Macrocellular iron foams: characterization and facile conversion into water splitting photoanodes
Amir
Kaplan
,
Eli
Korin
and
Armand
Bettelheim
*
Chemical Engineering Department, Ben-Gurion University of the Negev, P.O. Box 653, Beer-Sheva, 84105, Israel. E-mail: armandb@bgu.ac.il; Fax: +972-8-6477656; Tel: +972-8-6461799
First published on 20th August 2012
Abstract
A simple heat treatment of macrocellular iron foams forms an oxide layer consisting of ∼30 and 70% hematite and magnetite, respectively, with hematite positioned at the outer layer. A possible application of these foams as water splitting photoanodes is demonstrated.
Metal and carbon foams are a relatively new class of materials with unique combined properties such as high surface area, low density, and good electrical and thermal conductivity. These and the synthetic ability to control their pore size, make them attractive materials in many technological applications such as high power density batteries,1,2 hydrogen storage,3,4 and water treatment.1 Synthetic approaches for producing nanoporous metal foams have been reviewed recently,5 and these include combined templating and dealloying techniques,6 self assembly of prefabricated nanoparticles,7 nanosmelting of hybrid polymer–metal oxide aerogels,8 and combustion of either metal ion complexes 9 or sol–gels.10 Metallic sponges containing cells on the micrometer or even millimeter length-scale are more easily prepared, usually by use of a sacrificial template such as a polysaccharide.11 In this work we report some properties of macroporous iron foams and demonstrate a new application for these materials in the field of artificial photosynthesis.
The development of artificial photosynthesis systems using a large portion of the solar spectrum would be a major advance in hydrogen production and energy conversion. The energy required to drive the water splitting reaction to produce hydrogen and oxygen in this case is provided by light in the absence or presence of an external power source.12 The real problem is the half-reaction of water oxidation near the potential of the multielectron transfer process of oxygen evolution, +1.23 V vs. the normal hydrogen electrode (NHE).12,13 The photoelectrochemical (PEC) cell usually consists of a semiconductor electrode and a metal (or semiconductor) counter electrode immersed in an aqueous electrolyte. When the semiconductor at the anode is illuminated, it absorbs part of the light and generates charge carriers (electron and hole pairs). Holes migrate to the semiconductor/electrolyte interface and electrons to the counter electrode through the external circuit. This leads to oxidation of water to H+ and O2 by holes at the photoanode and reduction of H+ to H2 by electrons at the cathode.
The ability of iron oxide, and particularly α-Fe2O3 hematite, to absorb solar irradiation coupled with its abundance and non-toxicity make it an attractive photoanode material.14 However, there are a few challenges when using hematite for water splitting and these include low conductivity and high water oxidation overpotential. It has been reported that the photocurrent densities obtained with the currently used flat electrodes, transparent indium tin oxide (ITO) or fluorine doped tin oxide (FTO), can be significantly increased by optimizing the semiconductor morphology. Various methods for nanostructuring hematite have been reported and these include solution based techniques to obtain nanowires,15 electrochemical anodization to obtain nanotubes,16 electrodeposition,17 ultrasonic spray pyrolysis,18 and atmospheric pressure chemical vapor deposition (CVD).19 Photocurrents have also been increased by the incorporation of Sn4+ or Pt4+20 or SiO221 as dopants in hematite films. Also, various catalysts, such as Co2+,22 amorphous cobalt phosphate,23 and IrO219 have been attached to the surface of hematite photoanodes and these have been shown to enhance the kinetics of water oxidation.
We have previously investigated carbon based electrodes with high effective surface area, such as carbon foams (reticulated vitreous carbon, RVC) and aerogel carbon.24 These electrodes yield relatively high water splitting photocurrent densities after being coated with a hydrogel containing hematite and quinone (serving as an electron transfer agent). Moreover, the open structure of these electrodes allows for free pathways for the escape of evolved gas bubbles, which has a significant role in increasing stability of the catalytic layer. The present work deals with the characterization of iron foams. Their open 3D structure, which permits light penetration, and the simple procedures (heat treatment in air and adsorption from salt solutions) to coat them with a catalytic layer (hematite, dopant, and water oxidation catalyst) are exploited for the preparation of photoanodes in water splitting cells.
