Understanding dendritic polymer–hydrocarbon interactions for oil dispersion
Nicholas K.
Geitner
a,
Priyanka
Bhattacharya
a,
Muriel
Steele
b,
Ran
Chen
a,
David A.
Ladner
b and
Pu Chun
Ke
*a
aDepartment of Physics and Astronomy, Clemson University, Clemson, SC 29634, USA. E-mail: pcke11@clemson.edu
bDepartment of Environmental Engineering and Earth Sciences, Clemson University, Clemson, SC 29634, USA
First published on 22nd August 2012
Abstract
This study examines the interactions of poly(amidoamine) dendrimers and hyperbranched poly(ethyleneimine) polymers with model linear and polyaromatic hydrocarbons. Large-scale complexations were formed for both types of dendritic polymers hosting the linear but not the polyaromatic hydrocarbon. Furthermore, both types of dendritic polymers exhibited a strong and comparable hosting/dispersion capacity for the polyaromatic hydrocarbon, while the hyperbranched polymers at concentrations below 30 μM showed a consistently higher hosting capacity than the dendrimers for the linear hydrocarbon. Such complexity in hosting capacity of the two types of dendritic polymers is attributed to the more hydrophobic interior and less steric hindrance of the hyperbranched polymers for the partitioning of the hydrocarbons.
The 2010 Deepwater Horizon oil spill marks one of the greatest environmental disasters in recent history, releasing an estimated 200 million gallons of crude oil into the Gulf of Mexico.1 This disaster pales in comparison to the 420 million gallons spilled during the first Gulf War.2 Recent history also saw 11 million gallons during the 1989 Exxon Valdez spill3 and 140 million gallons in the 1979 IXTOC 1 spill.4 Once one includes spills from facility repairs and daily “normal” operation, an estimated 943 million gallons of oil was spilled globally between 1990 and 1999 alone.5 A number of measures are available to respond to these enormous quantities of crude oil, one of the most widely used being chemical oil dispersants.1,6 Studies by both the US EPA and FDA found that most dispersants, including the Corexit 9500A used during the Deepwater Horizon spill, are approximately as toxic (or even more toxic) as crude oil alone.7,8 For future oil spill mitigation, it is therefore highly desirable to develop oil dispersant alternatives that are both effective and environmentally responsible.
Dendritic polymers are a class of synthetic polymeric nanostructures that consist of a central core and a series of branches emanating from this core. Within this class of dendritic polymers are dendrimers (well-ordered and monodisperse) and hyperbranched polymers (a more random branching structure).9 Due to the flexibility of their structures, ample interior space, the ability to form several intermolecular interactions at the same time, high molecular weights and biocompatibility,10 dendritic polymers are promising alternatives for the practice of water purification.11–16 Poly(amidoamine) (PAMAM) dendrimers, the most studied and commercialized dendrimers, possess a hydrophobic core and positively charged surface groups at neutral pH. This unique physicochemistry makes dendrimers ideal for hosting hydrophobic substances in the aqueous phase, as exemplified by their applications in water purification,17in vitro drug delivery,18 and a host of other applications.19 Hyperbranched (HY) poly(ethyleneimine) is another class of dendritic polymers which have also shown potential in encapsulating guest species including metal ions20 and polyaromatics21 owing to the chelating properties of their amine groups and their hydrophobic interiors at neutral pH. Their small size, globular structure and low viscosity also enable their integration into ultrafiltration membranes allowing them to be operated at low pressures,22 thus making the process energetically favourable and thermodynamically spontaneous as opposed to reverse osmosis or nanofiltration.23 However, despite their similarity in structure to the more expensive dendrimers, there are only a few studies of HY in host–guest systems.23–26
The water solubility, globular structure, and ample hydrophobic interior voids give dendritic polymers significant advantages over other recently investigated oil remediation solutions including cellulosic fibres which are roughly linear and lose efficiency when exposed to water.27 The oil-dispersing capabilities are related to another promising application of dendritic polymers: their effective prevention of gas hydrate formation in oil pipelines,28 a problem that plagues the petroleum industry.29,30
Here we present a proof-of-concept study comparing the hosting capabilities of dendritic polymers – generation four (G4) PAMAM dendrimers and HY, towards both linear hexadecane (C16) and poly-aromatic phenanthrene (PN) hydrocarbons (see Fig. 1). The HY polymers (MW: 10 kDa) were chosen to match the size and molecular weight of the G4 PAMAM dendrimers (14 kDa). A stock concentration of 15 mM of HY was prepared by diluting 2.25 g of 99% HY (Polysciences) in 15 mL deionized (DI) water (18 MΩ cm). These polymers contain primary, secondary and tertiary amines in the ratio of 25/50/25. This ratio is similar to the ratio of amines in a G4-PAMAM dendrimer. The fundamental interactions between the chosen hydrocarbons and dendritic polymers were examined using UV-vis spectrophotometry (Biomate 3, Thermo Electric Corp.), dynamic light scattering (Zetasizer S90, Malvern Instruments), and static contact angle measurements (DSA20, Krüss).
