthesised in good yield using one and two equivalents, respectively, of NBS in DMF and subsequent N-alkylation using NaOH, a two-phase toluene–water solvent system and 1-bromooctane. Initially, we attempted to synthesise 1,3,6-tribromocarbazole and 1,3,6,8-tetrabromocarbazole by following the previous literature, using carbazole and NBS–SiO2 in CH2Cl2 at room temperature.11 However, after numerous attempts we could only synthesise 1,3,6-tribromocarbazole in less than 10% yield and we were unable to synthesise 1,3,6,8-tetrabromocarbazole. During the synthesis of Cz-2Br from 9-octylcarbazole using NBS (2 equivalents)-SiO2 in CH2Cl2, we isolated a small amount of 3,6,8-tribromo-9-octylcarbazole (Cz-3Br). This led us to believe that for the synthesis of Cz-3Br, 9-octylcarbazole (Cz-1), and not carbazole, was the more appropriate starting material. Indeed, Cz-3Br was finally synthesised in high yield (72%) using 5 equivalents of NBS–SiO2 and CHCl3 with stirring for almost 3 days. Interestingly, during the course of the reaction, no tetra-bromination was observed, despite the presence of excess NBS. The synthesis of 1,3,6,8-tetrabromocarbazole was accomplished by Br2/FeCl3 in CH2Cl2 with moderate yield (34%). The solubility of 1,3,6,8-tetrabromocarbazole was poor in common organic solvents and, therefore, was purified twice by recrystallisation from toluene without chromatography. The target material Cz-4Br was synthesised in the same way as Cz-1Br and Cz-2Br by N-alkylation. It is noteworthy that Cz-4Br can also be prepared by bromination of 9-octylcarbazole using Br2/FeCl3 in CH2Cl2. Standard Suzuki conditions were used to prepare the final oligomeric materials, Cz-2, Cz-3, Cz-4 and Cz-5 (Scheme 2).
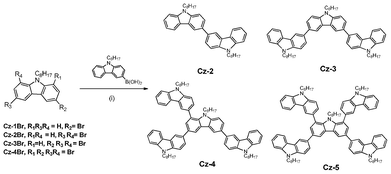 |
| | Scheme 2 Reagents and conditions: (i) Pd(PPh3)4 (10 mol%), K2CO3, phase transfer catalyst (PTC), toluene–H2O (2:1), 90 °C, 2 days. | |
Thermal properties
All differential scanning calorimetry (DSC) scans show first-order transitions on the first cooling cycle and second heating cycle. For all the carbazole oligomers, the first heating cycle shows an endothermic melting transition without an exothermic crystallisation peak upon cooling (Fig. 2 and Fig. 3), which suggests that all the carbazole oligomers are isolated as crystalline materials that can be vitrified after melting and subsequent cooling. The alkyl chains at the N-position function very well in enhancing the solubility and increasing the viscosity, in which the latter affect reduces the likelihood of crystallisation. As the carbazole core enlarges, from Cz-2 to Cz-5, the Tg value augments, which is most likely due to the increased rigidity and a subsequent decrease in the free volume. The same trend is also observed for both linear oligofluorenes12 and oligocarbazoles.8 Of course, the Tg can also be increased by decreasing the alkyl chain length at the N-position. However, we synthesised an analogous material to Cz-5, with methyl groups at all 5 N-positions and no Tg was observed. If the alkyl chain is too small, the viscosity is not high enough to prevent crystallisation after the initial melting and, therefore, the amorphous phase cannot be “frozen” upon cooling to room temperature. All carbazole oligomers show excellent thermal stability, reflected by their high thermal decomposition temperatures (Td) and are considerably higher than other reported oligocarbazoles and oligofluorenes.9,12
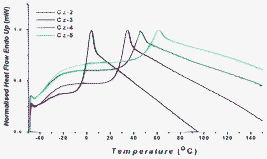 |
| | Fig. 2 A comparison of the glass transition temperature (Tg) of Cz(2–5) from normalised DSC scans on the second heating. | |
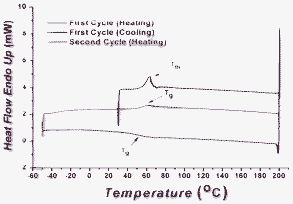 |
| | Fig. 3 A full DSC scan for Cz-5, which shows clearly the thermal behavior and transitions; Tm = melting point temperature (63 °C) and Tg = glass transition temperature (54 °C). | |
Optical and electrochemical properties
The effect of the number of bromine atoms on the absorption spectra of carbazole is shown in Fig. 4a. For Cz-1, using Platt notations,13 we can assign the peaks as follows: 332 nm/345 nm (1Lb–1A), 293 (1La–1A), 261 (1Ba–1A) and 236 (1Ca–1A). The lowest energy transition, 1Lb–1A, which is polarised along the short axis, undergoes a slight bathochromic shift with increased carbazole bromination, and a trend of decreasing fluorescence (quenching) due to the internal heavy atom effect. These 4 bands condense into 2 bands as the carbazole core enlarges, from Cz-1 to Cz-5 (Fig. 4b). The molar absorptivity increases from Cz-1 to Cz-5, and a slight red-shift from Cz-1 to Cz-3 is observed with a very slight blue shift from Cz-3 to Cz-5, which might indicate an increase and a decrease in conjugation, respectively. The large change in the optical band gap (Eg) from Cz-1 to Cz-2, compared to only slight variations for the other carbazole oligomers suggests that changes in the conjugation is greatest for these two analogues. In any case, the absorption wavelengths and Eg dispersions from Cz-2 to Cz-5 are very small and maybe negligible (Table 1). Cz-3 has previously been reported to possess limited conjugation along its carbazole backbone, behaving like a meta-linked polyphenylene.8 Further linking more carbazole units at the 1- and 8-positions, in the case of Cz-4 and Cz-5, seem to have little effect on the optical properties and are comparable to Cz-3 with negligible changes in the effective conjugation length. These trends are further supported by Raman spectra (see later).