Commercially available iron foams with cell diameters of 450 or 800 μm (IF450 and IF800, respectively), both with an average surface density of 500 g m−2 as determined by the supplier (Alantum Corp., Germany), were used. Heat treatment to coat the foams with an oxide layer was achieved in a tubular furnace (Carbolite, STF 16/180) at a temperature of 500 °C in air. The chemical structure of the oxide layers was determined by X-ray diffraction (XRD, Philips PW 1050/70). Morphology of the foams and the oxide coating thickness were examined using high resolution scanning electron microscopy (HR-SEM, JSM-7400F). Chemical characterization of the surface and deeper layers (depth profiling rate of ∼0.1 nm s−1) was achieved using X-ray photoelectron spectroscopy (XPS, ESCALAB 250) with an Al X-ray source and monochromator. Doping the oxide layers with silica was achieved by dipping the samples in a 24 mM tetraethyl orthosilicate (TEOS) solution in ethanol for a period of 10 min, drying in air, and subsequent heat treatment in air at 500 °C. Introduction of Co(II) ions in the oxide layers was obtained by dipping the heat treated iron foams in 0.1 M CoCl2 for ten minutes, followed by rigorous water rinsing and drying in air.
The electrochemical experiments were performed with an EG&G potentiostat/galvanostat model 263A. Photoelectrochemistry was conducted using a three-compartment glass cell and 0.1 M Na2CO3 at pH 12 as electrolyte, kept at 25 °C (Quantum Northwest TC 125 thermostat). The working electrode was a sample of foam IF450 or IF800, 2.0 ± 0.5 mm thick. The surface area of the electrode exposed to the electrolyte solution, 2.0 ± 0.5 cm2, was calculated from the surface density. This electrode faced a quartz window, through which it was illuminated (Newport Oriel product, 200 W Hg(Xe) lamp, 65 mW cm−2). The counter and reference electrodes in the other two compartments were a platinum wire and Ag/AgCl/KClsatd, respectively.
Fig. 1A shows an SEM image of the macrocellular structure of the foams, as observed for IF800. Although a 3D open architecture is observed, the cells are randomly distributed, thus limiting transmittance of light into deep electrode layers (∼6 and ∼10% transmittance for 2 mm thick IF450 and IF800 samples, respectively). SEM analysis of a single cell as well as of sites residing on the cell walls (Fig. 1B and 1C, respectively) reveals a morphology with a roughness peak to valley height of 3 ± 0.6 μm. Cross-section SEM images, such as that demonstrated in Fig. 1E for a 4 h treated IF800 sample, reveal that the oxide thickness layer obtained after 1 and 4 h heat treatment is 0.3 ± 0.1 and 3.0 ± 0.4 mm, respectively.
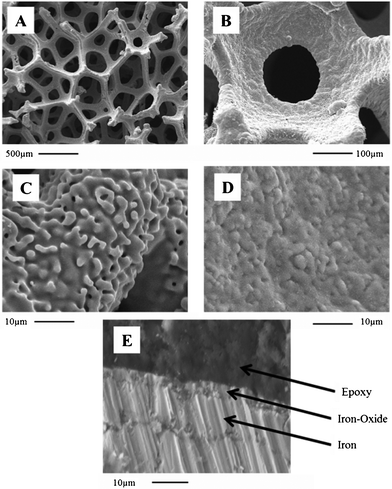 | ||
| Fig. 1 SEM images showing the macrocellular structure of IF800 (A) and morphology surrounding one cell (B). Images (C) and (D) show surface morphology for this sample, before and after being heat treated in air for 4 h, respectively. (E) SEM cross-section of a 4 h heat treated IF800 sample cast into epoxy resin. | ||
The nature of the oxide layer was examined by XRD. Fig. 2 shows diffractograms obtained for IF800 before (A), and after 1 and 4 h heat treatment (B and C, respectively). Analysis of the peaks attributed to α-F2O3 hematite and Fe3O4-magnetite indicates a mixture containing ∼30 and 70%, respectively, of the two oxides after a 1 h heat treatment. Moreover, although hematite is the most stable phase, the composition does not significantly change after extending the heat treatment to 4 h (∼33 and 67% of hematite and magnetite, respectively), thus indicating low diffusion rate of oxygen through the oxide layer.25,26
