Role of ionic liquids in protein refolding: native/fibrillar versus treated lysozyme
Sara
Mangialardo
*a,
Lorenzo
Gontrani
b,
Francesca
Leonelli
c,
Ruggero
Caminiti
c and
Paolo
Postorino
d
aDepartment of Physics, Università di Roma Sapienza, Italy. E-mail: saramangialardo@fastwebnet.it; Fax: +3906443158; Tel: 06499123502
bCNR-ISM Tor Vergata, Roma, Italy. E-mail: l.gontrani@caspur.it
cDepartment of Chemistry, Università di Roma Sapienza, Italy. E-mail: francesca.leonelli@uniroma1.it; r.caminiti@caspur.it
dDepartment of Physics, Università di Roma Sapienza, Italy. E-mail: paolo.postorino@roma1.infn.it; Fax: +3906443158; Tel: 06499123502
First published on 11th October 2012
Abstract
Several ionic liquids (ILs) are known to revert aggregation processes and to improve the in vitro refolding of denatured/fibrillar proteins. Here, Raman spectroscopy is exploited to verify the refolding capability of several ammonium-based ILs and to identify the microscopic signatures of the structural rearrangements induced by the interaction of ILs with fibrillar lysozyme. We collected and carefully analyzed spectra from native, fibrillar and ILs-treated fibrillar lysozyme to follow the microscopic process induced by ILs. These allowed us to identify different mechanisms of interaction depending on the length of the cation alkyl chain. A clear refolding effect was observed with EAN, as well as a tendency of the longer alkyl chain (PAN and BAN) of dissolving the fibril packing. A specific interaction mainly affecting the aromatic residues was identified for MEOAN (a long chain ILs with an ether group). The whole of the results, thus, provides new and detailed information on the ILs–protein interaction and shows Raman spectroscopy as a simple, reliable and effective diagnostic technique in this field.
Introduction
Protein aggregation is one of the most important problems in the production and storage of industrial processes and, therefore, among the causes of the major economic loss in biotechnology and pharmaceutical factories. Indeed in manufacturing commercial products, the goal is to obtain a stable and correct protein folding for allowing the full functionality.1 The problems of protein aggregation and structural stability are not limited to the manufacturing processes. Protein misfolding diseases are a well-known class of ailments, including Alzheimer, Parkinson and Huntington diseases. They all involve protein aggregation and share common features, such as the presence of insoluble fibrous protein aggregation in a specific structural motif characterized by a cross-β sheet structure.2 Key issues on aggregation are not yet fully addressed, such as the detailed microscopic mechanism leading to aggregation, the structure of the aggregates, how the environmental conditions can affect the rate and the amount of aggregation and how aggregation can be prevented and/or removed.Additives may promote the stabilization of the native state of the protein, accelerating the kinetics of the correct folding and removing/inhibiting the aggregation of denatured polypeptides and intermediates of the folding pathways.3
In recent years ionic liquids (ILs) have been used to stabilize the protein activity, to inhibit or reduce aggregation, and to improve the in vitro refolding of denatured proteins.4,5 ILs have numerous attractive characteristics, including their non-volatility, good solvating properties, thermal stability and recyclability, that render these compounds “environmentally green”.6–10 One of the most important qualities of these solvents is the high tunability of their chemical structure. Indeed, they can be designed to have specific physical and chemical qualities by acting on either the alkyl chains (i.e. modifying the length, the presence of hydrophobic groups, etc.) or the anion (i.e. varying the degree of the charge delocalization, its hydrogen bonding ability, etc.).11 ILs that have coordinating anions which are strong hydrogen bond acceptors (e.g. Cl−, NO3−, CH3COO− and (MeO)2PO2−) can dissolve many compounds, which are insoluble or sparingly soluble in water and in most of the organic solvents. Examples include cellulose12 and several compounds having specific pharmacological activity.13–15 As to the effects of the cation alkyl chain length, it has been proposed that the polarity of ILs decreases with increasing alkyl chain length.10 This can be an important chemical parameter, as the polarity of ILs has an impact on the enzyme stability and selectivity. In particular, the lengthening of the alkyl chain seems to have a positive effect on dissolving the aggregates and a negative effect on refolding the protein.16–19
In recent years the effects of ILs on several proteins have been extensively investigated by varying the ILs composition (anion, cation and alkyl chains).20–22 Many interesting studies on ILs–protein interaction have been carried out on lysozyme (Fig. 1), an enzyme often used as a model protein to study fibril formation in vitro.23,24 As a matter of fact, this protein can be easily converted to amyloid fibrils under high temperature and low pH environmental conditions and the fibrils formed share similar characteristics to ailments' amyloid.25
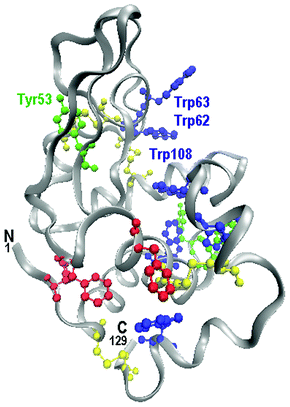 | ||
| Fig. 1 Hen Egg White Lysozyme (HEWL). N terminal and C terminal are labeled. The aromatic residues that were discussed in the text are highlighted: Phe (red), Tyr (green), Trp (blue). Among cysteine residues (yellow) the disulphide bridges take place. Tyr53, Trp62, Trp63 and Trp108 are in the active site of the enzyme. | ||
Recently N. Byrne and C. A. Angell26 have found that some protic ILs (PILs) have the property of partly recovering lysozyme functionality after severe denaturing procedures leading to fibril formation. Indeed, after the treatment with PILs, they observed the dissolution of the fibrillar aggregates and the recovery of the enzymatic activity. Among the others, ethyl ammonium nitrate (EAN) seems to be the most effective in refolding. Understanding the microscopic mechanism of interaction among protein fibrils and PILs is particularly interesting, since it is rather uncommon to find an additive able to dissolve aggregates and contemporaneously refold, at least partly, the protein.