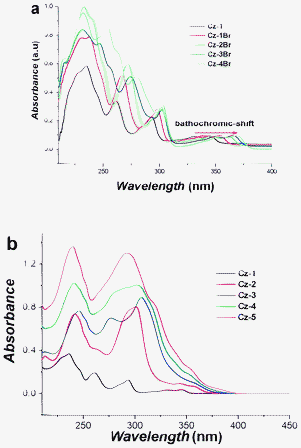 |
| | Fig. 4 (a) UV-Vis absorption of Cz-1 to Cz-4Br measured in THF solution, and (b) UV-Vis absorption of Cz-1 to Cz-5 measured in THF solution; concentration = 1 × 10−5 mol L−1. | |
In the solid-state, the emission peak wavelength of Cz-1 exhibits a large red shift compared to its monomeric emission in solution and Cz-2 shows a large tail, which should be due to intermolecular species, possibly an excimer (Fig. 5a, b). It is well known that other carbazole materials, such as PVK, also show excimer formation that subsequently reduces its fluorescence quantum yield (ΦF). The larger carbazole oligomers have less prominent tails in their solid-state emission, suggesting reduced intermolecular interactions. All carbazole materials emit deep blue light in the solid state, which is a highly desirable emission colour for OLEDs.14 However, these carbazole oligomers show low ΦF in solution, which should be less in the solid-state, therefore, excluding their use as efficient light-emitting materials in applications such as OLEDs. The ΦF of Cz-1 (29%) is in excellent agreement with a previous report concerning a similar material (30%, p-terphenyl as standard).15
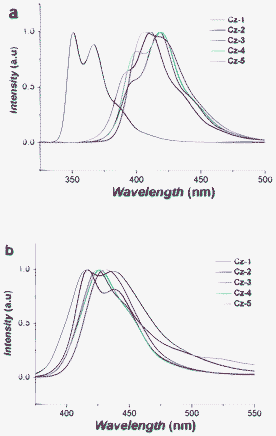 |
| | Fig. 5 (a) Normalised PL spectra of Cz-1 to Cz-5 measured in THF solution (1 × 10−5 mol L−1), excitation = 300 nm, (b) normalised PL spectra of Cz-1 to Cz-5 in the solid state, excitation = 300 nm. | |
Over the years, Raman spectroscopy has become a useful tool for the investigation of the extent of conjugation and delocalization length in conjugated materials, in which the basis of its use is the “effective conjugation coordinate” (ECC) theory.16 The effective conjugation vibrational coordinate ( ) involves the stretching of the C
) involves the stretching of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C bonds and, therefore, represents the delocalisation of π-electrons. ECC predicts the
C/C–C bonds and, therefore, represents the delocalisation of π-electrons. ECC predicts the  -mode to be the strongest peak in the Raman spectra.17 For example, with regards to oligopyrrole, Zerbi et al. demonstrated that there is considerable frequency shift of the
-mode to be the strongest peak in the Raman spectra.17 For example, with regards to oligopyrrole, Zerbi et al. demonstrated that there is considerable frequency shift of the  -mode with increasing oligomer length.17 This has also been recently demonstrated for a series of α-oligofurans.18 Therefore, for our series of carbazole oligomers, we were curious if their Raman spectra underwent sizeable dispersion both in frequency and intensity. The main feature in the Raman spectra of our oligocarbazoles is the intense band around 1600 cm−1 and within this region there is negligible dispersion of frequency and intensity (Fig. 6), which suggests negligible change in the effective conjugation length.
-mode with increasing oligomer length.17 This has also been recently demonstrated for a series of α-oligofurans.18 Therefore, for our series of carbazole oligomers, we were curious if their Raman spectra underwent sizeable dispersion both in frequency and intensity. The main feature in the Raman spectra of our oligocarbazoles is the intense band around 1600 cm−1 and within this region there is negligible dispersion of frequency and intensity (Fig. 6), which suggests negligible change in the effective conjugation length.
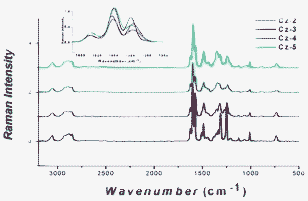 |
| | Fig. 6 Raman spectra in the solid state (powder); inset shows a closer view of the 1540–1650 cm−1 region, excitation = 1064 nm. | |
The electrochemistry of carbazole and its derivatives has been known for a long time, especially from the contributions by Ambrose (1968).19 In the simplest case, carbazole exhibits an irreversible redox process, in which single electron oxidation results in a highly unstable cationic radical, which couples to yield 3,3′-dicarbazole (see Fig. 7a). The steric hindrance at the 1- and 8- position prevents coupling at these locations. On the reverse scan, the cathodic peaks at about 0.84 V and 0.49 V are due to 3,3′-dicarbazole. In contrast, the dimer Cz-2 exhibits excellent electrochemical stability and it undergoes 2 reversible one-electron oxidations to finally yield a dicationic species via its radical monocation (Fig. 8).