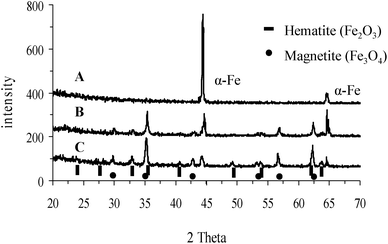 | ||
| Fig. 2 X-ray diffraction patterns for IF800 (A) before, and after (B) 1, and (C) 4 h of heat treatment. | ||
The iron foams, before and after being heat treated were further investigated by XPS. XPS spectra obtained for the foam surfaces revealed peaks which are characteristic of iron (Fe 2p: 706.4, 719.5 eV) and of oxygen (O 1s : 531.7 eV) and the absence of any significant impurity traces. Fe 2p spectra obtained during depth profiling for a heat treated IF800 sample are shown in Fig. 3A. While the intensities of the peaks for metallic iron increase, the main peaks for iron oxides (∼710, 723 eV) decrease as function of depth. Moreover, the satellite peaks at ∼716 and 730 eV, characterizing Fe2O3 rather than oxides of iron ion with lower oxidation state,27 disappear for depths >∼100 nm. The profile concentration for iron and oxygen as well as the O/Fe concentration ratio profile are shown in Fig. 3B. After a steep decrease of the concentration ratio in the first 12 nm, due to the decrease of surface adsorbed oxygen, a near constant value of 1.5 for the O/F concentrations ratio, as expected from an oxide of Fe(III), is obtained for depths up to ∼100 nm. The oxide thickness in this case is ∼300 nm according to SEM analysis and the two oxides detected by XRD are Fe2O3-hematite and Fe3O4-magnetite. This indicates that most of the hematite is situated at the outer oxide layer while the remaining inner oxide layer (∼70% of total oxides) consists mainly of magnetite. This is attributed to the low diffusion rate of oxygen through the oxide layer and the larger amount of oxygen needed to form hematite in comparison to magnetite.
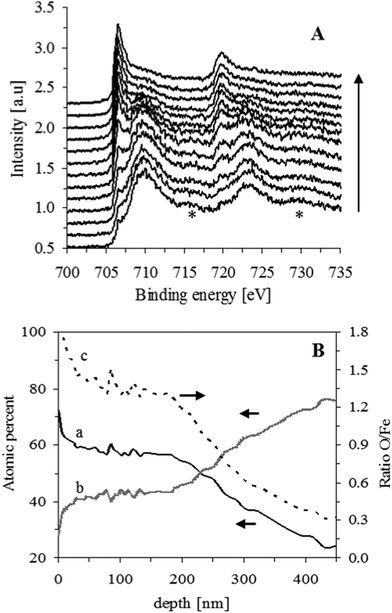 | ||
| Fig. 3 (A) XPS spectra obtained for IF800, after heat treatment for 1 h, at increasing depths (∼18 nm each, in the direction of the arrow). Satellite peaks of Fe2O3 are indicated by *. (B) Depth concentration profiles for oxygen and iron (curves a, and b, respectively, obtained by O 1s and Fe 2p main peaks) and their concentration ratio profile (curve c). | ||
The effect of light on water oxidation current density in 0.1 M Na2CO3 solution at pH 12 was examined at two potentials: +0.3 and +0.7 V vs. Ag/AgCl/KClsatd (∼+1.23 and +1.63 V vs. NHE). The chronoamperometric curves obtained at these potentials are depicted in Fig. 4 for IF800 (A and B, respectively) and IF450 (C and D, respectively) electrodes after being heat treated in air for 1 and 4 h (curves a and b, respectively). Comparison of the chronoamperometric curves at the same potential obtained for the two electrodes treated for 1 h (curves a in A and C, and curves a in B and D) indicates that the photocurrents for the two electrodes do not differ significantly. This can be expected from their similar effective surface area (area exposed to electrolyte as well as light). It can also be seen that for both electrodes, the shorter heat treatment yields higher photocurrents (at +0.3 V for IF800 after 1 and 4 h treatment: 40 and 10 μA cm−2).
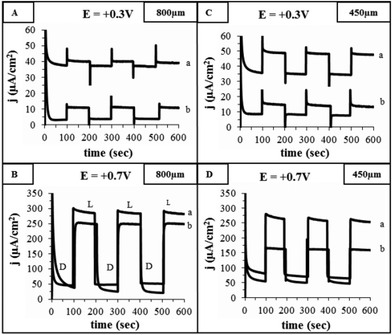 | ||
| Fig. 4 Dark (D) and photocurrents (L) obtained by chronoamperometry in 0.1 M Na2CO3 solution, pH 12, at +0.3 and +0.7 V for IF800 (A and B, respectively) and IF450 (C and D, respectively). Curves (a) and (b) represent the response obtained for iron foams which have been heat treated in air at 500 °C for 1 and 4 h, respectively. | ||
The photocurrents at high potentials do not differ significantly for these electrodes (at +0.7 V for IF800 after 1 and 4 h treatment: 300 and 250 μA cm−2). This is attributed to the higher resistance, as well of predominance of hole–electron recombination at low overpotential and in thicker oxide films.28
The effect of the presence of additives in the oxide layer on the water oxidation photoelectrochemical response was examined for an IF800 electrode after being heat treated for 1 h in air. Linear sweep voltammograms obtained in 0.1 M Na2CO3 solution at pH 12 for this electrode before being heat treated show that the onset of water oxidation is at ∼+0.85 V, and as expected, the electrode shows no photo-response (Fig. 5, curves a and b). After a 1 h heat treatment, the non-illuminated electrode shows considerably lower water oxidation currents, due to the high resistivity of the oxide layer29 (curve c). However, upon illumination the water oxidation potential onset is shifted to ∼+0.25 V and the currents are enhanced considerably (curve d). Adsorption of Co(II) ions, as water oxidation catalyst, in the oxide film not only causes an additional shift of the onset potential to ∼+0.15 V but also enhances the photocurrent in the entire potential range (curve e). The effect of dopant in the oxide layer was also examined. Curve f is a voltammogram obtained during illumination of a silica doped electrode. The photocurrent densities obtained at +0.7 V for the undoped and silica doped electrode (curves d and f) are ∼250 and 450 μA cm−2, respectively. The current increase is a result of the decrease of the oxide layer resistivity in the presence of dopant.14,28 The effect of the presence of both dopant and water oxidation catalyst in the oxide layer on the photoelectrochemical response of the electrode is illustrated in Fig. 5, curve g. At low potentials (E <∼+0.55 V), the effect of both additives considerably increases the current in comparison to that obtained with each additive separately (∼90, 70, and 200 μA cm−2 at +0.3 V, for electrode containing Co(II), SiO2, and both, respectively). However, at higher potentials, although the observed photocurrents for an electrode containing both additives are higher than that containing Co(II), they are lower than that containing only silica (∼380, 275, and 450 μA cm−2 at +0.7 V). The resistance decrease and water oxidation rate increase, provided by the dopant and catalyst, respectively, both enhance current density at low overpotential. However, it seems that an excess of Co(II) ions in the catalytic layer may have a masking effect which inhibits rapid charge transfer attained by the dopant at high overpotential.
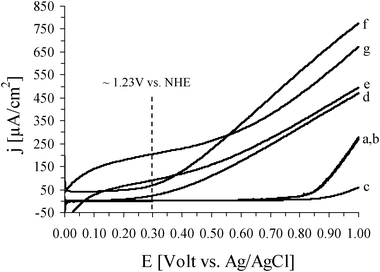 | ||
| Fig. 5 Linear sweep voltammograms obtained at a scan rate of 5 mV s−1 in 0.1 M Na2CO3, pH 12, for IF800. (a) and (b) are for a non-heat treated electrode before and during illumination, respectively. After being heat treated in air at 500 °C for 1 h, curves (c) and (d) are obtained before and during illumination, respectively. Curves (e), (f) and (g) are for this illuminated electrode after adsorption of Co(II) ions, after doping with silica, and after both doping with silica and adsorption of Co(II) ions, respectively. | ||
The maximum photocurrent density achieved in the present work for water oxidation, 200 μA cm−2 at +1.23 V vs. NHE, is within the photoactivity reported for ultrathin hematite films obtained by spray pyrolysis and coated on non-modified FTO electrodes and on ones which have been modified with nanostructured silica scaffolds (15–420 μA cm−2).30 However, it is lower than that reported for nanostructured silica doped hematite obtained by atmospheric pressure CVD with Co(II) or IrO2 as water oxidation catalyst (∼2.2 and 3.3 mA cm−2, respectively).14 Work is in progress to improve the performance of the iron foams by allowing light to penetrate into deeper layers of the electrodes and increase the catalytic activity of the hematite layer. Opening the 3D structure by chemical or electrochemical dissolution of iron from the pore walls, as well as optimization of the iron oxide layer structure and composition, are being examined in order to increase the photoelectrochemical activity of the foams.
To conclude, porous iron foams can be converted by simple methods into catalytic electrodes. This is expected to be an important step towards developing efficient, simple and inexpensive components of large scale electrochemical/photoelectrochemical cells.
Acknowledgements
The authors acknowledge the financial support from the Israel Ministry of National Infrastructures, Energy and Water Division.Notes and references
- D. R. Rolison, J. W. Long, J. C. Lytle, A. E. Fischer, C. P. Rhodes, T. M. McEvoy, M. E. Bourg and A. M. Lubers, Chem. Soc. Rev., 2009, 38, 226 RSC.
- J. S. Wang, P. Liu, E. Sherman, M. Verbrugge and H. Tataria, J. Power Sources, 2011, 196, 8714 CrossRef CAS.
- A. Martinez-Mesa, S. N. Yurchenko, S. Patchkovskii, T. Heine and G. Seifert, J. Chem. Phys., 2011, 135, 214701 CrossRef CAS.
- B. H. Kim, Y. J. Yun, W. G. Hong, C. H. Kim, S. M. Lee, H. Y. Yu and H. J. Kim, Int. J. Hydrogen Energy, 2011, 36, 12887 CrossRef CAS.
- B. C. Tappan, S. A. Steiner, III and E. P. Luther, Angew. Chem., Int. Ed., 2010, 49, 4544–4565 CrossRef CAS.
- G. W. Nyce, J. R. Hayes, A. V. Hamza and J. H. Satcher, Chem. Mater., 2007, 19, 344–346 CrossRef CAS.
- J. L. Mohanan, I. U. Arachchige and S. L. Brock, Science, 2005, 307, 397–400 CAS.
- N. Leventis, N. Chandrasekaran, A. G. Sadekar, C. Sotiriou-Leventis and H. Lu, J. Am. Chem. Soc., 2009, 131, 4576–4577 CrossRef CAS.
- B. C. Tappan, M. H. Huynh, M. A. Hiskey, D. E. Chavez, E. P. Luther, J. T. Mang and S. F. Son, J. Am. Chem. Soc., 2006, 128, 6589–6594 CrossRef CAS.
- Z. Hua, Y. Deng, K. Li and S. Yang, Nanoscale Res. Lett., 2012, 7, 129 CrossRef CAS.
- D. Walsh, L. Arcelli, T. Ikoma, J. Tanaka and S. Mann, Nat. Mater., 2003, 2, 386–390 CrossRef CAS.
- C. A. Grimes, O. K. Varghese and S. Ranjan, Light, water, hydrogen: the solar generation of hydrogen by water photoelectrolysis, Springer Verlag, 2008 Search PubMed.
- J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802 CrossRef CAS.
- K. Sivula, F. L. Florian and M. Grätzel, ChemSusChem, 2011, 4, 432 CrossRef CAS.
- N. Beermann, L. Vayssieres, S. Lindquist and A. Hagfeldt, J. Electrochem. Soc., 2000, 147, 2456 CrossRef CAS.
- S. K. Mohapatra, S. E. John, S. Banerjee and M. Misra, Chem. Mater., 2009, 21, 3048 CrossRef CAS.
- A. Kleiman-Shwarsctein, Y. Hu, A. J. Forman, G. D. Stucky and E. W. McFarland, J. Phys. Chem. C, 2008, 112, 15900 CAS.
- A. Duret and M. Grätzel, J. Phys. Chem. B, 2005, 109, 17184 CrossRef CAS.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405 CrossRef CAS.
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 7436 CrossRef CAS.
- S. Saremi-Yarahmadi, K. G. U. Wijayantha, A. A. Tahir and B. Vaidhyanathan, J. Phys. Chem. C, 2009, 113, 4768 CAS.
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714 CrossRef CAS.
- D. K. Zhong and D. R. Gamelin, J. Am. Chem. Soc., 2010, 132, 4202 CrossRef CAS.
- A. Kaplan, E. Korin, L. Soifer and A. Bettelheim, Electrochem. Solid-State Lett., 2012, 15, F1 CrossRef CAS.
- V. B. Trindade, R. Borin, B. Z. Hanjari, S. Yang, U. Krupp and H. Christ, Mater. Res., 2005, 8, 365 CrossRef CAS.
- B. Pujilaksono, T. Jonsson, M. Halvarsson, J. Svensson and L. Johansson, Corros. Sci., 2010, 52, 1560 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441 CrossRef CAS.
- F. L. Souza, K. P. Lopes, E. Longo and E. R. Leite, Phys. Chem. Chem. Phys., 2009, 11, 1215 RSC.
- R. M. Cornell and U. Schwertmann, Electronic, Electrical and Magnetic Properties and Colour, in The Iron Oxides, Wiley, Weinheim, 2003, pp. 111–137 Search PubMed.
- F. Le Formal, M. Grätzel and K. Sivula, Adv. Funct. Mater., 2010, 20, 1099 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |