Eugenol as a renewable feedstock for the production of polyfunctional alkenes via olefin cross-metathesis†
Hallouma
Bilel
ab,
Naceur
Hamdi
a,
Fethi
Zagrouba
a,
Cédric
Fischmeister
*b and
Christian
Bruneau
*b
aUniversity of Carthage, Higher Institute of Sciences and Technology of Environment of Borj Cedria, B.P. 1003, 2050, Hammam-Lif, Tunisia. E-mail: naceur.hamdi@isste.rnu.tn
bUMR6226: CNRS-Université de Rennes1, Institut des Sciences Chimiques, Campus de Beaulieu, 35042 Rennes Cedex, France. E-mail: christian.bruneau@univ-rennes1.fr
First published on 2nd August 2012
Abstract
The ruthenium-catalyzed cross-metathesis of eugenol derivatives with electron deficient olefins is reported. It is shown that in the presence of ruthenium catalysts, eugenol and its O-protected derivatives have a high tendency to undergo carbon–carbon double bond migration before and after metathesis leading to the formation of conjugated styrene derivatives. The addition of 1,4-benzoquinone suppresses these isomerization reactions and provides an efficient access to new polyfunctional phenol derivatives upon cross-metathesis of the biosourced eugenol with acrylates, acrylonitrile and acrylamides.
Introduction
Due to the foreseen depletion of fossil oil as a main feedstock for fine chemistry, chemists are more and more interested in transformations of renewable resources both towards large tonnage industry, especially for energy supplies and polymers, and for niche products with high added value.1 Catalysis, both heterogeneous and homogeneous, including organometallic, organo and biocatalysis, plays a crucial role in this field and forms the basis of biorefining processes.2 Several families of natural products contain carbon–carbon double bonds, which are particularly suited for their transformation via olefin metathesis processes.1f,g,3 Direct applications have been successfully developed in oleochemistry, especially for the cleavage of unsaturated fatty derivatives to produce shorter aliphatic esters and olefins via ethenolysis,4 α,ω-bifunctional molecules via cross-metathesis with functional olefins,5 and polymers via acyclic diene metathesis polymerization6 or ring opening metathesis polymerization.7 A few reports have recently appeared on the transformation by olefin metathesis of terpenes, another class of valuable natural products.8 Eugenol, featuring a phenolic allylbenzene structure, is representative of the kind of unsaturated compounds that can be extracted from lignocellulosic biomass and transformed via olefin metathesis. Only a few scarce examples of direct self-metathesis9 and cross-metathesis of eugenol with symmetrical internal olefins10 and electron deficient olefins10a,b,11 with ruthenium benzylidene and indenylidene catalysts have been reported. Beside these direct applications of eugenol, a bis(allyl)benzene diene constructed from eugenol has also been converted by acyclic diene metathesis polymerization.12We now report a more systematic study on cross-metathesis of eugenol, O-protected eugenol derivatives and ortho-eugenol with electron deficient olefins including methyl acrylate, methyl methacrylate, acrylonitrile and acrylamides13 leading to several new and bio-sourced polyfunctional phenol derivatives.
Results and discussion
The cross-metathesis reactions were investigated with the four second generation ruthenium catalysts I–IV (Fig. 1).14 | ||
| Fig. 1 Olefin metathesis catalysts. | ||
Cross-metathesis of eugenol with methyl acrylate was first investigated in toluene and the greener dimethyl carbonate15 (DMC) (Scheme 1). Whatever the catalyst used, complete conversion of eugenol 1 was obtained in relatively short reaction times at 80 °C (Table 1, entries 1–4 and 6–7) and even at room temperature (Table 1, entry 5). However, the reaction was not selective and other cross-metathesis products (Fig. 2) could be detected by gas chromatography and 1H NMR of the crude reaction mixtures. These products were identified via selected cross-metathesis transformations.