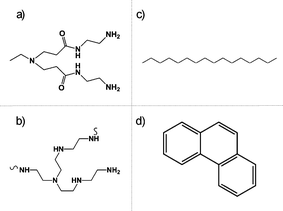 | ||
| Fig. 1 Chemical structures of single branches for both G4-PAMAM (a) and HY (b) dendritic polymers and C16 (c) and PN (d) hydrocarbons. The amide linkages present in PAMAM dendrimers cause the G4 core to be slightly less hydrophobic than the HY interior. | ||
All samples were prepared by first diluting stock dendritic polymers to a final concentration ranging from 12–65 μM in DI water and their pH adjusted to 8 using a stock 1 M NaOH solution. This choice of pH was to ensure that the dendritic polymers remained amphiphilic, which ensures a hydrophobic core as well as a hydrophilic exterior to maintain water solubility. A dose of 1.8 μL C16 or 1.0 mg PN was added to the dendritic polymers, ensuring concentrations that exceeded the hydrocarbons' solubilities. The experiment with PN was carried out in glass tubes rather than plastic to prevent adhesion of powdered PN to the container walls. The samples were bath-sonicated for 15 min and then mixed overnight on a rotor to reach equilibration. Each sample condition was prepared in triplicate for error analysis and all measurements were performed at room temperature.
Fig. 2 and 3 show the absorbance of dendritic polymers with the addition of PN and C16, respectively. In both cases the control dendritic polymer spectra were subtracted, and the resulting complex or hydrocarbon peak absorbance noted and plotted versus the concentration of dendritic polymers. The PN absorbance was read at 292 nm, corresponding to the absorbance peak of aqueous PN. The data point at 0 μM of dendritic polymer corresponded to PN dissolved in DI water (adjusted to pH 8). The peak PN absorbance increased linearly with increasing HY concentration (red circles, Fig. 2), whereas a saturation was observed for the PN absorbance in the case of G4-PAMAM (blue diamonds, Fig. 2). Initially, G4-PAMAM solubilized more PN in comparison with HY up to a concentration of 65 μM; however the absence of saturation in solubilisation of PN by HY indicates that HY can potentially solubilize more PN without loss of efficiency in the concentration range examined.