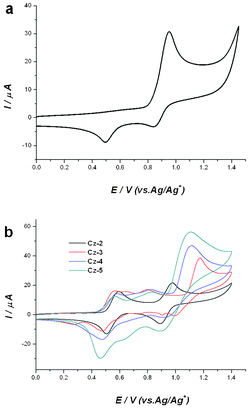 |
| | Fig. 7 Cyclic voltammograms of (a) Cz-1 and (b) Cz-2 to Cz-5 in dry CH2Cl2 solution (1 × 10−3 M) with Bu4NPF6 (0.1 M), scan rate 100 mVs−1 (Cz-1) and 50 mVs−1 (Cz-2 to Cz-5). The potentials were measured relative to the Ag/Ag+ reference electrode. | |
 |
| | Fig. 8 Scheme to illustrate the two reversible one-electron oxidation of 3,3′-bis(9-octylcarbazole). | |
It is clear from Fig. 7b that Cz-2 to Cz-5 exhibit similar electrochemical behaviours. The other CV scans showing the oxidation behaviour of Cz-3 to Cz-5 are most likely based upon a similar mechanism to Cz-2. As the carbazole core is enlarged, the oxidation potential decreases, which results in an increase of the HOMO level from Cz-1 to Cz-5; −5.58, −5.24, −5.21, −5.21 and −5.19, respectively. These values are in excellent agreement with other published electrochemical data regarding 3,6-linked ter-carbazoles (HOMO = −5.20 to −5.25 eV)8,9 and poly(3,6-carbazole) (−5.0 eV).20 The HOMO level of Cz-5 (−5.19 eV) is well matched compared to the work function of ITO (−5.0 eV), with only a 0.2 eV hole injection barrier, which suggests it can be used as a hole-transporting or host material in OLEDs.
Electroluminescence properties
The triplet energy level (ET) of Cz-2, Cz-3, Cz-4 and Cz-5 are 2.73 eV, 2.66 eV, 2.68 eV and 2.66 eV, respectively, which agree well with the literature value of 2.73 eV for Cz-3.6b All of the materials exhibit a higher ET than tris[2-(p-tolyl)pyridine]iridium(III) (Ir(mppy)3) (2.4 eV)21 and, therefore, triplet energy transfer is favoured. Also, Cz2–5 exhibit high HOMO levels and a low hole-injection barrier of ∼0.2 eV (compared to 5.0 eV of ITO). Therefore, electrophosphorescence using Cz2–5 as the host materials and Ir(mppy)3 as the green dopant was investigated. We fabricated solution processable multilayer OLED devices to explore the comparative hole-transporting and OLED performance properties of the various carbazole materials, with configuration ITO/PEDOT (40 nm)/Cz(2–5): 10% Ir(mppy)3 (30–35 nm)/TPBi (30 nm)/LiF (1 nm)/Al (100 nm). PEDOT functions as the hole-transporting layer and TPBi functions both as the hole-blocking and electron transporting layer. The current–brightness–voltage characteristics are shown in Fig. 9 and the detailed EL performances are listed in Table 2. From our data it is clear that the device with three carbazole units (Cz-3) exhibits the highest external quantum efficiency (EQE) due to its balanced hole and electron current. Cz-2 to Cz-5 have similar HOMO levels (5.21 ± 0.02) and ET values. Therefore, this might imply that the charge balance in the device is determined by the hole-transporting capability but not the PEDOT:PSS/host interfacial properties. Due to the negligible change in conjugation and electron delocalisation of the carbazole oligomers, the triplet energy level of all the carbazole oligomers are similar, around 2.7 eV, with negligible influence of the increasing carbazole substitution on the ET. As we know, an increase in the size of an oligomeric fluorene22 or carbazole8 core can increase the charge mobility. Therefore, Cz-3 might possess a higher hole mobility than Cz-2. However, further increasing the carbazole unit leads to the spirality of the spatial configuration, which might reduce the intermolecular packing and eventually hampers the hole hopping between molecules. This means Cz-4 and Cz-5 exhibit lower hole transport capability than Cz-3. Our interpretation is consistent with the device performance: unbalanced charge transport in Cz-2 device leads to the leakage of electron current and the lowest EQE. Balanced charge transport in the OLED device with Cz-3 results in the highest EQE. PVK is a carbazole-based material that has been used for solution processable phosphorescent OLEDs. However, it has been demonstrated that it is not such an attractive material for blue phosphorescent dopants due to dimer traps.23 Efficient solution processable host materials for blue phosphorescence still remains a challenge and the accessibility of the carbazole 1- and 8-positions could be beneficial.
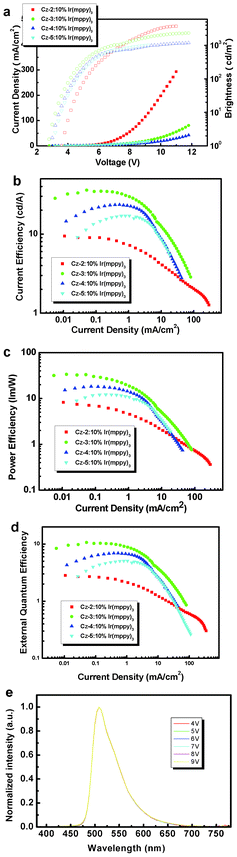 |
| | Fig. 9 (a) Change in luminance and current density with the applied bias, (b) current density versus current efficiency, (c) current density versus power efficiency, (d) current density versus external quantum efficiency, (e) EL spectra using Cz-3 host. | |
Table 1 Optical, electrochemical and thermal properties
| Material |
T
g (°C)a |
T
d (°C)b |
λ
abs (nm)c |
λ
em (nm)d |
HOMO (eV)e |
LUMO (eV)f |
E
g (eV)g |
ΦFh (%) |
| Solution |
Solid |
|
Abbreviations: Tg = glass transition temperature estimated from differential scanning calorimetry (DSC) using half Cp extrapolated on the second heating scan.