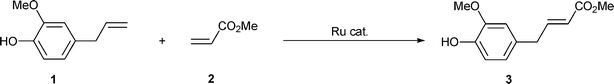 | ||
| Scheme 1 Cross-metathesis of eugenol with methyl acrylate. | ||
| Entry | Catalyst | Benzoquinone | t (h) | Conv. (%)b |
3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:6 GC area ratio :4:6 GC area ratio |
Yield of 3 (%)c |
|---|---|---|---|---|---|---|
| a Conditions: eugenol 1 (0.6 mmol), methyl acrylate 2 (1.2 mmol), solvent: dimethyl carbonate (2 ml), temperature: 80 °C, nd: not determined. b Conversion determined by GC. c Isolated yield. d Solvent: toluene. e Reaction carried out at room temperature. f Isolated yield for the mixture 3 + 4 + 6. | ||||||
| 1 | I (2 mol%) | no | 3 | 100 | 34:60:6 |
nd |
| 2 | II (2 mol%) | no | 3 | 100 | 36:42:22 |
65f |
| 3 | II (2 mol%)d | no | 3 | 100 | 46:42:12 |
50f |
| 4 | II (2 mol%) | no | 24 | 100 | 27:6:67 |
62f |
| 5 | II (0.5 mol%) | no | 3 | 100e | 57:43:nd |
nd |
| 6 | III (2 mol%) | no | 3 | 100 | 44:42:14 |
nd |
| 7 | IV (2 mol%) | no | 3 | 100 | 65:20:15 |
80f |
| 8 | II (1 mol%) | 5 mol% | 16 | 100 | 92:8:0 |
nd |
| 9 | IV (1 mol%) | 5 mol% | 8 | 100 | 91:9:0 |
73 |
| 10 | IV (1 mol%) | 5 mol% | 16 | 100 | 91:9:0 |
78 |
| 11 | IV (0.5 mol%) | 5 mol% | 3 | 74 | 94:6:0 |
nd |
 | ||
| Fig. 2 Undesired products from cross-metathesis reactions involving eugenol. | ||
The two major byproducts were compound 4, resulting from isomerization of the carbon–carbon double bond of 3, and to a lesser extent the styrene derivative 6, arising from cross-metathesis of isoeugenol, resulting from migration of the allylic bond prior to the cross-metathesis with 2 (Fig. 2).
Compound 4 was clearly identified by a tandem cross-metathesis (1 + 2) promoted by catalyst I followed by hydrogenation of the resulting reaction mixture. Whereas GC analysis of the cross-metathesis reaction mixture showed two major peaks, the GC trace of the hydrogenated reaction mixture showed only one major peak that was assigned by GC-MS as the saturated product 5 (Scheme 2, (a)).
 | ||
| Scheme 2 Side-products identification. | ||
Compound 6 was unambiguously identified after cross-metathesis of methyl acrylate 2 with the E-isoeugenol quantitatively obtained by treatment of eugenol 1 with 2 mol% of RuCl2(PPh3)3, an isomerization catalyst, in methanol at 60 °C during 17 h (Scheme 2, (b)). Compound 6 was isolated as the sole metathesis product and as the pure E-isomer.
Whereas it was reported that ruthenium indenylidene catalysts gave noticeable amounts of self-metathesis products, such as 8, with preferred formation of the E-stereoisomer during cross-metathesis of allylphenols and functional allylbenzene derivatives with electron deficient olefins such as acrylic acid, esters, amides, and conjugated enones,10b the reaction carried out with catalysts II, III and IV in dimethyl carbonate gave almost no self-metathesis product from 1. The formation of the self-metathesis product 8 was also less important when benzylidene ruthenium complexes featuring a bulky unsymmetrical N-heterocyclic carbene ligand were used.10a It was also reported that self-metathesis of eugenol without an olefinic partner occurred at 25 °C under neat conditions in the presence of Grubbs first generation catalyst RuCl2(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh)(PCy3)2in vacuo.9 In contrast, the treatment of eugenol 1 with catalyst II in dimethyl carbonate at 80 °C for 3 h provided complete conversion into the functional trans-stilbene 7 resulting from self-metathesis of isoeugenol.
CHPh)(PCy3)2in vacuo.9 In contrast, the treatment of eugenol 1 with catalyst II in dimethyl carbonate at 80 °C for 3 h provided complete conversion into the functional trans-stilbene 7 resulting from self-metathesis of isoeugenol.