In the last few decades Raman spectroscopy has become a common practice in protein structural investigations. This technique has many advantages: it is a non-destructive and non-invasive technique and it requires a small amount of sample. Moreover, reliable assignments between specific spectral features and local protein structures have been established, and clear, sensitive spectroscopic markers of the secondary structure27 and of the side-chain environments28 have been identified. A careful data analysis of Raman spectra can also provide direct information on the protein tertiary structure (e.g. on disulphide bridges29 and hydrogen bonds of the side chains30). The analysis of the amide bands is almost routinely exploited for the empirical quantitative estimate of the protein secondary structure31 with the accuracy comparable with that obtainable from circular dichroism experiments, as witnessed by the good agreement usually found with the analysis of X-ray structural data.32
In the present paper we report on a careful Raman study of hen egg white lysozyme (HEWL) in both native and fibrillar conformations. This study allowed us to get a full spectroscopic characterization of the protein in the two extreme conformations and, by comparison, to evaluate the refolding efficiency of the PILs based on the nitrate anion (NO3−) on lengthening the cation alkyl chain. Taking full advantages of the potentiality of the Raman spectroscopy as a structural diagnostic tool, we are able to achieve a deeper insight of PILs-induced structural modifications of lysozyme.
Experimental
Materials
Hen egg white lysozyme powder was purchased from Sigma-Aldrich (Fluka 62970) and used without further purification. HEWL fibrils were prepared by following a thermo-chemical protocol.33 A quantity of 14 mg of HEWL was dissolved in 1 ml of distilled, purified water and the pH of the solution was adjusted to 2 by adding HCl (Sigma-Aldrich). The temperature was then gradually increased with a rate of 10 °C h−1 up to 72 °C and the solution was kept at this temperature for 6 days, thus obtaining mature fibrils forming a gelatinous compound. Fibrils were separated from smaller species by centrifugation at 3800 RPM for 10 min obtaining a pellet of fibrils with only small residues of water + HCl.The PILs used in the present work are based on the nitrate anion, NO3−.34–36 Their typical formula is schematically depicted in Fig. 2, while their complete formulas are reported in Table 1. EAN and PAN were acquired from IoLiTec (Ionic Liquids Technologies), while BAN and MEOAN were synthesized in house.37
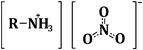 | ||
| Fig. 2 The general formula of the four PILs used in this work. | ||
| Name | Formula |
|---|---|
| 2-Methoxy Ethyl Ammonium Nitrate (MEOAN) | [CH3OCH2CH2N+H3][NO3]− |
| Ethyl Ammonium Nitrate (EAN) | [CH3CH2N+H3][NO3]− |
| Propyl Ammonium Nitrate (PAN) | [CH3CH2CH2N+H3][NO3]− |
| Butyl Ammonium Nitrate (BAN) | [CH3CH2CH2CH2N+H3][NO3]− |
Following ref. 26, weighted samples (1 mg) of fibril pellet were re-suspended in 1 ml of PILs. After 20 min at ambient conditions the solutions were centrifuged for 3 min (3800 RPM), decanted and washed with distilled purified water. The procedure was repeated 3 times. The applied treatment was sufficient to remove most of the ionic liquid from the sample (see the following). Finally, samples were dried to be measured by Raman spectroscopy.
Methods
Raman measurements were carried out using a confocal micro-Raman spectrometer by Jobin-Yvon, equipped with several objectives, a 20 mW He–Ne laser (632.8 nm wavelength) and a 1800 lines mm−1 grating. Raman spectra were collected in the back-scattering geometry and a notch filter was used to reject the elastic contribution, thus also preventing the collection of spectra close to the excitation line. Raman spectra were collected by means of a Peltier-cooled CCD (charge coupled device). Measurements were performed separately over four spectral ranges to cover the 200–3600 cm−1 wavenumbers region with a resolution better than 3 cm−1. A large confocal diaphragm of 400 μm has been used in order to obtain a good Raman signal. The absolute wavenumber calibration for each spectral range was obtained by collecting the emission lines of a neon lamp. Further experimental details can be found in ref. 38.Preliminary measurements on the protein powder placed onto a quartz slide were performed using available filters and objectives to find the best experimental conditions. Sample homogeneity and the absence of polarization effects were proved by repeating measurements on different sample regions. The typical acquisition time was 10 min for each frequency range. As a reference, the Raman spectra of the PILs were collected in a quartz cuvette (1 mm of optical path) after being de-hydrated in a controlled atmosphere under nitrogen flux.