T
d = thermal decomposition temperature measured using thermal gravimetric analysis (TGA).
λ
abs = absorption peaks at all intensities; concentration = 1 × 10−5 mol L−1.
λ
em = emission peaks at all intensities; solution (THF) at concentration = 1 × 10−5 mol L−1, solid state (powder, except liquid Cz-1), excitation = 300 nm.
HOMO (highest occupied molecular orbital) = −(Eonset-ox + 4.8 − Eferrocene), where Eferrocene = 0.062 V, estimated using cyclic voltammetry using Bu4NPF6 in dry DCM.
LUMO (lowest unoccupied molecular orbital) = (Eg + HOMO).
E
g = optical band gap estimated from the onset wavelength (λonset) of the absorption spectra.
ΦF = fluorescence quantum yield estimated using quinine sulphate (Cz-2 to Cz-5, ΦF = 0.54 in 0.1 M H2SO4) and anthracene (Cz-1, ΦF = 0.27 in ethanol).
|
| Cz-1 |
Liquid |
— |
236, 261, 293, 332, 345 |
351, 367 |
416, 438 |
−5.58 |
−2.07 |
3.51 |
29 |
| Cz-2 |
0 |
442 |
242, 301, 343 |
400, 411 |
416, 438 |
−5.24 |
−1.88 |
3.36 |
13.5 |
| Cz-3 |
30 |
490 |
246, 277, 307 |
418 |
424 |
−5.21 |
−1.90 |
3.31 |
11.9 |
| Cz-4 |
40 |
494 |
241, 302 |
419 |
426 |
−5.21 |
−1.89 |
3.32 |
16.2 |
| Cz-5 |
54 |
464 |
240, 293 |
406 |
432 |
−5.19 |
1.85 |
3.34 |
21.6 |
| Cz |
λ
EL (nm) |
V
on (V) |
L
max (cd m−2) |
η
P,max (lm/W) |
η
C,max (cd A−1) |
EQE (%) |
|
Abbreviations: λEL = EL max, Von = turn on voltage, Lmax = maximum brightness, ηP,max = maximum power efficiency, ηC,max = maximum current efficiency, EQE = external quantum efficiency.
|
| Cz-2 |
508 |
3.6 |
3720 |
8.2 |
9.1 |
2.8 |
| Cz-3 |
508 |
2.8 |
2310 |
33.6 |
35.5 |
10.6 |
| Cz-4 |
508 |
3.0 |
1180 |
18.2 |
22.1 |
6.9 |
| Cz-5 |
506 |
3.1 |
1020 |
12.1 |
18.2 |
4.8 |
Conclusions
We have reported the synthesis and a comparative study of carbazole oligomers substituted at the carbazole 1-, 3-, 6- and 8- positions. The X-shaped carbazole pentamer, that has carbazole groups substituted at all 4-positions (Cz-5), exhibits the highest Tg with excellent thermal stability. From Cz-2 to Cz-5, the effective conjugation length changes only very slightly, which allows the carbazole core to be enlarged without significant changes in optical and electrochemical properties. Moreover, from cyclic voltammetry, we have observed a lowering of the oxidation potential as the carbazole core enlarges, in which Cz-5 exhibits the highest HOMO level. This research work demonstrates the accessibility of the carbazole 1- and 8- positions by tri- and tetra-bromination and subsequent Suzuki cross-coupling reactions. Due to the great research interest in low molecular weight carbazole materials used as host materials for OLEDs, we hope that it would encourage the design of new materials, such as ambipolar materials, N-substituted carbazole oligomers and dendrimers, with improved properties.
Experimental
Carbazole (1, 15.0 g, 89.70 mmol) was dissolved in dry DMF (100 mL) in a 500 mL two necked round-bottom flask. The solution was cooled to 0 °C, and a solution of NBS (16.0 g, 89.70 mmol) in DMF (150 mL) was added dropwise under a nitrogen atmosphere. After the addition, the mixture was allowed to warm to RT and was then stirred for an additional 12 h. The reaction mixture was then poured into water (1 L) and the resulting white precipitate was filtered. The white powder was dissolved in DCM, washed with water to remove water soluble impurities, dried (MgSO4), filtered and concentrated under reduced pressure. The crude white solid was recrystallized from ethanol (×2) to yield white crystals (9.20 g 41.8%). 1H NMR (400 MHz, CDCl3): δ (ppm) 0.23–0.26 (m, 1H), 7.30 (d, 1H), 7.4–7.45 (m, 2H), 7.50 (d, 1H), 8.02 (d,1H), 8.07 (s, 1H), 8.19 (s, 1H). MS m/z (ACPI): 246.6 (M+), 245.7 (M − 1), 166.8 (M − Br). HPLC: 97%.
Carbazole (1, 1.50 g, 8.97 mmol) was dissolved in dry DMF (10 mL) in a 50 mL two necked round-bottomed flask. The solution was cooled to 0 °C, and a solution of NBS (3.20 g, 17.80 mmol) in DMF (15 mL) was added dropwise under a nitrogen atmosphere. After the addition, the mixture was allowed to warm to RT and was stirred for an additional 12 h. The reaction mixture was then poured into water (500 mL), and the resulting white precipitates was filtered. The white powder was dissolved in DCM, washed with water to remove water soluble impurities, dried (MgSO4), filtered and concentrated under reduced pressure. The crude white solid was recrystallized from ethanol to yield white crystals (1.60 g, 56.2%). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.12 (s, 3H), 7.51 (d,2H), 7.30 (d, 2H). MS m/z (ACPI): 324 (M − 1). HPLC: 99.7%.