From these preliminary tests, it appeared that catalysts bearing a chelating benzylidene ligand proved to be the most selective for the formation of 3. Of note, it was observed that a longer reaction time of 24 h led to higher formation of 6 (Table 1, entries 2, 4), which indicated that a double migration operated from 3 to give 4, and that cross-metathesis of 4 with the excess of methyl acrylate 2 also took place to give 6.
These results clearly indicated that pre- and post-cross-metathesis double bond migration reactions were the origin of undesired side reactions preventing efficient production of cross-metathesis products with electron deficient olefins.16 Limitation of this type of ruthenium-catalyzed olefin migration by the utilization of additives has already been investigated and the beneficial effect of 1,4-benzoquinone was found17 and already exploited in various metathesis transformations of natural products.18 We thus carried out cross-metathesis of eugenol 1 with 2 in the presence of 5 to 20 mol% of 1,4-benzoquinone. As a result, almost all isomerization products disappeared from the GC traces. This made the isolation of pure 3 possible by chromatography, whereas in the absence of 1,4-benzoquinone the mixture of cross-metathesis products could not be simply separated by chromatography over silica gel. The best results were obtained with 1 mol% of catalyst II or IV and 5 mol% of 1,4-benzoquinone in dimethyl carbonate at 80 °C. Under these conditions, complete conversion of 1 was achieved with highly selective formation of 3 isolated in 78% yield (Table 1, entry 10). By contrast with the self-metathesis reaction of eugenol performed without 1,4-benzoquinone, it is noteworthy that in the presence of 5 mol% of 1,4-benzoquinone and 2 mol% of catalyst II in DMC at 80 °C for 3 h, eugenol was converted in 92% yield into its self-metathesis product 8 without any trace of 7. However, in the presence of methyl acrylate and 1,4-benzoquinone, no self-metathesis product 8 was detected by GC analysis.
On the basis of these initial results obtained with methyl acrylate 2, other electron deficient olefins were successfully applied in cross-metathesis with eugenol 1 (Scheme 3, Table 2).
 | ||
| Scheme 3 Cross-metathesis of eugenol 1 with functional olefins. | ||
| Entry | Olefin | Solvent | T (°C) | t (h) | Conv. (%) | Isolated yield (%) |
|---|---|---|---|---|---|---|
|
a Conditions: eugenol (0.6 mmol), olefin (1.2 mmol), catalyst II (2 mol%), 1,4-benzoquinone (5 mol%), isolated yield based on 1, nd = not determined.
b 2 ml of methyl methacrylate (31 equiv.).
c Slow addition of the catalyst within 2 h at 0.5 ml h−1.
d
1:13 molar ratio = 1.
e Isolated yield based on 13.
f
1:13 molar ratio = 1.25.
g
1:15 molar ratio = 1.25.
h Isolated yield based on 15.
|
||||||
| 1 | 9 | Neatb | 90 | 16 | 100 | 60 |
| 2 | 11 | DEC | 100 | (2 + 3)c | 97 | 82 |
| 3 | 13 | DMC | 80 | (2 + 2)c | 93d | 40e |
| 4 | 13 | DMC | 80 | (2 + 2)c | 97f | 60e |
| 5 | 15 | DMC | 80 | (2 + 15)c | 98g | 70h |
Thus, under conditions inspired from cross-metathesis with terpene derivatives,8a when the more sterically hindered methyl methacrylate 9 was reacted with eugenol 1 in the presence of 2 mol% of catalyst II and 5 mol% of 1,4-benzoquinone without solvent, complete conversion of 1 was obtained after 16 h at 90 °C (Table 2, entry 1). No self-metathesis product 8 was formed, and compound 10 was isolated in 60% yield as the sole E-stereoisomer as shown by 2D NMR analysis.