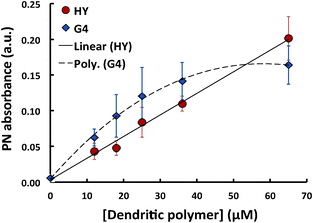 | ||
| Fig. 2 Absorbance for PN mixed with dendritic polymers at 292 nm. The G4 curve (blue diamonds) appears to exhibit saturation behaviour corresponding to a PN solubility of 35.3 mg L−1, unlike the linear curve for HY (red circles) which reaches 43.3 mg L−1 solubility without saturation. | ||
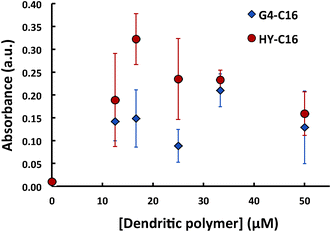 | ||
| Fig. 3 Absorbance for C16 mixed with dendritic polymers near 230 nm, corresponding to the new complex absorption peak. | ||
Atomistic MD simulations by Lin et al.31 showed that buried water molecules, i.e. water molecules in the interior of a PAMAM dendrimer, are thermodynamically unfavourable compared to that in the bulk water well outside the dendrimer at neutral pH. Hence the hydrophobic PN molecules would favourably partition into the interior of the dendrimers. This solubilisation is further augmented by the formation of charge-transfer complexes between the PN aromatics and the tertiary amines of the dendritic polymers.32 However, the HY polymers, despite their random interior structure, possess a greater internal hydrophobicity due to the absence of amide linkages (present in the PAMAM dendrimers) that readily form hydrogen bonds with water molecules (Fig. 1(a)). The interior tertiary amines remained deprotonated at neutral pH and hence offered more hydrophobicity to the core of the HY polymers (Fig. 1(b)). Thus, saturation in the hosting capacity towards PN was observed with PAMAM dendrimers, while the HY polymers displayed a greater solubilizing potential. Assuming the PN solubility in water of 1.29 mg L−1 (20 °C)33 corresponds to the PN absorbance observed when no dendritic polymers were present, we calculated the PN solubility for varying dendritic polymer concentrations using the corresponding PN absorbance levels. For G4 dendrimer, solubility saturation occurred at ∼35.3 mg L−1, which is a 27.3× improvement over the solubility of PN in pure water. HY polymers, however, elicited an effective PN solubility of 43.3 mg L−1 at 65 μM (a 33.6× improvement over water) without displaying any evident saturation behaviour. These appeared at first glance to be less favourable results than the PN loading capacity of surface-modified G4 “nanosponges” employed to remove PN from water,32 where highly hydrophobic films of dendrimers were created by modifying the surface groups of the dendrimers and such films were used to remove PN from the aqueous phase. That process is energetically more favourable than solubilizing PN in water, which is the aim of our study.
The absorbance of dendritic polymer-C16 samples (Fig. 3) suggests significant complexation between both types of the dendritic polymers and C16. The new absorbance peaks observed near 230 nm (after the subtraction of dendritic polymer control spectra) were likely due to non-covalent interactions between the C16 and the dendritic polymers, leading to conformational changes in G4-PAMAM and HY. The precise molecular nature of this complex is unclear and is a subject for our future work. However, we hypothesize that the partitioning of the linear hydrocarbon chains into the interiors of the dendritic polymers led to an overall mass redistribution of the polymers and consequently changes in their absorbance spectra. Here we also observed that there were statistical differences in the absorbance values of G4 and HY complexed with C16, with stronger complexation occurring for the host of HY polymers, particularly at concentrations below 30 μM. These differences could be attributed to the more hydrophobic core of HY polymers compared to that of the PAMAM dendrimers (as noted in the case with the aromatic PN) and the more open structure of the HY polymers that could have facilitated partitioning of the linear C16.
To understand the complexations of dendritic polymers and hydrocarbons, we further characterized the hydrodynamic sizes of pure dendritic polymers as well as those that had been incubated with either PN or C16, following the same mixing protocol described above. As shown in Table 1, both G4 and HY displayed a modest size increase after incubation with PN. This suggests that the PN molecules were partitioned or fully encapsulated within the dendritic polymers and that there were few inter-complex interactions present in the suspensions. Upon incubation with C16, however, there was a marked dependence of the size of polymer-hydrocarbon complexes on G4 or HY concentration, as indicated in Fig. 4.
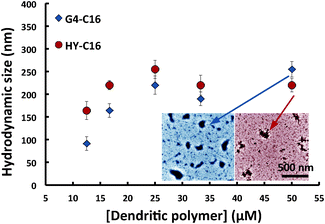 | ||
| Fig. 4 Hydrodynamic size of dendritic polymers mixed with C16. Both G4 (blue diamonds) and HY (red circles) saturate in size growth near 200 nm. This growth indicates strong inter-complex interactions. Inset: transmission electron microscopy images of G4-C16 (blue) and HY-C16 complexes (red). Scale bar: 500 nm. | ||
| Dendritic Polymer | PDI | With C16 | With PN | |
|---|---|---|---|---|
| HY | 5 ± 1 nm | 0.22 | * | 9 ± 2 nm |
| G4 | 4 ± 1 nm | 0.15 | * | 6 ± 1 nm |
Fig. 4 illustrates a range of sizes, all of which are much larger than seen for pure G4 or HY. This implies inter-complex interactions facilitated by the C16 incorporated into the polymers. For both G4 and HY incubated with C16, their hydrodynamic sizes increased with concentration of dendritic polymer and saturated near 200 nm. Consistent with Fig. 3, HY exhibited a higher hosting capacity than G4 for C16, at concentrations below 30 μM.