A solution of carbazole (1, 6.00 g, 35.88 mmol), C8H17Br (6.93 g, 35.88 mmol) and PTC (1.20 g, 3.53 mmol) in toluene (70 mL) was stirred in a 250 mL two necked round-bottomed flask. An aqueous NaOH solution (35.0 g with 70 mL H2O) was added and the reaction mixture was heated to 70 °C and stirred for 10 h. The reaction mixture was added to a separating funnel and the water layer removed and washed with toluene (70 mL). The combined toluene layers were combined and washed with water (140 mL). This was then dried (MgSO4), filtered and concentrated under reduced pressure. The crude brown oil was purified using a short chromatography column (silica gel) eluting with hexane to yield a colourless oil (7.20 g, 72.4%). 1H NMR (400 MHz, CDCl3): δ (ppm) 0.86 (t, 3H), 1.20–1.42 (m, 10H), 1.86 (quint, 2H), 4.29 (t, 2H), 7.22 (t, 2H), 7.39–7.48 (m, 4H), 8.10 (d, 2H). MS m/z (APCI): 280.1 (M + 1), 279.1 (M+). HPLC: 99.52%.
Bromine (4.60 mL, 0.0897 mol) was added slowly to a solution of carbazole (3.00 g, 0.0179 mol) and FeCl3 (0.09 g, 0.54 mmol) in dry dichloromethane (300 mL) at room temperature. The reaction mixture was stirred for 31 h and then poured into an acetone (100 mL) and ethanol (200 mL) solvent mixture and the precipitate filtered. The crude product was recrystallised from toluene (×2) to yield light brown crystals (2.90 g, 33.5%). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.75 (s, 2H), 8.05 (s, 2H), 8.33 (s, 1H). MS m/z (APCI): 481.9 (M − 1). HPLC: 100%.
3-Bromo-9-octylcarbazole (Cz-1Br)
A solution of 3-bromocarbazole (2, 7.00 g, 28.40 mmol), C8H17Br (6.10 g, 31.30 mmol) and PTC (0.60 g, 1.80 mmol) in toluene (70 mL) was stirred in a 250 mL two necked round-bottomed flask. An aqueous NaOH solution (35.0 g with 70 mL H2O) was added and the reaction mixture was heated to 70 °C and stirred for 10 h. The reaction mixture was added to a separating funnel and the water layer removed and washed with toluene (70 mL). The combined toluene layers were combined and washed with water (140 mL). It was then dried (MgSO4), filtered and concentrated under reduced pressure. The crude brown oil was purified using a short chromatography column (silica gel) eluting with hexane to yield a colourless oil (9.80 g, 96.7%). 1H NMR (400 MHz, CDCl3): δ (ppm) 0.85 (t, 3H), 1.22–1.36 (m, 10H), 1.82 (quint, 2H), 4.23 (t, 2H), 7.22 (t, 2H), 7.37 (d, 1H), 7.44–7.55 (m, 2H), 8.02 (d, 1H), 8.18 (s, 1H). MS m/z (APCI): 360.0 (M + 2), 358.2 (M+), 357.0 (M − 1), 245.9 (M − C8H17). HPLC: 100%.
A solution of 3,6-dibromocarbazole (3, 1.50 g, 4.60 mmol), C8H17Br (1.00 g, 5.07 mmol) and PTC (0.10 g, 0.28 mmol) in toluene (20 mL) was stirred in a 100 mL two necked round-bottomed flask. An aqueous NaOH solution (10.0 g with 20 mL H2O) was added and the reaction mixture was heated to 70 °C and stirred for 10 h. The reaction mixture was added to a separating funnel and the water layer removed and washed with toluene (20 mL). The combined toluene layers were combined and washed with water (40 mL). It was then dried (MgSO4), filtered and concentrated under reduced pressure. The crude light yellow solid was purified using a short chromatography column (silica gel) eluting with hexane to yield a white solid (1.20 g, 57%). 1H NMR (400 MHz, CDCl3): δ (ppm) 0.85 (t, 3H), 1.22–1.30 (m, 10H), 1.81 (quint, 2H), 4.23 (t, 2H), 7.26 (d, 2H), 7.55 (d, 2H), 8.13 (s, 2H). MS m/z (APCI): 437.1 (M+), 437.9 (M + 1) 357.0 (M − Br). HPLC: 100%.
3,6,8-Tribromo-9-octylcarbazole (Cz-3Br)
Silica gel (50.0 g) was added to a solution of 9-octylcarbazole (1.50 g, 0.0054 mol) in chloroform (150 mL) and stirred at room temperature for 5 mins. To this suspension was added a solution of N-bromosuccinimide (4.78 g, 0.0268 mol) in chloroform (100 mL) dropwise at room temperature. The resulting reaction mixture was stirred for an additional 65 h, the silica gel filtered and the chloroform removed under reduced pressure to yield a light brown powder (2.00 g, 72.2%). 1H NMR (400 MHz, CDCl3): δ (ppm) 0.87 (t, 3H), 1.25–1.39 (m, 10H), 1.83 (quint, 2H), 4.64 (t, 2H), 7.30 (d, 1H), 7.58 (d,1H), 7.73 (s, 1H), 8.08 (s, 1H), 8.11 (s, 1H). MS m/z (APCI): 515.9 (M+), 514.9 (M − 1). HPLC: 98%.
1,3,6,8-Tetrabromo-9-octylcarbazole (Cz-4Br)
A solution of 1,3,6,8-tetrabromo-carbazole (4, 1.00 g, 2.07 mmol), C8H17Br (0.40 g, 2.28 mmol) and PTC (0.10 g, 0.13 mmol) in toluene (20 mL) was stirred in a 50 mL two necked round-bottomed flask. An aqueous NaOH solution (10.0 g with 20 mL H2O) was added and the reaction mixture was heated to 70 °C and stirred for 10 h. The reaction mixture was added to a separating funnel and the water layer removed and washed with toluene (20 mL). The combined toluene layers were combined and washed with water (40 mL). It was then dried (MgSO4), filtered and concentrated under reduced pressure. The crude solid was recrystallised using a solvent mixture of toluene and ethanol (ratio = 7:3) to yield white crystals (1.00 g, 81.3%). 1H NMR (400 MHz, CDCl3): δ (ppm) 0.87 (t, 3H), 1.24–1.31 (m, 10H), 1.77 (quint, 2H), 5.10 (t, 2H), 7.78 (s, 2H), 8.04 (s, 2H). MS m/z (APCI): 596 (M + 1), 482.0 (M − C8H17). HPLC: 98.5%.
n-Butyllithium (2.5 M in hexanes, 12.3 mL, 30.75 mmol) was added slowly to a solution of 3-bromo-9-octyl-carbazole (Cz-1Br, 9.00 g, 25.10 mmol) in dry THF (100 mL) at approximately −78 °C in a dry 250 mL flask. The resulting reaction mixture was maintained at the same temperature for one hour and subsequently tri(isopropyl)borate (11.6 g, 61.50 mmol) was added to quench the lithium salt. The reaction was warmed to room temperature and stirred overnight. Dilute HCl (20% v/v, 100 mL) was added and stirred for one hour. The organics were extracted into diethylether (2 × 100 mL), washed with water (1 × 100 mL), dried (MgSO4), filtered and concentrated under reduced pressure. The crude boronic acid was purified by gravity column chromatography (silica gel) using DCM:MeOH (= 25:1) as the eluent to yield a white powder (7.00 g, 85.3%).
3,3′-Bis(9-octylcarbazole) (Cz-2)
3-Bromo-9-octylcarbazole (Cz-1Br, 0.30 g, 0.84 mmol), 3-bromo-9-octylcarbazole boronic acid (5, 0.32 g, 1.00 mmol), K2CO3 (0.35 g, 2.51 mmol) and PTC (0.03 g, 0.08 mmol) were added into a 50 mL flask and the system was evacuated, with the aid of a vacuum pump, and filled with nitrogen 3 times. Subsequently, Pd(PPh3)4 (0.05 g, 0.04 mmol) was added, then toluene (20 mL) and water (10 mL), also with vacuum/N2 (×3). After the reaction mixture was stirred at 90 °C for 2 days, water (20 mL) and toluene (10 mL) were added and the toluene layer separated and subsequently removed using a rotary evaporator. The crude product was purified by gravity column chromatography (silica gel) using DCM:hexane 20%:80% as the eluent to yield a white solid (0.20 g, 42.6%). 1H NMR (400 MHz, CD2Cl2): δ (ppm) 0.88 (t, 6H), 1.27–1.46 (m, 20H), 1.92 (quint, 4H), 4.36 (br, S, 4H), 7.16–7.32 (m, 2H), 7.48–7.64 (m, 6H), 7.77–7.96 (m, 2H), 8.19 (d, 2H, J = 7.6 Hz), 8.30–8.63 (m, 2H). MS m/z (APCI): 557.4 (M + 1), 556.4 (M+), 444.3 (M − C8H17). HPLC: 100%. Elemental analysis: expected N (5.03%), C (86.28%), H (8.69%); obtained N (5.14%), C (85.96%), H (8.414%).
3,6-Bis[9′-octylcarbazol-3′-yl]-9-octylcarbazole (Cz-3)
3,6-Dibromo-9-octylcarbazole (Cz-2Br, 0.30 g, 0.70 mmol), 3-bromo-9-octylcarbazole boronic acid (5, 0.50 g, 1.53 mmol), K2CO3 (0.50 g, 3.47 mmol) and PTC (0.10 g, 0.07 mmol) were added into a 50 mL flask and the system was evacuated, with the aid of a vacuum pump, and filled with nitrogen 3 times. Subsequently, Pd(PPh3)4 (0.05 g, 0.04 mmol) was added, then toluene (20 mL) and water (10 mL), also with vacuum/N2 (×3). After the reaction mixture was stirred at 90 °C for 2 days, water (20 mL) and toluene (10 mL) were added and the toluene layer separated and subsequently removed using a rotary evaporator. The crude product was purified by gravity column chromatography (silica gel) using DCM:hexane 20%:80% as the eluent to yield a white solid (0.40 g, 64.0%). 1H NMR (400 MHz, CD2Cl2): δ (ppm) 0.87 (overlapping t, 9H), 1.26–1.50 (m, 30H), 1.92 (overlapping quint, 6H), 4.34 (overlapping t, 6H), 7.16–7.32 (m, 2H), 7.42–7.53 (m, 8H), 7.84–7.87 (m, 4H), 8.20 (d, 2H, J = 7.6 Hz), 8.44 (overlapping d, 2H), 8.51 (overlapping d, 2H). MS m/z (APCI): 834.7 (M+), 833.5 (M − 1), 721.5 (M − C8H17). HPLC: 100%. Elemental Analysis: expected N (5.04%), C (86.38%), H (8.58%); obtained N (5.11%), C (85.91%), H (8.337%).
1,3,6-Tri(9′-octylcarbazol-3′-yl)-9-octylcarbazole (Cz-4)
3,6,8-Tribromo-9-octylcarbazole (Cz-3Br, 0.50 g, 0.97 mmol), 3-bromo-9-octylcarbazole boronic acid (5, 1.60 g, 3.86 mmol), K2CO3 (1.30 g, 9.66 mmol) and PTC (0.10 g, 0.10 mmol) were added into a 50 mL flask and the system was evacuated, with the aid of a vacuum pump, and filled with nitrogen 3 times. Subsequently, Pd(PPh3)4 (0.10 g, 0.10 mmol) was added, then toluene (20 mL) and water (10 mL), also with vacuum/N2 (×3). After the reaction mixture was stirred at 90 °C for 3 days, water (20 mL) and toluene (10 mL) were added and the toluene layer separated and subsequently removed using a rotary evaporator. The crude product was purified by gravity column chromatography (silica gel) using DCM:hexane 20%:80% as the eluent to yield a white solid (0.80 g, 78.0%). 1H NMR (400 MHz, CD2Cl2): δ (ppm) 0.60–1.50 (m, 54H), 1.93 (overlapping quint, 6H), 3.87 (t, 2H), 4.31–4.42 (overlapping t, 6H), 7.21–7.28 (m, 3H), 7.40–7.54 (m, 10H), 7.72 (dd, 1H, J = 1.6 Hz, 8.3 Hz), 7.80 (d, 1H, J = 1.8 Hz), 7.84 (dd, 1H, J = 1.6 Hz, 8.3 Hz), 7.89 (dd, 1H, J = 1.7 Hz, 8.5 Hz), 7.93 (dd, 1H, J = 1.6 Hz, 8.5 Hz), 8.11–8.23 (3H, m), 8.33 (d, 1H, J = 1.5 Hz), 8.47 (d, 1H, J = 1.5 Hz), 8.50 (d, 1H, J = 1.5 Hz), 8.56 (m, 2H). MS m/z (APCI): 1111.8 (M+), 1110.9 (M − 1). HPLC: 100%. Elemental analysis: expected N (5.04%), C (86.44%), H (8.52%); obtained N (5.13%), C (86.13%), H (8.299%).
1,3,6,8-Tetra(9′-octylcarbazol-3′-yl)-9-octylcarbazole (Cz-5)
1,3,6,8-Tetrabromo-9-octylcarbazole (Cz-4Br, 0.70 g, 1.17 mmol), 3-bromo-9-octylcarbazole boronic acid (5, 2.40 g, 5.87 mmol), K2CO3 (1.60 g, 11.70 mmol) and PTC (0.10 g, 0.12 mmol) were added into a 50 mL flask and the system was evacuated, with the aid of a vacuum pump, and filled with nitrogen 3 times. Subsequently Pd(PPh3)4 (0.10 g, 0.12 mmol) was added, then toluene (20 mL) and water (10 mL), also with vacuum/N2 (×3). After the reaction mixture was stirred at 90 °C for 3 days, water (20 mL) and toluene (10 mL) were added and the toluene layer separated and subsequently removed using a rotary evaperator. The crude product was purified by gravity column chromatography (silica gel) using DCM:hexane 30%:70% as the eluent to yield a white solid (0.70 g, 42.9%). 1H NMR (400 MHz, CD2Cl2): δ (ppm) 0.46–1.00 (m, 26H), 1.23–1.47 (m, 41H), 1.90 (overlapping quint, 8H), 3.44 (t, 2H), 4.31 (t, 4H), 4.35 (t, 4H), 7.25–7.29 (m, 4H), 7.39–7.52 (m, 12H), 7.74 (dd, 2H, J = 1.5 Hz, 8.3 Hz), 7.81 (d, 2H, J = 1.8 Hz), 7.96 (dd, 2H, J = 1.6 Hz, 8.5 Hz), 8.12 (d, 2H, J = 7.7 Hz), 8.20 (dd, 2H, J = 7.7 Hz), 8.35 (s, 2H), 8.54 (d, 2H, J = 1.4 Hz), 8.59 (d, 2H, J = 1.4 Hz). MS m/z (APCI): 1389.1 (M+), 1276.8 (M − C8H17). HPLC: 100%. Elemental analysis: expected N (5.04%), C (86.47%), H (8.49%); obtained N (5.18%), C (86.10%), H (8.261%).
Acknowledgements
This work was financially supported by the NSFC (20874025, 21174045), NSFC Research Fellowship for International Young Scientists (21150110141), the Key Fellowship of China Post-doctoral Science Foundation, the Open Program for Beijing National Laboratory for Molecular Sciences (BNLMS) and the Fundamental Research Funds for the Central Universities (HUST2010MS101). We also thank the Analytical and Testing Center of Huazhong University of Science and Technology.
References
- K. Y. Law, Chem. Rev., 1993, 93, 449 CrossRef CAS.
-
(a) S. Beaupre, P. L. T. Boudreault and M. Leclerc, Adv. Mater., 2010, 22, E6 CrossRef CAS;
(b) Z. C. He, C. Zhang, X. F. Xu, L. J. Zhang, L. Huang, J. W.Chen, H. B. Wu and Y. Cao, Adv. Mater., 2011, 23, 3086 CrossRef CAS;
(c) N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295 CrossRef CAS.
-
(a) C. H. Chen, J. Shi and C. W. Tang, Macromol. Symp., 1997, 125, 1 CrossRef;
(b) C. Adachi, K. Nagai and N. Tamoto, Appl. Phys. Lett., 1995, 66, 2679 CrossRef CAS;
(c) C. Giebler, H. Antoniadis, D. D. C. Bradley and Y. Shirota, J. Appl. Phys., 1999, 85, 608 CrossRef;
(d) H. Bassler, Adv. Mater., 1993, 5, 662 CrossRef.
-
(a) Y. D. Zhang, L. Wang, T. Wada and H. Sasabe, Appl. Phys. Lett., 1997, 70, 2949 CrossRef CAS;
(b) S. Grigalevicius, Synth. Met., 2006, 156, 1 CrossRef CAS and references therein..
- R. H. Partridge, Polymer, 1983, 24, 733 CrossRef CAS.
-
(a) S. Salman, D. Kim, V. Coropceanu and J-L. Bredas, Chem. Mater., 2011, 23, 5223 CrossRef CAS;
(b) V. Jankus and A. Monkman, Adv. Funct. Mater., 2011, 21, 3350 CrossRef CAS;
(c) K. Brunner, A. v-Dijken, H. Borner, J. J. A. M. Bastiaansen, N. M. M. Kiggen and B. M. W. Langeveld, J. Am. Chem. Soc., 2004, 126, 6035 CrossRef CAS;
(d) A. v-Dijken, J. J. A. M. Bastiaansen, N. M. M. Kiggen, B. M. W. Langeveld, C. Rothe, A. Monkman, I. Bach, P. Stossel and K. Brunner, J. Am. Chem. Soc., 2004, 126, 7718 CrossRef;
(e) Y. C. Chen, G. S. Huang, C. C. Hsiao and S. A. Chen, J. Am. Chem. Soc., 2006, 128, 8549 CrossRef CAS;
(f) D. Kim, V. Coropceanu and J-L. Bredas, J. Am. Chem. Soc., 2011, 133, 17895 CrossRef CAS;
(g) M. H. Tsai, Y. H. Hong, C. H. Chang, H. C. Su, C. C. Wu, A. Matoliukstyte, S. Grigalevicius, J. V. Grazulevicius and C. P. Hsu, Adv. Mater., 2007, 19, 862 CrossRef CAS.
-
(a) Q. Liu, W. M. Liu, B. Yao, H. K. Tian, Z. Y. Xie, Y. H. Geng and F. S. Wang, Macromolecules, 2007, 40, 1851 CrossRef CAS;
(b) C. Y. Chi, C. Im, V. Enkelmann, A. Ziegler, G. Lieser and G. Wegner, Chem.–Eur. J., 2005, 11, 6833 CrossRef CAS;
(c) R. E. Martin and F. Diederich, Angew. Chem., Int. Ed., 1999, 38, 1350 CrossRef;
(d) D. Rueda, M. G. Zolotukhin, G. Nequlqueo, C. Garcia, J. G. de la Campa and J. de Abajo, Macromol. Chem. Phys., 2000, 201, 427 CrossRef CAS;
(e) C. Z. Zhou, T. X. Liu, J. M. Xu and Z. K. Chen, Macromolecules, 2003, 36, 1457 CrossRef CAS.
- O. Paliulis, J. Ostrauskaite, V. Gaidelis, V. Jankauskas and P. Strohriegl, Macromol. Chem. Phys., 2003, 204, 1706 CrossRef CAS.
- M. Sonntag and P. Strohriegl, Chem. Mater., 2004, 16, 4736 CrossRef CAS.
- W. Jiang, L. Duan, J. Qiao, G. F. Dong, D. Q. Zhang, L. D. Wang and Y. Qiu, J. Mater. Chem., 2011, 21, 4918 RSC.
- K. Smith, D. M. James, A.G. Mistry, M. R. Bye and D. J. Faulkner, Tetrahedron, 1992, 48, 7479 CrossRef CAS.
- L. S. Chinelatto Jr, J. D. Barrio, M. Pinol, L. Oriol, M. A. Matranga, M. P. De Santo and R. Barberi, J. Photochem. Photobiol., A, 2010, 210, 130 CrossRef.
- J. R. Platt, J. Chem. Phys., 1949, 17, 484 CrossRef CAS.
- H. Park, J. Lee, I. Kang, H. Y. Chu, J. I. Lee, S. K. Kwon and Y. H. Kim, J. Mater. Chem., 2012, 22, 2695 RSC.
- I. Imae, Y. Kawakami, Y. Ooyama and Y. Harima, Macromol. Symp., 2007, 249 Search PubMed.
-
(a) B. Horovitz, Solid State Commun., 1982, 41, 729 CrossRef CAS;
(b) C. Castiglioni, M. Gussoni, J. T. Lopez Navarrete and G. Zerbi, Solid State Commun., 1988, 65, 625 CrossRef CAS.
- G. Zerbi, M. Veronelli, S. Martina, A. D. Schluter and G. Wegner, J. Chem. Phys., 1994, 100, 978 CrossRef CAS.
- C. C. Ferron, M. C. R. Delgado, O. Gidron, S. Sharma, D. Sheberla, Y. Sheynin, M. Bendikov, J. T. L. Navarrete and V. Hernandez, Chem. Commun., 2012, 48, 6732 RSC.
- J. F. Ambrose and R. F. Nelson, J. Electrochem. Soc., 1968, 115, 1159 CrossRef CAS.
- Y. C. Chen, G. S. Huang, C. C. Hsiao and S. A. Chen, J. Am. Chem. Soc., 2006, 128, 8549 CrossRef CAS.
- J. J. Park, T. J. Park, W. S. Jeon, R. Pode, J. Jang, J. H. Kwon, E. S. Yu and M. Y. Chae, Org. Electron., 2009, 10, 189 CrossRef CAS.
- T. Yasuda, K. Fujita, T. Tsutsui, Y. H. Geng, S. W. Culligan and S. H. Chen, Chem. Mater., 2005, 17, 264 CrossRef CAS.
- V. Jankus and A. P. Monkman, Adv. Funct. Mater., 2011, 21, 3350 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 