The cross-metathesis with acrylonitrile and acrylamide turned out to be more difficult to perform with high efficiency probably due to the fast degradation of the catalytic species under the experimental conditions used. We have already shown that important efficiency improvements in the cross-metathesis of unsaturated fatty acid derivatives with acrylonitrile were obtained when the catalyst was slowly added to the reaction mixture.5d,e Following this procedure almost complete conversion of 1 was achieved in diethyl carbonate (DEC) when 2 mol% of catalyst II was introduced during 2 h (0.5 ml h−1) and the reaction maintained at 100 °C for 3 additional hours. Under these conditions, 12 was isolated in 82% yield as a mixture of (Z)- and (E)-isomers in a 2:1 respective ratio, and no self-metathesis product 8 or other by-products resulting from pre- or post-isomerization were detected (Table 2, entry 2). The cross-metathesis of eugenol 1 with 2 equivalents of acrylamide 13 proved to be more difficult. When the reaction was carried out for 16 h in the presence of 2 mol% of catalyst II in either dimethyl carbonate at 80 °C or toluene at 100 °C it led to low 66% and 25% respective conversions of 1. Furthermore, it was found that the separation of the two primary acrylic amides 13 and 14 by silica gel chromatography was very difficult. To overcome these problems, catalyst II was slowly added to the reaction mixture and a slight excess of eugenol (1:13 molar ratio = 1.25) was used to facilitate purification. With this protocol a conversion of 97% of 13 was obtained and 14 was isolated as the (E)-isomer in 60% yield after column chromatography over basic alumina (Table 2, entry 4). Transferring these conditions to the cross-metathesis with isopropylacrylamide 15, 98% conversion of 15 was obtained and (E)-16 was isolated in 70% yield (Table 2, entry 5).
O-Protected eugenol derivatives were then applied in cross-metathesis reactions with the same electron deficient olefins under the best conditions found for the cross-metathesis with eugenol (Scheme 4). The results obtained with the phenol functionality protected as an isopropoxy ether (compound 17) are collected in Table 3. Conditions could always be found to reach complete conversion of the substrate 17 and in all cases the addition of 1,4-benzoquinone was found beneficial and improved the isolated yields of the expected products. The isolation of amide derivatives 21 and 22 was again more difficult and moderate isolated yields were obtained even after chromatography over basic alumina, but starting from the olefins 2, 9 and 11, products 18, 19 and 20 were isolated in good yields ranging from 75–85%. From methyl methacrylate the best yields were obtained in neat conditions, and for the cross-metathesis with acrylonitrile and acrylamides the slow addition of the catalyst proved to be the most convenient procedure. The effect of 1,4-benzoquinone was much less pronounced when the reaction was performed in neat methyl methacrylate, which indicated that the excess of methacrylate disfavored the isomerization and self-metathesis of 16. The (E)-isomers of products 18, 19, 21 and 22 were formed as the major products with very high stereoselectivity, whereas 20 was isolated as a mixture of (Z) and (E)-isomers in a 2:1 ratio.
 | ||
| Scheme 4 Cross-metathesis of eugenol derivative 17 with functional olefins. | ||
| Entry | Olefin | Solvent | T (°C) | t (h) | Conv. (%) | Isolated yield (%) |
|---|---|---|---|---|---|---|
|
a
17 (0.6 mmol), olefin (1.2 mmol), catalyst II (2 mol%), 1,4-benzoquinone (5 mol%), conversion based on 17.
b Catalyst IV.
c 2 ml of methyl methacrylate (39 equiv.).
d Slow addition of the catalyst within 2 h at 0.5 ml h−1.
e
17:13 molar ratio = 1.25.
f
17:15 molar ratio = 1.25.
g Yield based on 17.
h Yield based on the electron deficient olefin.
|
||||||
| 1 | 2 | DMC | 80 | 3 | 100 | 18 (75)g |
| 2b | 2 | DMC | 80 | 8 | 100 | 18 (85)g |
| 3 | 9 | Neatc | 90 | 16 | 100 | 19 (75)g |
| 4 | 9 | Neatc | 90 | 16 | 100 | 19 (78)g |
| 5 | 11 | DEC | 100 | (2 + 3)d | 98 | 20 (80)g |
| 6 | 13 | DMC | 80 | (2 + 15)d | 98e | 21 (57)h |
| 7 | 15 | DMC | 80 | (2 + 15)d | 98f | 22 (53)h |
Compound 23, obtained by O-acylation of eugenol, was also submitted to cross-metathesis under the best conditions identified during the experiments with eugenol for each olefin partner (Scheme 5). The best results are gathered in Table 4. As depicted for the cross-metathesis of 17, all reactions proceeded with high to full conversion and the cross-metathesis products were isolated in moderate to good yields. Again, the slow addition of catalyst was necessary to ensure high conversion with acrylonitrile and acrylamide derivatives. Products 24, 25, 27 and 28 were obtained as (E)-isomers whereas 26 was obtained as a mixture of (Z)/(E) isomers in a 3:1 ratio.