The formation of G4 or HY-C16 complexes was confirmed by transmission electron microscopy (TEM, Hitachi H7600), where G4 (50 μM) or HY (50 μM) were incubated overnight at room temperature with C16 (1.8 μL) following the protocol described above, and were subsequently negatively stained with uranyl acetate for 10 min prior to imaging. The average sizes of G4 or HY-C16 complexes ranged between 200 and 280 nm (Fig. 4, inset), in agreement with the hydrodynamic size measurement.
To better understand the differential hosting capacity of G4 and HY polymers, the above samples were subjected to a static contact angle experiment, during which a 2 μL droplet of each sample was placed on a Teflon substrate and the static contact angle was measured. Pure dendritic polymer samples (also at pH 8) were used as controls and were observed to have static contact angles equal to that of pure water. The parameter of interest was the change in contact angle, once the G4 or HY had been incubated with a hydrocarbon (see Fig. 5). Since Teflon is highly hydrophobic, a very high contact angle (136°) was yielded for hydrophilic samples (i.e. water and pure dendritic polymers) while smaller static angles were seen for more hydrophobic samples (i.e. those containing hydrocarbons). Fig. 5 shows an interesting behaviour of these host–guest systems: we first observed less negative changes in contact angle as the concentrations of polymer increased, indicating the increasing hydrophilicity of the suspensions. At higher polymer concentrations, however, increasingly more negative changes in contact angle were recorded, implying increasing hydrophobicity of the samples.
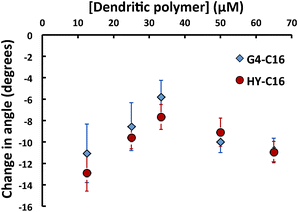 | ||
| Fig. 5 Change in contact angle for dendritic polymers mixed with C16. Increases in contact angle at low concentration (from −13° to −8° for HY and −11° to −6° for G4) imply more hydrophilic behaviour, while decreases in contact angle (returning to −11° for both G4 and HY) at higher concentrations imply a more hydrophobic behaviour. Note that no change in contact angle was observed for dendritic polymers incubated with PN. | ||
The contact angle measurement trends were in excellent agreement with the trends observed in the hydrodynamic size experiment summarized in Table 1 and Fig. 4 and the absorbance data shown in Fig. 3. One plausible interpretation here is that it was the available hydrocarbons, i.e., hydrocarbons that had at least one end available for interaction with the solution and Teflon surface that affected the contact angle. The implication of combining the hydrodynamic size and contact angle results is twofold – first, they suggest that PN molecules, which measured 1.17 nm × 0.80 nm,34 were partitioned within the dendritic polymers35 and had few exposed moieties available for initiating additional interactions with other polymer-PN complexes. Second, they implied the formation of “super-complexes” of dendritic polymers and C16 as a result of the linearity and rigidity of the hydrocarbon. The proposed schemes for the latter implication are illustrated in Fig. 6. Because C16 measures at 2.2 nm long36 compared to the hydrodynamic radii of 2.0 nm and 2.5 nm for G4 and HY, respectively, we conclude that C16 must not be partitioned completely to the core of the dendritic polymer. Therefore, there should be one end of C16 protruding from the dendritic polymer.