All of the spectra were fitted using Levenberg-Marquardt minimization algorithm (LM algorithm) and Lorentzian-Gaussian pseudo-Voigt functions as peak profiles.39–42
Results and discussion
Raman spectra of native and fibrillar lysozyme
Before discussing our spectroscopic data we want to recall that the protein fibrillation pathway starts with the destabilization and partial unfolding of the native protein induced (in the present case) by a high temperature and low pH environment. Partial unfolded proteins are converted into intermediate oligomers, which subsequently are turned into protofibrils and finally into amyloids.43 The sequence of these structural transitions can be monitored by Raman spectroscopy, which allows us to follow the protein tertiary structure by means of the skeletal bending and the C–C–N stretching frequencies of the peptide backbone, and by the S–S and the C–S stretching frequencies of the disulphide bonds. The spectra of the native powder and fibrillar HEWL, as shown in Fig. 3, reveal remarkable differences indeed. The peaks ascribed to the S–S stretching vibrations between 500–550 cm−1 (ν(S–S) in Fig. 3) are still present after the thermo-chemical process, even if their shapes are clearly modified. The correlation between the band frequencies and the conformers of the disulfide bonds has been well established through normal coordinate analysis and extensive experimental investigation.44,45 As shown in Fig. 3, the four S–S bridges in native lysozyme give rise to three Raman bands at 507, 526 and 540 cm−1 indicating that the intra-molecular S–S bonds in native lysozyme are in GGG, GGT and TGT conformations,46 which is in agreement with the results of Van Wart et al.29 After fibril formation, the intensities of ν(S–S) vibrations near 530 and 540 cm−1 greatly decreased, clearly indicating a distortion of the dihedral angles with respect to the native structure.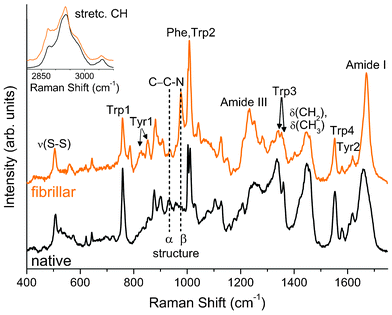 | ||
| Fig. 3 Raman spectra of HEWL in native and fibrillar conformations. The region of CH stretching mode is in the inset. Spectra were normalized in the frequency region of the Phe and Trp2 peaks (990–1025 cm−1). | ||
On the other side, the C–C–N stretching peaks are centered around 930 cm−1 when the protein structure is mainly α helix and at higher frequencies (around 960–980 cm−1) when it is mainly in a β sheet conformation (see the vertical dashed lines in Fig. 3).47,48 A close inspection of Fig. 3 clearly reveals that the band intensities ascribed to α helix and a β sheet conformations are markedly unbalanced towards the β structure on going from the native to the fibrillar protein.
An in-depth analysis of the Raman spectra allows us to monitor the environment experienced by side chains and, consequently, to get information on the protein folding. This is the case in the analysis of the relative intensities of the peaks forming the Fermi doublets49 arising from Tyr (830–850 cm−1) and Trp residues (1340–1360 cm−1) clearly detectable in the Raman spectra of both native and fibrillar HEWL (Tyr1 and Trp3 in Fig. 3, respectively).30,50
The peak intensity ratio I850/I830 of Tyr1 is a sensible marker of the hydrogen bonding of the Tyr phenoxyl group. If the ratio is around 0.3 the phenolic hydroxyl is the proton donor in a strong hydrogen bond, if I850/I830 is around 1.3 the phenolic oxygen is both an acceptor and a donor of a weak hydrogen bond, and if the ratio is around 2.5 the phenolic oxygen is the acceptor of a strong hydrogen bond.51 Decreases of the I850/I830 ratio reflect increases in buried residues, while a ratio around 1.3 is typical of solvent exposed residues. The analysis of our data shows I850/I830 ≅ 1.2 for the native sample accordingly with a conformation where Tyr residues are exposed and able to participate in moderate or weak hydrogen bonding.52 After the thermo-chemical treatment the ratio exceeds 2.0 suggesting the onset of strong hydrogen bonds and a conformation with Tyr more exposed to the solvent. This can be seen as a further spectroscopic signature of the unfolding process occurring after the thermo-chemical treatment.53,54 It is worth noting that the displacement of Tyr side chains in lysozyme during the unfolding of the tertiary structure can be strictly related to a change in the activity of the enzyme. Indeed, Tyr53 (see Fig. 1) is hydrogen bonded with the amino acid group of Asp66 and it is adjacent to the catalytic residue Asp52. A change of the Tyr53 position could, thus, affect the enzymatic active site of lysozyme.55
The peak intensity ratio I1360/I1340 of the Trp3 doublet is a marker of the hydrophobic/hydrophilic environment of the Trp indole ring.56 If the relative intensity ratio I1360/I1340 is smaller than 1.0, the indole ring is considered to be in a hydrophilic environment (or exposed to aqueous medium), whereas if the ratio is greater than 1.0 it is considered to be in a hydrophobic environment (or in contact with aliphatic side chains). The analysis of the present measurements provides I1360/I1340 = 0.4 for the native and I1360/I1340 = 2.5 for the fibrillar sample. The Trp side chains, thus, pass from a hydrophilic to a hydrophobic environment when going from the native to fibrillar state. Changes in the Raman spectra in the regions of Trp residues are of great importance because 3 of the 6 Trp residues (Trp62, Trp63 and Trp108, see Fig. 1) are in the active site of the enzyme,57 thus, changes in their environment should play an important role in the enzymatic activity.
Important information on lysozyme environment and structure can be obtained by analyzing the other bands associated with the Trp residues at 1550 and 1011 cm−1 (Trp4 and Trp2 in Fig. 3). The latter shows a frequency shift from 1011 cm−1 (native) to 1008 cm−1 (fibrillar)58 although, in this case, the conspicuous redistribution of the spectral intensities between the Trp2 and the close Phe peaks (∼1003 cm−1) is the most remarkable effect. This finding can be ascribed to a decrease of the intensity of the Phe peak rather than to an increase of the Trp2 peak, thus suggesting a larger exposure of Phe residue to the solvent after the thermal treatment.59
HEWL is a globular protein consisting of approximately 40–45% of α helix and around 20% of β sheets in its native conformation.60 Since it is well known that different secondary structures give rise to different components in the Raman amide bands,61 a standard analysis was carried out to obtain quantitative information on the secondary structures of HEWL. The results of the LM fitting procedure of the amide I band for the native structure are in good agreement with the literature values62 and are reported together with those obtained for the fibrillar sample in Table 2 (the spectral deconvolution of the amide band of the untreated fibrillar sample is shown in the following section in Fig. 5A). For the secondary structure characterization, since we are mainly interested in the α helix and β sheet contributions, we categorized the rest of the protein conformations as unordered structures. The fibrillation process decreases the α helix and increases the β components. In the first stage of the fibrillation, the native structure falls in a disordered structure and then develops into an organized β inter-chain structure.63 It is important to recall that the peak at around 1670 cm−1 is the marker of ordered β sheet structures in the aggregated sample and that this feature together with the narrowing of the amide I band is the most clear markers of fibril formation in the Raman spectra.64 Shifts at lower frequencies can be observed in the amide III region coherently with the above mentioned changes in the secondary structure from α helix (around 1300 cm−1) toward ordered β sheet (around 1230 cm−1) (see Fig. 3).40,65
| Native HEWL | Fibrillar HEWL | ||
|---|---|---|---|
| Unordered (random + turns) | 1644, 1693 | 1648, 1689 | Wavenumber (cm−1) |
| 32 | 19 | Area (%) | |
| α helix | 1660 | 1659 | Wavenumber (cm−1) |
| 45 | 19 | Area (%) | |
| β sheet inter-chain | — | 1670 | Wavenumber (cm−1) |
| — | 39 | Area (%) | |
| β sheet intra-chain | 1679 | 1633, 1679 | Wavenumber (cm−1) |
| 23 | 23 | Area (%) |
Raman spectra of HEWL treated with PILs
The Raman spectra of fibrillar lysozyme after the treatment with PILs are shown in Fig. 4A together with the spectra of the protein in the native and in the pristine fibrillar conformations. The spectra of the four pure PILs are shown Fig. 4B where it can be noticed that the symmetric stretching mode of NO3− is largely the most prominent spectral feature. Despite three successive centrifugations and re-dilutions in water, this peak is still present in the Raman spectra of the treated samples (see Fig. 4A). Nevertheless by comparing the spectra in Fig. 4A and B it is rather clear that the presence of a small amount of PIL in the treated samples does not significantly affect the Raman signal arising from lysozyme, at least away from the frequency of the NO3− stretching peak. In particular, the Raman contribution originating from residual PIL can be safely neglected over the frequency regions where the most relevant lysozyme spectral markers are found. By comparing the spectra of the treated samples in Fig. 4A with those collected from the protein in the fibrillar and the native conformations we can highlight and discuss the effects of the PILs different treatment.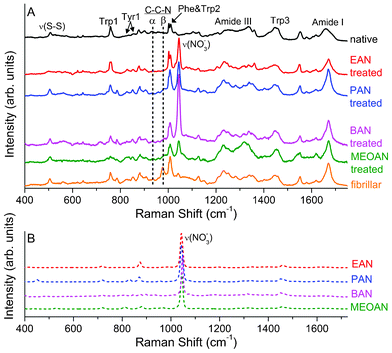 | ||
| Fig. 4 (A) Raman spectra of the HEWL in native and fibrillar conformations, and of the fibrillar sample treated with: EAN, PAN, BAN and MEOAN. (B) Raman spectra of EAN, PAN, BAN and MEOAN. | ||
From a qualitative point of view, the Raman spectra of PAN and BAN treated samples appear very similar to the one collected from pristine fibrillar HEWL. In particular, looking at the disulphide bonds we notice that the two ν(S–S) vibrations at the highest frequency are still lacking, suggesting the same disposition of the dihedral angle as the fibrillar sample, i.e. distorted with respect to the native structure. Similar conclusions can be drawn looking at the intensity ratio found for the Tyr1 and Trp3 doublets (see Table 3). The values found for PAN and BAN treated samples show indeed that Tyr experienced a strong hydrogen bond interaction (I850/I830 = 1.9 and 1.8, respectively) and that Trp was still in a hydrophobic environment (I1360/I1340 = 2.0 and 1.6, respectively), as well as in the pristine fibrillar sample (I850/I830.= 2.1 and I1360/I1340 = 2.5). Some differences with the pristine fibrillar sample can be found in the C–C–N stretching peak region where a broadening and an intensity reduction of the spectral structure around 977 cm−1 (β anti-parallel configuration) is observed in the two treated samples.47 These findings reflect slight changes in the tertiary structure and reveal an increased disorder in the β structures of the treated samples (see also the quantitative analysis of the amide I band reported in the following text).