 | ||
| Scheme 5 Cross-metathesis of eugenol derived 23. | ||
| Entry | Olefin | Solvent | T (°C) | t (h) | Conv. (%) | Isolated yield (%) |
|---|---|---|---|---|---|---|
|
a
23 (0.6 mmol), olefin (1.2 mmol), catalyst II (2 mol%), 1,4-benzoquinone (5 mol%).
b 2 ml of methyl methacrylate (39 equiv.).
c Yield based on 23.
d Slow addition of the catalyst within 2 h at 0.5 ml h−1.
e Yield based on the electron deficient olefin.
f
23:13 molar ratio = 1.25.
g
23:15 molar ratio = 1.25.
|
||||||
| 1 | 2 | DMC | 80 | 8 | 100 | 24 (81)c |
| 2 | 9 | Neatb | 90 | 16 | 99 | 25 (72)c |
| 3 | 11 | DEC | 100 | (2 + 3)d | 96 | 26 (79)c |
| 4 | 13 | DMC | 80 | (2 + 15)d | 92f | 27 (65)e |
| 5 | 15 | DMC | 80 | (2 + 3)d | 98g | 28 (53)e |
The best conditions identified for the cross-metathesis of eugenol with methyl acrylate, acrylonitrile and acrylamide were used to perform the cross-metathesis with 2-allyl-6-methoxy phenol 29 (o-eugenol), an isomer of eugenol (Scheme 6). Complete conversion of 29 was obtained with 2 equivalents of methyl acrylate 2 and the product 30 was isolated in 60% yield when the reaction was carried out in the presence of 2 mol% of catalyst II and 5 mol% of 1,4-benzoquinone in dimethyl carbonate at 80 °C for 24 h.
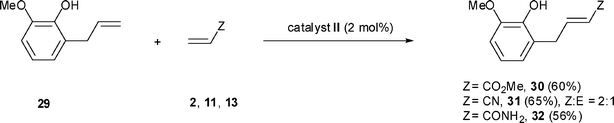 | ||
| Scheme 6 Cross-metathesis of o-eugenol 29. | ||
The reactions with acrylonitrile and acrylamide were conducted in the presence of 5 mol% of 1,4-benzoquinone using the slow addition of 2 mol% of catalyst II within 2 h followed by 3 h at the reaction temperature. The reaction with acrylonitrile 11 performed at 100 °C in diethyl carbonate led to 95% conversion of 29 and 31 was isolated in 65% yield as a 2:1 mixture of (Z) and (E)-isomers, respectively. From acrylamide, the reaction carried out at 80 °C in dimethyl carbonate with a 29:13 molar ratio of 1.25 produced a lower conversion of the amide (75%) and the acrylic amide 32 was isolated as the (E) isomer in 56% yield.
The cross-metathesis reaction with 29 required more severe conditions than with eugenol 1. This is probably due to the presence of the OH group in ortho-position of the reactive allyl group. A steric interaction can take place but the coordinating ability of the hydroxyl group that can interact with a catalytic metal intermediate might also have an inhibiting effect.
Conclusion
We have shown that cross-metathesis of eugenol (4-allyl-2-methoxyphenol) with electron deficient olefins in the presence of second generation ruthenium catalysts is subject to concurrent isomerization reactions catalyzed by the metathesis catalyst precursor, which hampers the production of the expected products. This problem has been solved by using 5 mol% of 1,4-benzoquinone, which inhibits the isomerization of the allyl group of eugenol derivatives before cross-metathesis, and also the double bond migration from the formed metathesis products. Putting in action different procedures such as reactions under neat conditions with methyl methacrylate or slow addition of the catalyst in the case of acrylonitrile and acrylamides, it was possible to prepare a variety of functionalized phenol derivatives. Cross-metathesis from the eugenol isomer 2-allyl-6-methoxyphenol proved to be less productive. All these transformations were carried out without solvent or in green solvents, mainly dialkyl carbonates, and they open the route to the preparation of a variety of polyfunctional products from renewable feedstocks.Acknowledgements
The authors are grateful to the French-Tunisian joint project PHC-UTIQUE n° 09G1203 for financial support to HB.References
- (a) P. N. R. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502 CrossRef; (b) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538 RSC; (c) A.-L. Marshall and P. Alaimo, Chem.–Eur. J., 2010, 16, 4970 CrossRef CAS; (d) C. H. Christensen, J. Raas-Hansen, C. C. Marsden, E. Taarning and K. Egeblad, ChemSusChem, 2008, 1, 283 CrossRef CAS; (e) R. Diercks, J.-D. Arndt, S. Freyer, R. Geier, O. Machhammer, J. Schwartze and M. Volland, Chem. Eng. Technol., 2008, 5, 631 CrossRef; (f) A. Corma, S. Iborra and A. P. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS; (g) U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schaefer, Angew. Chem., Int. Ed., 2011, 50, 3854 CrossRef CAS.
- (a) M. R. L. Furst, R. Le Goff, D. Quinzler, S. Mecking, C. H. Botting and D. J. Cole-Hamilton, Green Chem., 2012, 14, 472 RSC; (b) L. Montero de Espinosa and M. A. R. Meier, Eur. Polym. J., 2011, 47, 837 CrossRef CAS; (c) D. J. Cole-Hamilton, Angew. Chem., Int. Ed., 2010, 49, 8564 CrossRef CAS; (d) A. Behr and J. Perez Gomes, Eur. J. Lipid Sci. Technol., 2010, 112, 31 CrossRef CAS; (e) M. A. R. Meier, Macromol. Chem. Phys., 2009, 210, 1073 CrossRef CAS.
- A. Rybak, P. A. Fokou and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2008, 110, 797 CrossRef CAS.
- (a) C. P. Park, M. M. Van Wingerden, S.-Y. Han, D.-P. Kim and R. H. Grubbs, Org. Lett., 2011, 13, 2398 CrossRef CAS; (b) R. M. Thomas, B. K. Keitz, T. M. Champagne and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 7490 CrossRef CAS; (c) S. C. Marinescu, R. R. Schrock, P. Müller and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 10840 CrossRef CAS; (d) C. Thurier, C. Fischmeister, C. Bruneau, H. Olivier-Bourbigou and P. H. Dixneuf, ChemSusChem, 2008, 1, 118 CrossRef CAS; (e) Y. Schrodi, T. Ung, A. Vargas, G. Mkrtumyan, C. W. Lee, T. M. Champagne, R. L. Pederson and S. H. Hong, Clean, 2008, 36, 669 CAS; (f) G. S. Forman, R. M. Bellabarba, R. P. Tooze, A. M. Z. Slawin, R. Karch and R. Winde, J. Organomet. Chem., 2006, 691, 5513 CrossRef CAS; (g) K. A. Burdett, L. D. Harris, P. Margl, B. R. Maughon, T. Mokhtar-Zadeh, P. C. Saucier and E. P. Wasserman, Organometallics, 2004, 23, 2027 CrossRef CAS; (h) M. B. Dinger and J. C. Mol, Adv. Synth. Catal., 2002, 344, 671 CrossRef CAS.
- (a) A. Rybak and M. A. R. Meier, Green Chem., 2008, 10, 1099 RSC; (b) A. Rybak and M. A. R. Meier, Green Chem., 2007, 9, 1356 RSC; (c) A. Behr, J. Perez Gomes and Z. Bayrak, Eur. J. Lipid Sci. Technol., 2011, 113, 189 CrossRef CAS; (d) U. Biermann, M. A. R. Meier, W. Butte and J. O. Metzger, Eur. J. Lipid Sci. Technol., 2011, 113, 39 CrossRef CAS; (e) X. Miao, R. Malacea, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 2911 RSC; (f) X. Miao, P. H. Dixneuf, C. Fischmeister and C. Bruneau, Green Chem., 2011, 13, 2258 RSC; (g) C. Bruneau, C. Fischmeister, X. Miao, R. Malacea and P. H. Dixneuf, Eur. J. Lipid Sci. Technol., 2010, 112, 3 CrossRef CAS; (h) R. Malacea, C. Fischmeister, C. Bruneau, J.-L. Dubois, J.-L. Couturier and P. H. Dixneuf, Green Chem., 2009, 11, 152 RSC; (i) T. Jacobs, A. Rybak and M. A. R. Meier, Appl. Catal., A, 2009, 353, 32 CrossRef CAS.
- (a) J. Trzaskowski, D. Quinzler, C. Bährle and S. Mecking, Macromol. Rapid Commun., 2011, 32, 1352 CrossRef CAS; (b) H. Mutlu, L. Montero de Espinosa and M. A. R. Meier, Chem. Soc. Rev., 2011, 40, 1404 RSC; (c) K. L. Opper and K. B. Wagener, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 821 CrossRef CAS; (d) J. C. Ronda, G. Lligadas, M. Galià and V. Cadiz, Eur. J. Lipid Sci. Technol., 2011, 113, 46 CrossRef CAS; (e) D. Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893 RSC; (f) M. A. R. Meier, Macromol. Chem. Phys., 2009, 210, 1073 CrossRef CAS; (g) F. Pardal, S. Salhi, B. Rousseau, M. Tessier, S. Claude and A. Fradet, Macromol. Chem. Phys., 2008, 209, 64 CrossRef CAS; (h) M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788 RSC.
- (a) Y. Lu and R. C. Larock, ChemSusChem, 2009, 2, 136 CrossRef CAS; (b) P. Henna and R. C. Larock, J. Appl. Polym. Sci., 2009, 112, 1788 CrossRef CAS.
- (a) H. Bilel, N. Hamdi, F. Zagrouba, C. Fischmeister and C. Bruneau, Green Chem., 2011, 13, 1448 RSC; (b) H. Borrée, T. H. Dinh, F. Caijo, C. Crévisy and M. Mauduit, Synthesis, 2011, 2125 Search PubMed; (c) K. Yoshikai, T. Hayama, K. Nishimura, K.-I. Yamada and K. Tomioka, Green Chem., 2011, 13, 1448 RSC; (d) S. Gutierrez and M. A. Tlenkopatchev, Polym. Bull., 2011, 66, 1029 CrossRef CAS; (e) H. A. Meylemans, R. L. Quintana, B. R. Goldsmith and B. G. Harvey, ChemSusChem, 2011, 4, 465 CrossRef CAS; (f) S. Wolf and H. Plenio, Green Chem., 2011, 13, 2008 RSC; (g) S. Monfette, K. D. Camm, S. I. Gorelsky and D. E. Fogg, Organometallics, 2009, 28, 944 CrossRef CAS; (h) R. T. Mathers, K. C. McMahon, K. Damodaran, C. J. Retarides and D. J. Kelley, Macromolecules, 2006, 39, 8982 CrossRef CAS; (i) J. C. Conrad, H. H. Parnas, J. L. Snelgrove and D. E. Fogg, J. Am. Chem. Soc., 2005, 127, 11882 CrossRef CAS; (j) T. R. Hoye and H. Zhao, Org. Lett., 1999, 1, 1123 CrossRef CAS; (k) D. C. Bradock and A. Matsuno, Tetrahedron Lett., 2002, 43, 3305 CrossRef; (l) B. De Clercq and F. Verpoort, Tetrahedron Lett., 2001, 42, 8959 CrossRef CAS; (m) W. A. Nugent, J. Feldman and J. C. Calabrese, J. Am. Chem. Soc., 1995, 117, 8992 CrossRef CAS.
- H. E. Blackwell, D. J. O’Leary, A. K. Chatterjee, R. A. Washenfelder, D. A. Bussmann and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 58 CrossRef CAS.
- (a) L. Vieille-Petit, H. Clavier, A. Linden, S. Blumentritt, S. P. Nolan and R. Dorta, Organometallics, 2010, 29, 775 CrossRef CAS; (b) J. Broggi, C. A. Urbina-Blanco, H. Clavier, A. Leitgeb, C. Slugovc, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2010, 16, 9215 CrossRef CAS; (c) D. F. Taber and K. J. Frankowski, J. Chem. Educ., 2006, 83, 283–284 CrossRef CAS.
- J. Moïse, S. Arseniyadis and J. Cossy, Org. Lett., 2007, 9, 1695–1698 CrossRef.
- S. Günther, P. Lamprecht and G. A. Luinstra, Macromol. Symp., 2010, 293, 15 CrossRef.
- Functional olefins are obtained from fossil sources, however, recent examples show that they can also be obtained from renewable materials. For acrylonitrile, see: (a) J. Le Nôtre, E. L. Scott, M. C. R. Franssen and J. P. M. Sander, Green Chem., 2011, 13, 807 RSC; (b) V. Calvino-Casilda, M. O. Guerrero-Pérez and M. A. Bãnares, Green Chem., 2009, 11, 939 RSC; (c) J.-L. Dubois, US 2010/0048850 A1, 2010. For acrylic derivatives, see: (d) J.-L. Dubois, C. Duquennex and W. Holderich, US 7,910,771,B2, 2011; (e) A. Witsuthammakul and T. Sooknoi, Appl. Catal., A, 2012, 413, 109 CrossRef.
- (a) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS; (b) S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168 CrossRef CAS; (c) Z. Y. Zhan, PCT WO 2007/003135, A1, 2007; (d) D. Arlt, M. Bieniek, R. Karch, WO 2008034552, 2008.
- (a) X. Miao, C. Fischmeister, C. Bruneau and P. H. Dixneuf, ChemSusChem, 2008, 1, 813 CrossRef CAS; (b) V. Le Ravalec, C. Fischmeister and C. Bruneau, Adv. Synth. Catal., 2009, 351, 1115 CrossRef CAS; (c) V. Le Ravalec, A. Dupé, C. Fischmeister and C. Bruneau, ChemSusChem, 2010, 3, 1291 CrossRef CAS; (d) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554 CrossRef.
- (a) B. Alcaide, P. Almendros and A. Luna, Chem. Rev., 2009, 109, 3817 CrossRef CAS; (b) A. Fürstner, O. R. Thiel, L. Ackermann, H.-J. Schanz and S. P. Nolan, J. Org. Chem., 2000, 65, 2204 CrossRef; (c) A. E. Sutton, B. A. Seigal, D. F. Finnegan and M. L. Snapper, J. Am. Chem. Soc., 2002, 124, 13390 CrossRef CAS; (d) J. C. Sworen, J. H. Pawlow, W. Case, J. Lever and K. B. Wagener, J. Mol. Catal. A: Chem., 2002, 194, 69 CrossRef; (e) D. Bourgeois, A. Pancrazi, S. P. Nolan and J. Prunet, J. Organomet. Chem., 2002, 643, 247 CrossRef; (f) B. Schmidt, Eur. J. Org. Chem., 2004, 1865 CrossRef CAS; (g) B. Schmidt, J. Mol. Catal. A: Chem., 2006, 254, 53 CrossRef CAS; (h) S. S. Kinderman, J. H. van Maarseveen, H. E. Schoemaker, H. Hiemstra and F. P. J. T. Rutjes, Org. Lett., 2001, 3, 2045 CrossRef CAS.
- (a) S. H. Hong, D. P. Sanders, C. W. Lee and R. H. Grubbs, J. Am. Chem. Soc., 2005, 127, 17160 CrossRef CAS; (b) I. O’Doherty, J. J. Yim, E. A. Schmelz and F. C. Schroeder, Org. Lett., 2011, 13, 5900 CrossRef.
- (a) G. B. Djigoué and M. A. R. Meier, Appl. Catal., A, 2009, 368, 158 CrossRef; (b) P. A. Fokou and M. A. R. Meier, Macromol. Rapid Commun., 2010, 31, 368 CrossRef CAS; (c) H. Mutlu, L. Montero de Espinosa, O. Tütünç and M. A. R. Meier, Beilstein J. Org. Chem., 2010, 6, 1149 CrossRef CAS; (d) O. Kreye, T. Toth and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2011, 113, 31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra21638h |
| This journal is © The Royal Society of Chemistry 2012 |