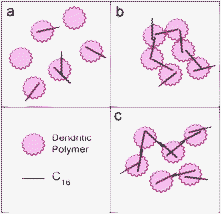 | ||
| Fig. 6 Proposed schemes of C16 complexation with dendritic polymers. C16 molecules are partitioned into dendritic polymers (a). Potential interactions leading to super-complexes include (b) C16 capping by dendritic polymers and (c) C16 end-to-end interactions, both through hydrophobic interactions. | ||
If C16 did penetrate completely to the core we would not observe the nearly identical size behaviour of super-complexes for G4 and HY. This protruding end either partitioned into another G4 or HY (Fig. 6b), or paired with an additionally captured C16 through hydrophobic interactions (Fig. 6c). Energetically the former configuration is more favourable than the latter due to the reduced exposure of hydrocarbon to the aqueous environment. In both scenarios the C16 molecule would have both ends engaged and become unavailable for further interaction with either the suspension or the Teflon surface. This cross-linking of complexes could continue until super-complexes were formed, reaching the approximate 200 nm in mean size as observed. At this critical size the super-complexes no longer grew any further but instead simply formed in greater numbers. This likely occurred as a result of the decreasing number of hydrocarbons per unit surface area for larger super-complexes, thus making further growth sterically less feasible. Indeed, once this critical size was reached, an increasingly hydrophobic behaviour was observed for the suspensions. Because of the random nature of the interactions, some C16 molecules inevitably remained on the outer surfaces of the super-complexes. We believe that it was these outer, uncapped hydrocarbons that directly affected the hydrophobicity and thus the contact angle of the dendritic polymer-hydrocarbon samples. No change in static contact angle on Teflon was seen for PN complex samples with dendritic polymers at any concentration, suggesting stable hydrophobicity associated with such samples and further confirms that PN molecules have been completely encapsulated by G4 and HY.
Conclusions
In summary, both G4 and HY dendritic polymers possessed a strong capacity for forming complexes with both linear and poly-aromatic hydrocarbons, as evidenced by distinct UV-vis signatures of hydrocarbons in aqueous solution, growth in polymer hydrodynamic size, as well as significant decreases in contact angle on Teflon to manifest a more hydrophobic solution. The 27.3× improvement in the solubility of PN in water for G4 and 33.6× improvement (without observed saturation) for HY are strong indicators of their potential as oil dispersants. In consideration of the significantly lower cost of HY than G4 as well as the comparable (for polyaromatic PN) or more favourable hosting capacity (for linear C16) of HY as shown in this study, HY polymers seem to be a better choice than PAMAM dendrimers for the practice of oil dispersion. This is understandable because of the more hydrophobic interior and more open structure of the HY polymers, which cater for a greater accessibility of linear and nonlinear hydrocarbons.Further study will include computational modelling to better understand the nature of the hydrocarbon – dendritic polymer interactions. In addition to hosting capacity, future studies will also be conducted to verify the biocompatibility of dendritic polymers employed as oil dispersants.
Acknowledgements
This work was supported by NSF CAREER award CBET-0744040 and NSF grant CBET-1232724 to Ke and US EPA grant RD835182 to Ladner and Ke. Steele acknowledges an NSF graduate fellowship. Bhattacharya is becoming a Linus Pauling Distinguished Postdoctoral Fellow with the Pacific Northwest National Laboratory. The authors thank Pengyu Chen for assistance with TEM imaging.References
- Restore The Gulf: http://www.restorethegulf.gov.
- N. Tawfiq and D. A. Olsen, Mar. Pollut. Bull., 1993, 27, 333 Search PubMed.
- J. Johnson and M. Torrice, BP's Ever-Growing Oil Spill, Chem. Eng. News, 2010, 88, 15–24.
- J. S. Patton, M. W. Rigler, P. D. Boehm and D. L. Fiest, Nature, 1981, 290, 235 Search PubMed.
- D. S. Etkin, Analysis of Oil Spill Trends in the United States and Worldwide. International Oil Spill Conference, 2001, 1291 Search PubMed.
- International Tanker Owners Pollution Federation: http://www.itopf.com.
- M. J. Hemmer, M. G. Barron and R. M. Greene, Environ. Toxicol. Chem., 2011, 30, 2244 Search PubMed.
- O. J. Bandele, M. F. Santillo, M. Ferguson and P. L. Weisenfeld, Food Chem. Toxicol., 2012, 50, 1653 Search PubMed.
- M. J. Frechet and D. A. Tomaliain, Dendrimers and other Dendritic Polymers, Wiley and Sons; New York, 2011 Search PubMed.
- B. Helms and E. W. Meijer, Science, 2006, 313, 929 CrossRef CAS.
- M. S. Diallo, L. Balogh, A. Shafagati, J. H. Johnson, W. A. Goddard III and D. A. Tomalia, Environ. Sci. Technol., 1999, 33, 820 CrossRef CAS.
- M. S. Diallo, S. Christie, P. Swaminathan, J. H. Johnson Jr and W. A. Goddard III, Environ. Sci. Technol., 2005, 39, 1366 CrossRef.
- M. S. Diallo, K. Falconer, J. H. Johnson Jr and W. A. Goddard III, Environ. Sci. Technol., 2007, 41, 6521 CrossRef CAS.
- M. S. Diallo, W. Arasho, J. H. Johnson Jr and W. A. Goddard III, Environ. Sci. Technol., 2008, 42, 1572 CrossRef CAS.
- M. Arkas, D. Tsiourvas and C. M. Paleos, Chem. Mater., 2003, 15, 2844 CrossRef CAS.
- P. Chen, Y. Yang, P. Bhattacharya, P. Wang and P. C. Ke, J. Phys. Chem. C, 2011, 115, 12789 CrossRef CAS.
- N. A. Kahn, in Water and Wastewater Treatment using Nano-Technology, Springer-Verlag; Berlin, 2012 Search PubMed.
- P. Daftarian, A. E. Kaifer, W. Li, B. B. Blomberg, D. Frasca, F. Roth, R. Chowdhury, E. A. Berg, J. B. Fishman, H. A. Al Sayegh, P. Blackwelder, L. Inverardi, V. L. Perez, V. Lemmon and P. Serafini, Cancer Res., 2011, 71, 7452 Search PubMed.
- S. Parveen, R. Misra and S. K. Sahoo, Nanomed.: Nanotechnol., Biol. Med., 2012, 8, 147 Search PubMed.
- M. Mortimer, K. Kasemets, M. Heinlaan, I. Kurvet and A. Kahru, Toxicol. in Vitro, 2008, 22, 1412 CrossRef CAS.
- Z. Shen, PhD Dissertation, Johannes Guttenberg University, 2009 Search PubMed.
- M. Arkas and L. Eleades et al., J. Appl. Polym. Sci., 2005, 97, 2299 Search PubMed.
- M. S. Diallo, US Patent No. 7,758,755 B2, 2010 Search PubMed.
- S. P. Sun, T. A. Hatton and T. S. Chung, Environ. Sci. Technol., 2011, 45, 4003 Search PubMed.
- A. Kichler, C. Leborgne, E. Coeytaux and O. Danos, J. Gene Med., 2001, 3, 135 CrossRef CAS.
- Y. Liu, T. Steele and T. Kissel, Macromol. Rapid Commun., 2010, 31, 1509 Search PubMed.
- K. C. Payne and C. D. Jackson et al., Environ. Sci. Technol., 2012, 46, 7725 Search PubMed.
- K. Pasupathy, N. W. Suek, J. L. Lyons, J. Ching, A. Jones, Q. Lu, M. H. Lamm and P. C. Ke, The Open Nanoscience Journal, 2008, 2, 47 Search PubMed.
- P. J. Froehling, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3110 CrossRef CAS.
- B. Halford, Chem. Eng. News, 2005, 83, 30.
- S. T. Lin, P. K. Maiti and W. A. Goddard, J. Phys. Chem. B, 2005, 109, 8663 CrossRef CAS.
- M. Arkas, D. Tsiourvas and C. Paleos, Chem. Mater., 2003, 15, 2844 CrossRef CAS.
- D. Mackay and W. Y. Shiu, J. Chem. Eng. Data, 1977, 22, 399 CrossRef CAS.
- L. C. Sander and S. A. Wise, Polycyclic Aromatic Hydrocarbon Structure Index. NIST Special Publication, 1997 Search PubMed.
- P. C. Ke and M. H. Lamm, Phys. Chem. Chem. Phys., 2011, 13, 7273 RSC.
- Y. He, T. Ye and B. Borguet, J. Phys. Chem. B, 2002, 106, 11264 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |