Superior high rate performance of core–shell Li4Ti5O12/carbon nanocomposite synthesized by a supercritical alcohol approach†
Agung
Nugroho
ab,
Wonyoung
Chang
c,
Su
Jin Kim
c,
Kyung
Yoon Chung
c and
Jaehoon
Kim
*abd
aSupercritical Fluid Research Laboratory, Clean Energy Research Center, Korea Institute of Science and Technology (KIST), Hwarangno 14-gil 5, Seongbuk-gu, Seoul 136-791, Republic of Korea. E-mail: jaehoonkim@kist.re.kr; Fax: +82-2-958-5205; Tel: +82-2-958-5874
bClean Energy and Chemical Engineering, University of Science and Technology, 113 Gwahangno, Yuseong-gu, Daejeon 305-333, Republic of Korea
cCenter for Energy Convergence, Korea Institute of Science and Technology (KIST)
dGreen School, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 136-701, Republic of Korea
First published on 17th September 2012
Abstract
A facile, fast, and effective method for the preparation of a core–shell LTO/C nanocomposite via a supercritical alcohol route is presented. Organic-modified LTO nanocrystals were first synthesized in supercritical methanol at a short reaction time of 15 min using oleylamine as a surface-modifier and as a carbon precursor. Subsequent calcination under inert conditions resulted in a highly crystalline LTO core with a size of 5–15 nm and highly graphitic carbon shell with a thickness of 0.7–2.3 nm. The obtained core–shell LTO/C nanocomposite exhibited superior high rate performance, long-term cyclability, and low-temperature discharge capacity.
Because of the current issues of global warming and fossil fuel depletion, extensive efforts have been made to develop renewable and sustainable energy sources and energy storage materials. Large-scale Li-ion batteries (LIBs) have received considerable attention not only as power sources for plug-in hybrid electric vehicles (PHEV) and electric vehicles (EV), but also for energy storage applications.1,2 For such applications, it is essential that the LIB retains a high level of safety, high energy and power densities, excellent cyclability, and low cost. Spinel lithium titanate (Li4Ti5O12, LTO) is considered to be one of the most promising anode materials for the large-scale LIBs, for use in applications such as electric vehicles and storage devices for renewable energy.2,3 LTO is a very safe anode material because of its high voltage plateau at around 1.5 V (vs. Li+/Li), avoiding electrolyte decomposition and lithium metal deposition. For the same reason, a solid electrolyte interface is less likely to form on the surface of LTO, which can mean an improvement in electrochemical performance.1 In addition, LTO is known to exhibit remarkable long-term cyclability and structural stability because of negligibly small volume changes during lithium ion insertion/deinsertion.4,5 These properties make it a highly promising anode material for large-scale LIB applications. However, LTO has an inherent drawback; it has extremely poor electrical conductivity (∼10−13 S cm−1)6 and sluggish lithium ion diffusion (∼10−8 cm2 s−1),7 therefore coarse LTO often exhibits unsatisfactory rate performance and poor cyclability for large-scale applications.
Several approaches have been suggested to overcome the significant drawbacks associated with LTO. The most common methods involve a reduction in particle size to decrease the lithium diffusion pathway8–16 and the formation of a conductive coating (e.g., carbon, surface nitridation, phosphates, metals, etc.) on the LTO surface in order to enhance electronic conductivity between the particles and the current collector.17–34 Both of these strategies have been shown to enhance the rate performance. However, the previous works have considered “reduction in particle size” and “coating of carbon layer” as separate operations, rather than merging both of the operations together. It is a great challenge to produce nano-sized and highly crystalline LTO particles, because of the preferential formation of a titanium oxide phase from the titanium precursors. Once this phase is formed, a very-high-temperature (above 800 °C) calcination is required to form phase-pure LTO with high crystallinity, which inevitably leads to interparticle agglomeration and therefore increased particle size.13,18,35 Attempts to prepare nano-sized LTO using low-temperature synthetic procedures and a short annealing period often produce compound particles with low crystallinity that contain impurities, thereby the compounds would exhibit low structural integrity and low charge–discharge cycling performance.
In addition to the difficulties involved in synthesizing highly crystalline, nano-sized LTO, it is extremely challenging to obtain uniform carbon coverage on the entire surface of nano-sized particles. Table S1† summarizes the various morphologies of carbon-coated LTO (LTO/C) produced using different methods, and the results of electrochemical performance testing. Thermal vapor decomposition or wet mixing followed by subsequent calcination are the most widely studied approaches for the preparation of LTO/C. These methods, however, often result in non-uniform, partial coverage of the carbon layer on the LTO particles, which can lead to polarization of the electrode and poor electrochemical performance. Furthermore, to ensure graphitization from the carbon precursors, the calcination has to be carried out at elevated temperatures, which inevitably leads to particle growth to the detriment of the rate performance. Based on previous findings, the ideal LTO/C for a high rate performance and long-term cyclability would be nano-sized LTO particles with high crystallinity, completely and uniformly covered with a highly conductive carbon film of nanometer-scale thickness, namely a core–shell LTO/C structure. Such a synthetic strategy would be a great challenge using the conventional methods summarized in Table S1.†
Supercritical fluids can offer environmentally benign and facile synthetic conditions for the production of nanomaterials owing to their unique physical properties, including low viscosity, fast diffusion, zero surface tension, and tunable physical properties.36–38 These factors make them promising media for overcoming the barriers associated with other techniques, which include the generation of toxic reaction waste, difficulty in producing nano-sized materials, nonuniform coating, and transport limitations. Herein, we report a facile and effective supercritical methanol (scMeOH) approach to synthesizing a core–shell LTO/C nanocomposite. As shown in Fig. 1, organic-modified LTO nanocrystals were first synthesized in scMeOH using oleylamine as a surface-modifier and as a carbon precursor in a short reaction time of 15 min. Subsequent calcination under inert conditions resulted in both an increase in LTO crystallinity and the formation of a uniform carbon layer on the individual LTO nanocrystals. In contrast to the conventional carbon coating methods, an interparticle agglomeration-free LTO/C nanocomposite could be obtained, even after the high-temperature annealing, because the formation of carbon from the surface capping agent could prevent the nanocrystals from aggregating. As a result, a highly crystalline LTO core with a size of 5–15 nm and highly graphitic carbon shell with a thickness of 0.7–2.3 nm could be obtained. By adjusting the surface-modifier coverage on the LTO particles, the thickness of the carbon layer and the carbon content could be easily controlled. The synthesized core–shell LTO/C nanocomposite exhibited an excellent rate performance, long-term stability and low-temperature discharge capacity as a result of both high electronic conductivity and a short lithium ion diffusion pathway. This simple synthetic route for producing carbon-coated nanocrystals may be easily extended to other anode or cathode materials, where the effects of the nanoscale particles and electronic conductivity are essential for improving their electrochemical performance.
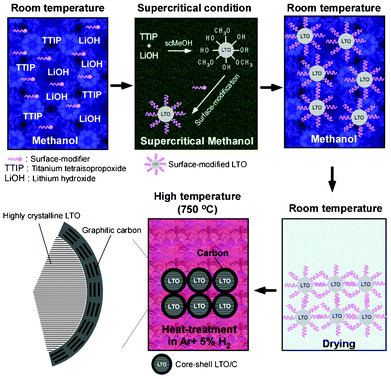 | ||
| Fig. 1 Preparation scheme for core–shell LTO/C nanocomposite in supercritical methanol and subsequent calcination. | ||
The powder X-ray diffraction (PXRD) patterns in Fig. S1† show the LTO phase formed in scMeOH using titanium tetraisopropoxide (TTIP) and lithium hydroxide as the precursors. In the presence of oleylamine, surface-modified LTO was produced, as shown in the Fourier-transform infrared (FT-IR) spectra of the samples (Fig. S2†). The morphologies of the synthesized samples were examined using high-resolution transmission electron microscopy (HR-TEM), as shown in Fig. 2. Unmodified LTO (LTO-scMeOH) showed a slightly agglomerated structure and particle sizes were in the range 5–10 nm. The surface-modified LTO at the low oleylamine concentration (LTO-OA1, TTIP/oleylamine molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) showed a very similar size and morphology to those of LTO-scMeOH (Fig. S3†). An increase in oleylamine concentration to a molar ratio of TTIP/oleylamine of 10:8 caused a decrease in particle size to 2–4 nm (LTO-OA2, Fig. 2b and Fig. S4†). This reduction in particle size in the presence of a high concentration of surface-modifier is probably caused by the covering of the surfaces of the nanoparticles, resulting in inhibition of particle growth. After the calcination, a slight increase in particle size was observed in the LTO/C nanocomposites; the particle size of LTO-OA1-750C was in the range 5–15 nm and that of LTO-OA2-750C was 5–10 nm. The crystallite sizes for LTO-OA1-750C and LTO-OA2-750C estimated using the PXRD patterns were 9.2 nm and 5.0 nm, respectively. Considering the significant increase in particle size during calcination of LTO without surface-modifiers,7,34,36 the HR-TEM and PXRD results clearly suggest that particle growth was highly restricted during the heat-treatment step at 750 °C for 5 h. This indicates that the formation of the carbon layer from oleylamine molecules attached to the surface of LTO nanocrystals effectively restricted the particle growth, possibly by interparticle agglomeration. A close investigation of the HR-TEM images of the LTO/C samples clearly revealed that each LTO nanocrystal was completely and uniformly covered by a carbon layer, thereby forming a core–shell structure. One to five graphene layers were observed in the LTO/C samples, and Raman spectra of the samples showed graphitic carbon formation (Fig. S5†). The carbon thickness and carbon content of LTO-OA1-750C were 0.7–1.5 nm and 6.4%, respectively, and those of LTO-OA2-750C were 1.3–2.3 nm and 14.0%, respectively (Fig. S6†). This indicates that the carbon layer thickness and carbon content can be controlled by adjusting the surface-modifier concentration. In addition, each core–shell LTO/C particle was well–connected to its neighboring particle, as shown in Fig. S7,† which can provide good electron path continuity around the LTO nanocrystals.
:1) showed a very similar size and morphology to those of LTO-scMeOH (Fig. S3†). An increase in oleylamine concentration to a molar ratio of TTIP/oleylamine of 10:8 caused a decrease in particle size to 2–4 nm (LTO-OA2, Fig. 2b and Fig. S4†). This reduction in particle size in the presence of a high concentration of surface-modifier is probably caused by the covering of the surfaces of the nanoparticles, resulting in inhibition of particle growth. After the calcination, a slight increase in particle size was observed in the LTO/C nanocomposites; the particle size of LTO-OA1-750C was in the range 5–15 nm and that of LTO-OA2-750C was 5–10 nm. The crystallite sizes for LTO-OA1-750C and LTO-OA2-750C estimated using the PXRD patterns were 9.2 nm and 5.0 nm, respectively. Considering the significant increase in particle size during calcination of LTO without surface-modifiers,7,34,36 the HR-TEM and PXRD results clearly suggest that particle growth was highly restricted during the heat-treatment step at 750 °C for 5 h. This indicates that the formation of the carbon layer from oleylamine molecules attached to the surface of LTO nanocrystals effectively restricted the particle growth, possibly by interparticle agglomeration. A close investigation of the HR-TEM images of the LTO/C samples clearly revealed that each LTO nanocrystal was completely and uniformly covered by a carbon layer, thereby forming a core–shell structure. One to five graphene layers were observed in the LTO/C samples, and Raman spectra of the samples showed graphitic carbon formation (Fig. S5†). The carbon thickness and carbon content of LTO-OA1-750C were 0.7–1.5 nm and 6.4%, respectively, and those of LTO-OA2-750C were 1.3–2.3 nm and 14.0%, respectively (Fig. S6†). This indicates that the carbon layer thickness and carbon content can be controlled by adjusting the surface-modifier concentration. In addition, each core–shell LTO/C particle was well–connected to its neighboring particle, as shown in Fig. S7,† which can provide good electron path continuity around the LTO nanocrystals.
 | ||
| Fig. 2 HR-TEM images of (a) LTO-scMeOH, (b) LTO-OA2, (c) LTO-OA1-750C, and (d) LTO-OA2-750C. White dashed lines delineate LTO nanocrystals. White numbers indicate carbon layer thickness. | ||
The obtained core–shell LTO/C nanocomposite exhibited excellent rate performance, as shown in Fig. 3a. For comparison purposes, the electrochemical properties of micron-sized LTO particles synthesized using a solid-state method (LTO-SS), submicron-sized LTO particles synthesized in supercritical water and subsequently calcined at 700 °C (LTO-scH2O-700C),36 and mesoporous nano/microspheres synthesized in scMeOH and subsequently calcined at 600 °C (LTO-scMeOH-600C) are also shown in the figure. The morphologies of LTO-SS, LTO-scH2O-700C and LTO-scMeOH-600C are shown in Fig. S8.† The discharge capacity of LTO-OA2-750C at 0.1 C was 176.0 mA h g−1, which is very close to that of LTO-scMeOH-600C, and is higher than those of LTO-scH2O-700C and LTO-SS. At higher C-rates, LTO-OA2-750C exhibited a much higher specific discharge capacity; the capacity was 139.0 mA h g−1 at 20 C and 101.0 mA h g−1 at 50 C. As shown in Fig. 3b and listed in Table S1,† the core–shell LTO/C nanocomposite exhibited a remarkable rate performance, which was superior to those of other carbon-coated LTOs,17–24,27–31,33 surface-nitrided LTOs,25,26,32 and nanostructured LTOs reported in the literature.9,11,16 To the best of our knowledge, the highest discharge capacity ever reported for LTO at 50 C is 102 mA h g−1.9 However, the composition of the conductor (i.e., carbon black) in the electrode was much higher (15%) than the one used in this study (8%), indicating a lower volumetric capacity in their composite electrode. The galvanostatic charge–discharge curves for lithium insertion–extraction of each sample at 1 C are shown in Fig. S9.† The voltage differences in the charge–discharge curves of LTO-OA2-750C are much smaller than those of the other LTO samples, indicating low polarization of the core–shell LTO/C sample. The significant improvement in the rate performance of LTO-OA2-750C and lower polarization could be mainly caused by the short lithium diffusion path and the high electronic conductivity associated with the core–shell structure.
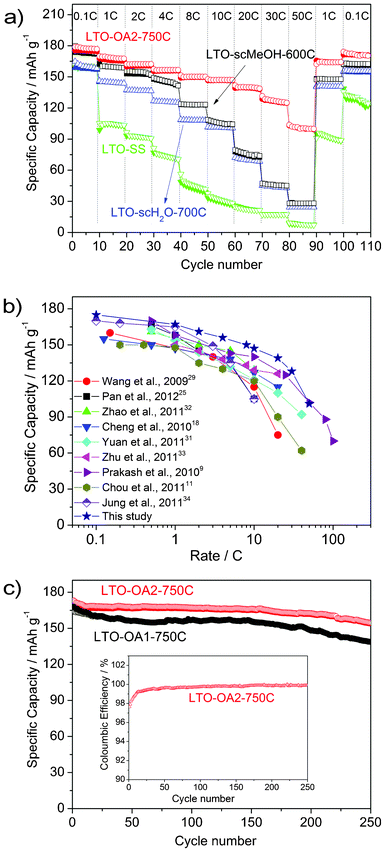 | ||
| Fig. 3 (a) High rate performance of core–shell LTO/C nanocomposite with variable C-rates compared to LTO synthesized using other methods. The samples were progressively charged and discharged at 0.1–50 C in the potential range 1.0–2.5 V (vs. Li+/Li). (b) Specific capacity of core–shell LTO/C nanocomposite compared to the other LTO/C or LTO/N materials as a function of C-rate. (c) Cycling performance and coulombic efficiency (inset) of the core–shell LTO/C nanocomposite at a rate of 1 C up to 250 cycles in the potential range 1.0–2.5 V (vs. Li+/Li). | ||
The core–shell LTO/C nanocomposite exhibited excellent capacity retention, as shown in Fig. 3c. The discharge capacity after 250 cycles at 1 C was 154.6 mA h g−1 for LTO-OA2-750C, which is 89% of the initial discharge capacity (173.2 mA h g−1). The coulombic efficiency, shown in the inset in Fig. 3b, was almost 100% after the first 13 cycles. The sample with a low carbon content (LTO-OA1-750C) also exhibited excellent long-term cyclability. Furthermore, LTO-OA2-750C exhibited superior initial capacity and capacity retention even at the low temperature of −20 °C, as shown in Fig. S10,† with an initial capacity of 152.5 mA h g−1 at 1 C and 144.8 mA h g−1 after 100 cycles. The low-temperature capacity was only ∼10% lower compared to the capacity at 25 °C and ∼15% lower compared to the capacity at 55 °C.
Electrochemical impedance spectroscopy revealed that the improved electrochemical performance of the core–shell LTO/C nanocomposite was mainly the result of enhanced electronic conductivity, as inferred from the much lower resistance than was seen for the uncoated sample (Fig. S11† and Table S2†). LTO-OA2-750C exhibited a charge-transfer resistance (Rct) two orders of magnitude lower (4.6 Ω) than that of LTO-scMeOH-600C (142.9 Ω), which is the main reason for the improved rate performance of the core–shell LTO/C nanocomposite. The uniform and complete coverage of the carbon layer on each LTO nanocrystal facilitates electron transport on the surface, and lithium ion transport occurs smoothly as a result of the small particle size.
In summary, we have demonstrated a facile, fast, and effective method for the preparation of a core–shell LTO/C nanocomposite via surface-modified LTO synthesis in scMeOH in the very short reaction time of 15 min with subsequent calcination. This is the first method reported for obtaining uniform and complete coverage of a carbon layer on individual LTO nanocrystals. The obtained core–shell LTO/C nanocomposite exhibited superior high rate performance, long-term cyclability, and low-temperature discharge capacity compared to all previously reported LTO/C composite materials. The outstanding electrochemical performance can be attributed to the unique core–shell structure, high electrical conductivity, good structural integrity, and high crystallinity of the core–shell LTO/C nanocomposite. Currently, various types of carbon-coated cathode and carbon-coated anode materials are under development in our laboratory, using the supercritical alcohol route. Because of its simplicity, fast reaction rate, and ease in scale-up by employing a continuous reaction system, supercritical-alcohol-based synthesis is a highly promising technique for the production of functional materials for large-scale LIB applications.
Acknowledgements
This research was supported by the Global Research Lab (GRL) Program of the Ministry of Education, Science and Technology. The authors also acknowledge support from the Green City Technology Flagship Program funded by the Korea Institute of Science and Technology (KIST-2012-2E23337) and the National Research Foundation of Korea Grant funded by the Korean Government (MEST) (2012, University-Institute cooperation program).References
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS.
- T. Ohzuku, A. Ueda and N. Yamamoto, J. Electrochem. Soc., 1995, 142, 1431–1435 CrossRef CAS.
- K. M. Colbow, J. R. Dahn and R. R. Haering, J. Power Sources, 1989, 26, 397–402 CrossRef CAS.
- C. H. Chen, J. T. Vaughey, A. N. Jansen, D. W. Dees, A. J. Kahaian, T. Goacher and M. M. Thackeray, J. Electrochem. Soc., 2001, 148, A102–A104 CrossRef CAS.
- K. Zaghib, M. Simoneau, M. Armand and M. Gauthier, J. Power Sources, 1999, 81–82, 300–305 CrossRef CAS.
- W. J. H. Borghols, M. Wagemaker, U. Lafont, E. M. Kelder and F. M. Mulder, J. Am. Chem. Soc., 2009, 131, 17786–17792 CrossRef CAS.
- A. S. Prakash, P. Manikandan, K. Ramesha, M. Sathiya, J. M. Tarascon and A. K. Shukla, Chem. Mater., 2010, 22, 2857–2863 CrossRef CAS.
- L. Kavan and M. Gratzel, Electrochem. Solid-State Lett., 2002, 5, A39–A42 CrossRef CAS.
- S. L. Chou, J. Z. Wang, H. K. Liu and S. X. Dou, J. Phys. Chem. C, 2011, 115, 16220–16227 CAS.
- T. Doi, Y. Iriyama, T. Abe and Z. Ogumi, Chem. Mater., 2005, 17, 1580–1582 CrossRef CAS.
- L. Cheng, H. J. Liu, J. J. Zhang, H. M. Xiong and Y. Y. Xia, J. Electrochem. Soc., 2006, 153, A1472–A1477 CrossRef CAS.
- Y. Li, G. L. Pan, J. W. Liu and X. P. Gao, J. Electrochem. Soc., 2009, 156, A495–A499 CrossRef CAS.
- E. M. Sorensen, S. J. Barry, H. K. Jung, J. R. Rondinelli, J. T. Vaughey and K. R. Poeppelmeier, Chem. Mater., 2006, 18, 482–489 CrossRef CAS.
- J. Kim and J. Cho, Electrochem. Solid-State Lett., 2007, 10, A81–A84 CrossRef CAS.
- L. Cheng, X. L. Li, H. J. Liu, H. M. Xiong, P. W. Zhang and Y. Y. Xia, J. Electrochem. Soc., 2007, 154, A692–A697 CrossRef CAS.
- L. Cheng, J. Yan, G.-N. Zhu, J.-Y. Luo, C.-X. Wang and Y.-Y. Xia, J. Mater. Chem., 2010, 20, 595–602 RSC.
- X. B. Hu, Z. J. Lin, K. R. Yang, Y. J. Huai and Z. H. Deng, Electrochim. Acta, 2011, 56, 5046–5053 CrossRef CAS.
- Z. L. Jian, L. Zhao, R. Wang, Y. S. Hu, H. Li, W. Chen and L. Q. Chen, RSC Adv., 2012, 2, 1751–1754 RSC.
- E. Kang, Y. S. Jung, G.-H. Kim, J. Chun, U. Wiesner, A. C. Dillon, J. K. Kim and J. Lee, Adv. Funct. Mater., 2011, 21, 4349–4357 CrossRef CAS.
- X. Li, M. Z. Qu, Y. J. Huai and Z. L. Yu, Electrochim. Acta, 2010, 55, 2978–2982 CrossRef CAS.
- X. Li, M. Z. Qu and Z. L. Yu, Solid State Ionics, 2010, 181, 635–639 CrossRef CAS.
- H. Liu, Y. Feng, K. Wang and J. Y. Xie, J. Phys. Chem. Solids, 2008, 69, 2037–2040 CrossRef CAS.
- H. Pan, L. Zhao, Y.-S. Hu, H. Li and L. Chen, ChemSusChem, 2012, 5, 526–529 CrossRef CAS.
- K. S. Park, A. Benayad, D. J. Kang and S. G. Doo, J. Am. Chem. Soc., 2008, 130, 14930–14931 CrossRef CAS.
- G. J. Wang, J. Gao, L. J. Fu, N. H. Zhao, Y. P. Wu and T. Takamura, J. Power Sources, 2007, 174, 1109–1112 CrossRef CAS.
- J. Wang, X. M. Liu, H. Yang and X. D. Shen, J. Alloys Compd., 2011, 509, 712–718 CrossRef CAS.
- Y. G. Wang, H. M. Liu, K. X. Wang, H. Eiji, Y. R. Wang and H. S. Zhou, J. Mater. Chem., 2009, 19, 6789–6795 RSC.
- T. Yuan, R. Cai and Z. P. Shao, J. Phys. Chem. C, 2011, 115, 4943–4952 CAS.
- T. Yuan, X. Yu, R. Cai, Y. K. Zhou and Z. P. Shao, J. Power Sources, 2010, 195, 4997–5004 CrossRef CAS.
- L. Zhao, Y.-S. Hu, H. Li, Z. Wang and L. Chen, Adv. Mater., 2011, 23, 1385–1388 CrossRef CAS.
- G.-N. Zhu, H.-J. Liu, J.-H. Zhuang, C.-X. Wang, Y.-G. Wang and Y.-Y. Xia, Energy Environ. Sci., 2011, 4, 4016–4022 CAS.
- H.-G. Jung, S.-T. Myung, C. S. Yoon, S.-B. Son, K. H. Oh, K. Amine, B. Scrosati and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 1345–1351 CAS.
- T. Yuan, R. Cai, R. Ran, Y. Zhou and Z. Shao, J. Alloys Compd., 2010, 505, 367–373 CrossRef CAS.
- A. Nugroho, S. J. Kim, K. Y. Chung, B.-W. Cho, Y.-W. Lee and J. Kim, Electrochem. Commun., 2011, 13, 650–653 CrossRef CAS.
- T. Adschiri, Y.-W. Lee, M. Goto and S. Takami, Green Chem., 2011, 13, 1380–1390 RSC.
- J. Kim, Y.-S. Park, B. Veriansyah, J.-D. Kim and Y.-W. Lee, Chem. Mater., 2008, 20, 6301–6303 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Materials and methods, Fig. S1–S10, Tables S1 and S2. See DOI:10.1039/c2ra21653a |
| This journal is © The Royal Society of Chemistry 2012 |