| Native HEWL | EAN treated | PAN treated | BAN treated | MEOAN treated | Fibrillar HEWL | |
|---|---|---|---|---|---|---|
| Tyr1 (I850/I830) | 1.2 (2) | 1.0 (2) | 1.9 (1) | 1.8 (1) | 0.7 (2) | 2.1 (2) |
| Trp3 (I1360/I1340) | 0.4 (1) | 0.9 (1) | 2.0 (1) | 1.6 (1) | 1.3 (6) | 2.5 (2) |
A similar peak broadening and intensity decrease can be observed in the spectrum of the MEOAN-treated sample, albeit in this case other spectral modifications are also evident from the comparison with the spectrum of the pristine fibrillar lysozyme. In particular, MEOAN modifies the lysozyme tertiary structure mainly on Phe, Tyr and Trp residues. The interaction between MEOAN and the aromatic residues is indeed well evidenced in the Raman spectra by the modifications of the frequencies, shapes and intensities of the Trp peaks at 757 cm−1 (Trp1), 1340/1360 cm−1 (Trp3), 1550 cm−1 (Trp4) and of the Tyr peaks at 830/850 cm−1 (Tyr1), 1610 cm−1 (Tyr2). The frequency values reported here refer to the native conformation. To explain these changes in the Raman spectra we hypothesize a cation–π interaction between the NH+ cation of MEOAN and the electrostatic negative charge of the aromatic ring of Phe, Tyr and Trp residues.66,67 MEOAN is the only PILs among those studied here that exhibits such a behavior, we can thus address this peculiar interaction to the presence of an ether group in the alkyl chain, which makes MEOAN more polar than the others. On the other hand, this type of interaction is quite common in proteins68 and we have observed similar spectral modifications in a sample of fibrillar insulin treated with MEOAN (data not shown). Moreover, the Raman spectrum of the MEOAN-treated lysozyme shows changes with respect to that of the pristine fibrillar sample, also in the peaks involved in the NH modes (see around 1317 and 1540 cm−1), reinforcing the hypothesis of a cation–π interaction. The strong interaction among the MEOAN and the aromatic residues likely causes changes in the environment and relative position of the Tyr residues. Indeed, the low value of the intensity ratio of Tyr1 (see Table 3) is typical of residues buried inside the protein.
Among those presently investigated, EAN is the ionic liquid showing the most remarkable differences between the spectra collected from the EAN-treated and the pristine fibrillar lysozyme. These changes are mostly compatible with a PIL-induced refolding process. Looking at Fig. 4A, it can be noticed that the C–C–N stretching mode, related to the β anti-parallel structure (around 977 cm−1) totally vanishes in the spectrum of the EAN treated sample closely resembling the spectrum of the native protein. Indications of the ongoing refolding process can be also found looking at the Phe–Trp2 doublet just above 1000 cm−1. As it was already observed before, the decreasing in intensities of the Phe peaks (see Fig. 4A) is due to the solvent exposure of the Phe induced by the fibrillation process.33 Further indications of the refolding process can be obtained by the analysis of the Raman spectra in the S–S stretching region and from the Tyr Fermi doublet. A restoration of the native dihedral angle is revealed by the ν(S–S) vibrations, while the value of the Tyr1 ratio (I850/I830 = 1.0) is close to the native one, displaying a character in the hydrogen bond that is both donor and acceptor. Finally, the value obtained by the analysis of the ratio of the Trp3 Fermi doublet (I1360/I1340= 0.9) shows a partial restoration of the hydrophilic environment around the Trp residues, resembling the case for the native lysozyme. We also would like to note that the peak around 786 cm−1 (see Fig. 4A), albeit not clearly assigned, can be found in all of the spectra, except the native and the EAN treated ones.
Looking at Fig. 4A we notice that the amide I band is actually unaffected by any PILs treatments except for the EAN. The latter causes a detectable broadening of the band: the FWHM (full width half maximum) goes from 25 cm−1 to 30 cm−1 after the EAN treatment of the fibrillar sample. As mentioned above, more quantitative information about the refolding process can be obtained by a careful shape analysis of the amide band. A standard band fitting procedure was carried out for all of the investigated samples. In Fig. 5 the best fit curves obtained using the LM algorithm for the EAN-treated and the pristine fibrillar samples are compared with the experimental data. The different components used in the fitting procedure associated to different secondary structures of the HEWL conformation are also shown. In Table 4 the percentages for the secondary structures considered are reported. Also in this case, the analysis shows the peculiarity of the EAN treatment, which apparently induces modification of the secondary structure of fibrillar HEWL different from those induced by the other PILs. In particular, in Table 4 going from the fibrillar to the EAN-treated sample, the percentage of the α helix grows (from 19% to 31%) and that of the β sheet inter-chain diminishes (from 39% to 30%). It is important to stress once more that the concomitant diminishing of the β sheet inter-chain structure and the broadening of the amide I band (see Fig. 5) in the EAN-treated sample are clear signs of the ongoing processes of fibril dissolution and protein refolding. Looking at Table 4, it is also important to notice that, albeit weak, a systematic decrease of the unordered components simultaneous to the increase of the β sheet intra-chain trend can be observed going from PAN to BAN to MEOAN, i.e. actually on increasing the alkyl chain.
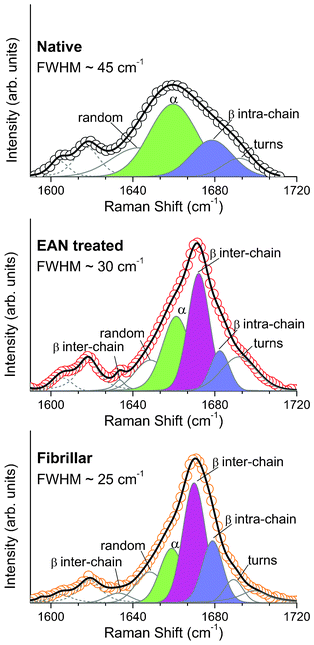 | ||
| Fig. 5 Raman spectra of the native HEWL (black circles), fibrillar HEWL (orange circles) and fibrillar HEWL treated with EAN (red circles) fitted through the LMA (black line in every panels). In solid grey, the different conformations of the amide I band are shown, and in dashed grey are the Tyr2 and Phe contributions. The three main conformations of the protein are coloured to be easily compared among the panel (green, α helix; blue, β sheet intra-chain; magenta, β sheet inter-chain). | ||
| Fibrillar HEWL | Fibrillar HEWL treated with MEOAN | Fibrillar HEWL treated with BAN | Fibrillar HEWL treated with PAN | Fibrillar HEWL treated with EAN | Native HEWL | ||
|---|---|---|---|---|---|---|---|
| Unordered (random + turns) | 19 | 11 | 20 | 20 | 27 | 32 | Area (%) |
| α helix | 19 | 16 | 14 | 14 | 31 | 45 | Area (%) |
| β sheet inter-chain | 39 | 39 | 35 | 35 | 30 | — | Area (%) |
| β sheet intra-chain | 23 | 34 | 33 | 31 | 12 | 23 | Area (%) |
Conclusions
We report on a complete Raman investigation of lysozyme in the native and fibrillar conformations. In addition to the standard shape analysis of the amide I band, the backbone deformation and the Fermi doublets of Tyr and Trp were analyzed in depth, thus obtaining a comprehensive spectral characterization of the native and fibrillar lysozyme. This preliminary investigation provided us with the fundamental basis for an analysis of the refolding effect of several PILs on fibrillar lysozyme. We carried out a systematic Raman investigation of four PILs-treated fibrillar lysozyme samples on lengthening the PIL alkyl chain and through the comparison with the native and the untreated fibrillar sample we were able to evaluate the refolding efficiency of the solvents. Different mechanisms of interaction among the PILs and the fibrillar lysozyme were observed.A schematic representation of the protein conformations induced by the thermo-chemical and by the subsequent PIL treatments is shown in Fig. 6. We noticed that the fibrillar conformation (Fig. 6b) is mainly characterized by a large extent of β aggregates (β inter-chain, red segment), which, according to the results of the quantitative analysis of the secondary conformations, is significantly reduced by the treatment with the shortest chain PIL studied here (EAN) (Fig. 6c). This progress towards the native conformation (Fig. 6a) induced by the EAN treatment is evidenced by the refolding of several protein structures (blue and green segments in Fig. 6c) consistently with the indications obtained from several spectroscopic markers related to specific residues. In particular, changes in the environment of Phe, Tyr and Trp residues induced by fibrillation were almost completely removed after the EAN treatment. These changes are particularly relevant since Tyr53, Trp62, Trp63 and Trp108 are in the lysozyme active site and have an important role in its enzymatic activity.54,56 This result provides new information about the refolding microscopic mechanism and is in fairly good agreement with the recovery of the lysozyme functionality reported in ref. 26.
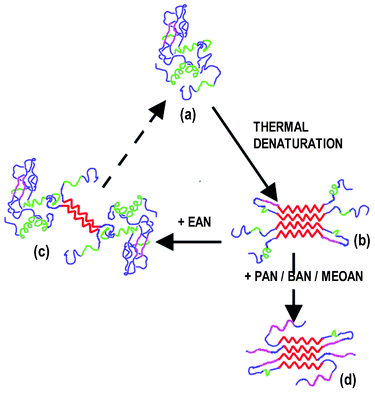 | ||
| Fig. 6 A schematic representation of HEWL conformations: (a) native, (b) fibrillar, (c) partially refolded after EAN treatment and (d) average configuration after PAN, BAN and MEOAN treatments. α helix segments are in green, β intra-chain in magenta, β inter-chain in red and unordered segments in blue. | ||
Treatments with long chain PILs (PAN, BAN, and MEOAN) do not drive fibrillar lysozyme toward the native conformation, but, on the contrary, they increase the percentage of β intra-chain (magenta segments) with respect to both fibrillar and native conformations. Generally speaking the interaction with long chain PILs leads to a protein conformation markedly β (around 70% on average) far away from the mainly α helix structure of the native protein (45% α and 23% β), but also from the fibrillar (∼60% β). In particular, the comparative spectral analysis shows that the tertiary structure of the fibrillar protein was only partially affected by PAN and BAN. The analysis of the spectroscopic markers related to Phe, Tyr and Trp residues shows that their environment is still the same as that in the fibrillar conformation, thus preventing any recovery of the enzymatic activity. Thus, these two long chain PILs seems to act on the packing of fibrils partly dissolving it and leaving the protein in a partly β un-aggregated conformation. No evidences of a real refolding process have been found also in the fibrillar sample treated with MEOAN (a long chain PIL with an ether group). In this case, likely due to the presence of the ether group, we observed a clear indication of a different PIL-protein interaction mainly affecting the aromatic residues. Our results show that only the treatment with EAN (the shortest chain PIL) induces a significant refolding of fibrillar lysozyme.
The possibility offered by the Raman spectroscopy to gain a deeper insight into the structural modifications induced by refolding additives opens the way to a systematic use of Raman spectroscopy as a simple, fast and effective diagnostic tool in refolding studies. Indeed the parallel investigation of the secondary (amide bands) and the tertiary (Tyr and Trp Fermi doublets) structures seems to be very useful in quantifying and in understanding the steps followed along the refolding path.
On concluding, our results show a significant dependence of the PIL refolding properties from the cation structure. The long alkyl chain PILs affect in a certain extent the ordered β aggregates, a peculiar aspect of the fibrils conformation, and prevent any conversion of the β sheets into α helices.On the contrary the shortest alkyl chain compound (EAN) is able to induce both the fibril melting and the refolding of the secondary structure. Finally, our results unravel the conformational changes that involve the microscopic mechanism at the origin of the lysozyme functionality recovery and, in principle, allow for the search of task-specific ILs.
Acknowledgements
L. G. acknowledges support from FIRB “Futuro in Ricerca” research project RBFR086BOQ_001, “Structure and dynamics of ionic liquids”.References
- H.-C. Mahler, W. Friess, U. Grauschopf and S. Kiese, J. Pharm. Sci., 2009, 98, 2909–2934 CrossRef CAS.
- R. Nelson, M. R. Sawaya, M. Balbirnie, A.Ø. Madsen, C. Riekel and R. Grothe& D. Eisenberg, Nature, 2005, 435, 773–778 CrossRef CAS.
- C. A. Summers and R. A. Flowers II, Protein Sci., 2000, 9, 2001–2008 CrossRef CAS.
- N. Byrne, L.-M. Wang, J.-P. Belieres and C. A. Angell, Chem. Commun., 2007, 2714–2716 RSC.
- C. Lange, G. Patil and R. Rudolph, Protein Sci., 2005, 14, 2693–2701 CrossRef CAS.
- M. Moniruzzamana, K. Nakashimab, N. Kamiyaa and M. Goto, Biochem. Eng. J., 2010, 48, 295–314 CrossRef.
- H. Tokuda, K. Ishii, M. A. B. H. Susan, S. Tsuzuki, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2006, 110, 2833–2839 CrossRef CAS.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC.
- P. Bonhôte, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef.
- S. Zhang, N. Sun, X. He, X. Lu and X. Zhang, J. Phys. Chem. Ref. Data, 2006, 35, 1475–1517 CrossRef CAS.
- (a) P. D'angelo, A. Zitolo, V. Migliorati, E. Bodo, G. Aquilanti, J. L. Hazemann, D. Testemale, G. Mancini and R. Caminiti, J. Chem. Phys., 2011, 135, 074505 CrossRef; (b) K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351–356 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS.
- D. M. Anderson, Patent WO000057-A1, 2003.
- Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2008, 10, 44–46 RSC.
- J. L. Anderson, J. Ding, T. Welton and D. W. Armstrong, J. Am. Chem. Soc., 2002, 124, 14247–14254 CrossRef CAS.
- S. H. Schofer, N. Kaftzik, P. Wasserscheid and U. Kragl, Chem. Commun., 2001, 425–426 RSC.
- S. N. Baker and T. M. Mc Cleskey, et al. , Chem. Commun., 2004, 940–942 RSC.
- K. Fujita, D. R. MacFarlane and M. Forsyth, Chem. Commun., 2005, 4804–4806 RSC.
- N. Byrne and C. A. Angell, J. Mol. Biol., 2008, 378, 707–714 CrossRef CAS.
- E. Yamamoto, S. Yamaguchi& and T. Nagamune, Appl. Biochem. Biotechnol., 2011, 164, 957–967 CrossRef CAS.
- R. M. Lau, M. J. Sorgedrager, G. Carrea, F. van Rantwijk, F. Secundo and R. A. Sheldon, Green Chem., 2004, 6, 483–487 RSC.
- R. Buchfink, A. Tischer, G. Patil, R. Rudolph and C. Lange, J. Biotechnol., 2010, 150, 64–72 CrossRef CAS.
- M. R. Krebs, D. K. Wilkins, E. W. Chung, M. C. Pitkeathly, A. K. Chamberlain, J. Zurdo, C. V. Robinson and C. M. Dobson, J. Mol. Biol., 2000, 3, 541–549 CrossRef.
- L. A. Morozova-Roche, J. Zurdo, A. Spencer, W. Noppe, V. Receveur, D. B. Archer, M. Joniau and C. M. Dobson, J. Struct. Biol., 2000, 2-3, 339–351 CrossRef.
- A. Cao, D. Hu and L. Lai, Protein Sci., 2004, 2, 319–324 CrossRef.
- N. Byrne and C. A. Angell, Chem. Commun., 2009, 1046–1048 RSC.
- J. T. Peltonand and L. R. McLean, Anal. Biochem., 2000, 277, 167–176 CrossRef.
- S. A. Overman and G. J. Thomas Jr, J. Raman Spectrosc., 1998, 29, 23–29 CrossRef CAS.
- H. E. Van Wart, A. Lewis, H. A. Scheraga and F. D. Saevat, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 2619 CrossRef CAS.
- M. N. Siamwiza, R. C. Lord, M. C. Chen, T. Takamatsu, I. Harada, H. Matsuura and T. Shimanouchi, Biochemistry, 1975, 14, 4870 CrossRef CAS.
- N. C. Maiti, M. M. Apetri, M. G. Zagorski, P. R. Carey and V. E. Anderson, J. Am. Chem. Soc., 2004, 126, 2399 CrossRef CAS.
- S. U. Sane, S. M. Cramer and T. M. Przybycien, Anal. Biochem., 1999, 269, 255–272 CrossRef CAS.
- M. Xu, V. V. Ermolenkov, V. N. Uversky and I. K. Lednev, J. Biophotonics, 2008, 1, 215–229 CrossRef CAS.
- R. Atkin and G. G. Warr, J. Phys. Chem. B, 2008, 112, 4164–4166 CrossRef CAS.
- T. L. Greaves, A. Weerawardena, I. Krodkiewska and C. J. Drummond, J. Phys. Chem. B, 2008, 112, 896–905 CrossRef CAS.
- E. Bodo, P. Postorino, S. Mangialardo, G. Piacente, F. Ramondo, F. Bosi, P. Ballirano and R. Caminiti, J. Phys. Chem. B, 2011, 115, 13149–13161 CrossRef CAS.
- J.-P. Belieres and C. A. Angell, J. Phys. Chem. B, 2007, 111, 4926–4937 CrossRef CAS.
- P. Fattibene, A. Carosi, V. De Coste, A. Sacchetti, A. Nucara, P. Postorino and P. Dore, Phys. Med. Biol., 2005, 50(6), 1095–1108 CrossRef CAS.
- S. Mangialardo, F. Piccirilli, A. Perucchi, P. Dore and P. Postorino, J. Raman Spectrosc., 2012, 43, 692–700 CrossRef CAS.
- N. C. Maiti, M. M. Apetri, M. G. Zagorski, P. R. Carey and V. E. Anderson, J. Am. Chem. Soc., 2004, 126, 2399 CrossRef CAS.
- M. Jackson and H. H. Mantasch, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 95 CrossRef CAS.
- K. Huang, N. C. Maiti, N. B. Philips, P. R. Carey and M. A. Weiss, Biochemistry, 2006, 45, 10278 CrossRef CAS.
- H. R. Kalhor, M. Kamizi, J. Akbari and A. Heydari, Biomacromolecules, 2009, 10, 2468–2475 CrossRef CAS.
- H. Sugeta, Spectrochim. Acta, 1975, 31A, 1729–1737 CrossRef CAS.
- W. Qian, W. Zhao and S. Krimm, J. Mol. Struct., 1991, 250, 89–102 CrossRef CAS.
- A. T. Tu, Spectroscopy of Biological System, ed., R. J. H. Clark, R. E. Hester, John Wiley and Sons,New York, 1986, p 47 Search PubMed.
- A. Hedoux, Y. Guinet and L. Paccou, J. Phys. Chem. B, 2011, 115, 6740–6748 CrossRef CAS.
- S. Ngarize, A. Adams and N. K. Howell, Food Hydrocolloids, 2004, 18, 49–59 CrossRef CAS.
- Fermi resonance occurs between normal and overtone modes, if they are nearly coincident in energy. Indeed the resonance in the aromatic residue is between an in plane fundamental vibration and one or more combination bands due to out of plane vibrations of the aromatic ring. Tyr doublet arises from the normal mode ν1 (ring breathing fundamental) and the second harmonic 2ν16a (ring deformation overtone) of the para-substituted phenolic side chain. Trp doublet arises from the normal mode at 1340 cm−1 and a combination of two out-of-plane modes involving the benzene and pyrrole rings of Trp.
- I. Harada, T. Miura and H. Takeuchi, Spectrochim. Acta, Part A, 1986, 42, 307–312 CrossRef.
- Z.-Q. Wen, X. Cao and A. Vance, J. Pharm. Sci., 2008, 97(6), 2228 CrossRef CAS.
- W. Zhao and R. Yang, J. Phys. Chem. B, 2010, 114, 503–510 CrossRef CAS.
- A. Torreggiani, M. D. Foggia, I. Manco, A. D. Maio, S. A. Markarian and S. Bonora, J. Mol. Struct., 2008, 891, 115 CrossRef CAS.
- R. Ionov, A. Hedoux, Y. Guinet, P. Bordat, A. Lerbret, F. Affouard, D. Prevost and M. Descamps, J. Non-Cryst. Solids, 2006, 352, 4430 CrossRef CAS.
- C. C. F. Blake, G. A. Mair, A. C. T. North, D. C. Phillips and V. R. P. Sarma, Proc. R. Soc. London, Ser. B, 1967, 167, 365 CrossRef CAS.
- H. Takeuchi and I. Harada, Spectrochim. Acta, 1986, 42A, 1069–1078 CrossRef CAS.
- C. Formoso and L. S. Forster, J. Biol. Chem., 1975, 250(10), 3738–3745 CAS.
- M. C. Chen, R. C. Lord and R. Mendelshon, Biochim. Biophys. Acta, Protein Struct., 1973, 328, 252–260 CrossRef CAS.
- M. Xu, V. V. Ermolenkov, W. He, V. N. Uversky, L. Fredriksen and I. K. Lednev, Biopolymers, 2005, 79, 58–61 CrossRef CAS.
- T. Y. Cho, N. Byrne, D. J. Moore, B. A. Pethica, C. A. Angell and P. G. Debenedetti, Chem. Commun., 2009, 4441–4443 RSC.
- R. W. Williams, Methods Enzymol., 1986, 130, 311–31 CrossRef CAS.
- http://www.rcsb.org/pdb/explore/explore.do?structureId=2lyz .
- M. Xu, V. A. Shashilov, V. V. Ermolenkov, L. Fredriksen, D. Zagorevski and I. K. Lednev, Protein Sci., 2007, 16, 815–832 CrossRef CAS.
- W. S. Gosal, A. H. Clark and S. B. Ross-Murphy, Biomacromolecules, 2004, 5, 2408–2419 CrossRef CAS.
- L.A. Popova, R. Kodali, R. Wetzel and I. K. Lednev, J. Am. Chem. Soc., 2010, 132, 6324–6328 CrossRef CAS.
- D. A. Dougherty, Science, 1996, 271, 163–168 CAS.
- Z.-Q. Wen, X. Cao and A. Vance, J. Pharm. Sci., 2008, 97(6), 2228 CrossRef CAS.
- R. M. Johnson, K. Hecht and C. M. Deber, Biochemistry, 2007, 46, 9208–9214 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |