DOI:
10.1039/C2RA21587J
(Paper)
RSC Adv., 2012,
2, 9091-9099
A carbanion induced ring switching synthesis of spiranes: an unprecedented approach†
Received
27th July 2012
, Accepted 30th July 2012
First published on 31st July 2012
Abstract
An unique approach to the synthesis of heterocyclic spiranes through ring switching transformation of suitably functionalized 2H-pyran-2-ones, benzo[h]chromene and thiochromeno[4,3-b]pyrans has been developed. The spirane dimer 11d displayed interesting halogen-hydrogen bonding to generate an elliptical cavity and may be relevant to the generation of a host guest assembly for specific cations and also a low helimerization energy barrier of spirane 17a.
Introduction
The unique topography and interesting conformational features of spiranes and their structural implication on biological systems have drawn the attention of organic chemists towards the development of their chemistry. There are numerous biologically active natural products1,2 in which spiranes feature in the sub-structure. However, in the spiranes the plane of both rings are mutually perpendicular along the tetrahedral spiro carbon. The asymmetric characteristic of the spiro carbon also gives the pronounced pharmacological properties.3 Fridericamycin (1),4 Pronuciferine (2),5 and Spirofomabuxine (3)6 are some of the representatives of this large family of compounds. Besides natural products, synthetic carbocyclic and heterocyclic spiranes have demonstrated various medicinal properties.7 Amongst these (+)-3-[4-(1-piperidinoethoxy)phenyl]spiro[indene-1,1′-indane]-5,5′-diol (4)8 is one of the most potent nonsteroidal estrogen agonists (Fig. 1). Due to steric constraints, spiranes exhibit various photochemical phenomena9 and are used as building blocks in the thermotropic liquid crystals, employed in optical displays and screens.10
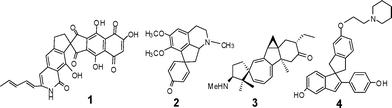 |
| | Fig. 1 Naturally occurring and synthetic spiranes. | |
Results and discussion
Chemistry
Amongst numerous approaches for the construction of spiranes, the most prominent are through alkylation,11 transition metal catalyzed reactions,12 rearrangement based reactions,13 ring expansion and contraction reactions,14 photochemical reactions,15 Diels–Alder cyclo-addition reactions,16 radical cyclization,17 and metathesis.18 However, these procedures suffer with limitations of functional group incompatibility and restriction to substitution patterns. A comprehensive literature survey revealed that there are only a few instances where useful functional groups attached to spiranes are transformed to newer molecules through synthetic manipulation. We envisaged a novel approach to the synthesis of functionalized spiro systems, free from the limitations of earlier protocols. This approach also opens a new avenue for functional group transformation to generate molecular diversity under mild reaction conditions.
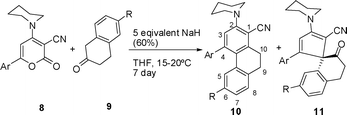
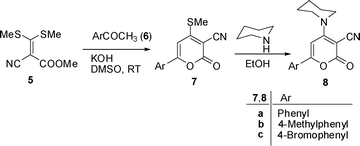
We report herein an unprecedented synthesis through ring switching transformation of suitably functionalized 6-aryl-4-sec-amino-2H-pyran-2-ones (8) by 2-tetralone (9) in dry THF using NaH as a base at 15–20 °C to produce spiranes. 6-Aryl-4-(piperidin-1-yl)-2H-pyran-2-one-3-carbonitriles (8) used as precursors for the synthesis of spiranes were prepared19 in two steps. The first step was the synthesis of 6-aryl-4-methylthio-2H-pyran-2-ones (7) from the reaction of methyl 2-cyano-3,3-dimethylthioacrylate (5) and aryl methyl ketone (6). Amination of 7 with piperidine in boiling ethanol afforded 8 as depicted in Scheme 1.
 |
| | Scheme 1 Synthesis of the 2H-pyran-2-ones, precursors for the synthesis of phenanthrenes 10 and spiranes 11. | |
Thus, a reaction of an equivalent amount of 6-phenyl-4-methylthio-2H-pyran-2-one-3-carbonitrile19 (7) and 2-tetralone (9a) in dry THF in the presence of 5 equivalent NaH (60%) as a base at 15–20 °C for 7 days led to yield a complex mixture with recovery of major unreacted starting lactone (7). However, the reaction of 6-aryl-4-sec-amino-2H-pyran-2-one-3-carbonitriles19 (8) with 2-tetralones (9) under analogous reaction conditions gave two compounds in which one was characterized as a phenanthrene derivative (10) and the other major product as a spirane (11), Scheme 2 (Table 1). The structure of 11d has been confirmed by X-ray diffraction analysis24 (Fig. 2).
| Entry |
8,10,11 |
Ar |
R |
Yields (%) |
|
10
|
11
|
|
Absolute configuration represented on the basis of X-ray analysis.24
|
| 1 |
a
|
Phenyl |
H |
10 |
40 |
| 2 |
b
|
4-Methylphenyl |
H |
11 |
34 |
| 3 |
c
|
4-Methylphenyl |
OMe |
19 |
31 |
| 4 |
d
|
4-Bromophenyl |
OMe |
17 |
29a,24 |
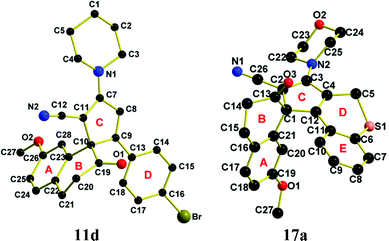 |
| | Fig. 2 Perspective view of 11d and 17a with atom numbering. | |
The reaction is possibly initiated with a Michael addition and liberation of carbon dioxide followed by ring switching to give the spirane (11) and highly functionalized phenanthrene (10) following path A and path B respectively. A plausible mechanism for the formation of 10 and 11 is depicted in Scheme 3.
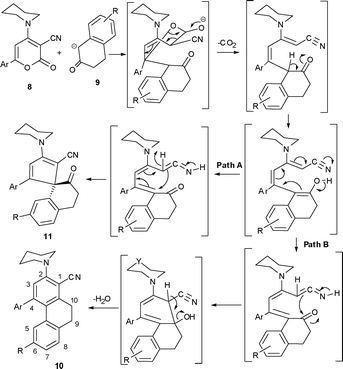 |
| | Scheme 3 A plausible mechanism for the formation of 10 and 11. | |
In order to optimize the reaction conditions, a model experiment was performed using 8a and 9a as reactants in different solvent, base, temperature and time to improve the yields and these results are summarized in Table 2. As is evident from Table 2, the increase of the reaction time beyond 7 days did not enhance the yield. However, reaction is very sensitive to temperature and above 20 °C, there was almost complete loss of yield. Change of solvent from THF to DMF and base from NaH to KOH/NaOH at room temperature exclusively gave phenanthrene derivatives (10).
Table 2 Optimization of reaction conditions of phenanthrene 10a and spiranes 11a
| Entry |
Basea |
Solvent |
Temperature/°C |
Time |
Products (%) |
|
10a
|
11a
|
|
NaH (60%) was used.
|
| 1 |
KOH/NaOH |
DMF |
22–30 |
24 h |
65 |
— |
| 2 |
NaH(1.5 equi) |
THF |
22–30 |
24 h |
40 |
1–2 |
| 3 |
NaH (3 equi) |
THF |
15–20 |
3 d |
32 |
15 |
| 4 |
NaH (5 equi) |
THF |
15–20 |
5 d |
10 |
30 |
| 5 |
NaH (5 equi) |
THF |
15–20 |
7 d |
10 |
40 |
| 6 |
NaH (5 equi) |
THF |
15–20 |
10 d |
10 |
40 |
Further, to generalize this reaction for the construction of fused polycyclic spiro systems, 5,6-dihydro-4-sec-amino-2-oxo-2H-benzo[h]chromene-3-carbonitrile20 were prepared from the reaction of 1-tetralone and thiochromen-4-ones (14a) and 4-sec.amino-2-oxo-2,5-dihydrothiochromeno-[4,3-b]pyran-3-carbo- nitriles (14b-e) with methyl 2-cyano-3,3-dimethyl thioacrylate21 (5) followed by amination with sec.amine in boiling ethanol, afforded 4 in good to excellent yields, Scheme 4.23
![Synthesis of the substituted 5,6-dihydro-2-oxo-2H-benzo[h] chromene-3-carbonitriles (13a,14a) and 2,5-dihydro-2-oxo-thiochromeno [4,3-b]pyran-3-carbonitriles (13b–e,14b-e).23](/image/article/2012/RA/c2ra21587j/c2ra21587j-s4.gif) |
| | Scheme 4 Synthesis of the substituted 5,6-dihydro-2-oxo-2H-benzo[h] chromene-3-carbonitriles (13a,14a) and 2,5-dihydro-2-oxo-thiochromeno [4,3-b]pyran-3-carbonitriles (13b–e,14b-e).23 | |
It was quite interesting that the ring transformation of 14 by 2-tetralones in the presence of 1.5 equivalents of NaH in THF at room temperature gave two products which were characterized as 5,6,9,10-tetrahydro-7-oxa[5]helicene (15) as the major product and the 8-sec-amino-5,6,9,10-tetrahydro[5]helicene (16) as the minor product,22Scheme 5.
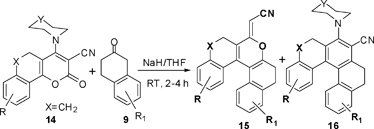 |
| | Scheme 5 Synthesis of tetrahydrooxahelicenes (15) and (16). | |
However, when this reaction was carried out by stirring an equimolar mixture of 14 and 9 using 5 equivalent NaH (60%) in dry THF at 15–20 °C for 7 days, it led to 3-sec-amino-2′-oxo-3′,4′-dihydro-2′H,4H-spiro[cyclopenta[c]chromene/thiochromene-1,1′-naphthalene]-2-carbonitriles (17) along with recovery of starting material 14, Scheme 6. Optimized reaction conditions of 17 performed under various temperatures are shown in Table 3. The structure of isolated product (17) was confirmed by single crystal X-ray diffraction24 (Fig. 2).
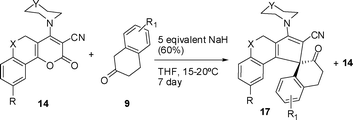 |
| | Scheme 6 Synthesis of spiranes 17 (Table 4). | |
Table 3 Optimized reaction conditions of spirane 17a under various temperatures
| Entry |
Time/d |
Temperature/°C |
Yield (%) (17a) |
Sa |
|
Starting lactone (14) recovered (%).
Under light.
Other complex unidentified products.
|
| 1 |
5 |
5–10 |
0 |
98 |
| 2 |
7 |
15–20 |
45 |
50 |
| 3 |
10 |
15–20 |
45 |
45 |
| 4 |
7 |
25–30 |
25 |
45c |
| 5 |
4 |
32–35 |
1–2 |
40c |
| 6 |
4 |
40–45b |
1 |
20c |
Our attempts failed to obtain spiro compounds by the ring transformation of 7 and 13 with 2-tetralone (9) under analogous reaction conditions. This indicated that the amino substituent has an immense electronic contribution in the formation of spiro compounds. It is also conspicuous that the synthesis of spiranes (17) is dependent on the reaction conditions (Table 3). It was interesting to note that the increase of temperature above 22 °C not only decreases the yield but leaves unreacted starting material and a non-separable complex mixture.
The reaction is possibly initiated with a Michael addition and liberation of carbon dioxide followed by framework rearrangement and cyclization to give spiranes (17) as shown in Scheme 6 (Table 4). A plausible mechanism for this reaction is also depicted in Scheme 7.
Table 4 Yields and melting points of spiranes 17 (Scheme 6)
| Entry |
R |
R1 |
X |
Y |
Mp/°C |
Yield (%) |
|
Absolute configuration represented on the basis of X-ray analysis.24
|
|
17a
a
|
H |
7-OMe |
S |
O |
270 |
45 |
|
17b
|
H |
7-OMe |
S |
CH2 |
290 |
39 |
|
17c
|
H |
6-OMe |
S |
CH2 |
256 |
40 |
|
17d
|
Cl |
6-OMe |
S |
CH2 |
258 |
42 |
|
17e
|
H |
6-OMe |
CH2 |
CH2 |
246 |
37 |
|
17f
|
H |
H |
S |
CH2 |
278 |
35 |
|
17g
|
H |
6-OMe |
S |
N–Ph |
216 |
31 |
![A plausible mechanism for the formation of 3-sec-amino-2′-oxo-3′,4′-dihydro-2′H,4H-spiro[cyclopenta[c] thiochromene-1,1′-naphthalene]-2-carbonitriles (17).](/image/article/2012/RA/c2ra21587j/c2ra21587j-s7.gif) |
| | Scheme 7 A plausible mechanism for the formation of 3-sec-amino-2′-oxo-3′,4′-dihydro-2′H,4H-spiro[cyclopenta[c] thiochromene-1,1′-naphthalene]-2-carbonitriles (17). | |
Crystal structure and quantum chemical studies
Crystal structure analysis.
Crystals of X-ray quality for 11d was obtained by slow evaporation of solvent (EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane; 1:25) and 17a was obtained by slow evaporation of solvent (DCM:THF; 10:3) at room temperature (15–18 °C). The molecular structures of the compounds with arbitrary numbering are presented in Fig. 2 illustrating the non-planar, helically distorted conformation of the molecule.
:hexane; 1:25) and 17a was obtained by slow evaporation of solvent (DCM:THF; 10:3) at room temperature (15–18 °C). The molecular structures of the compounds with arbitrary numbering are presented in Fig. 2 illustrating the non-planar, helically distorted conformation of the molecule.
The compounds 11d and 17a crystallize in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group having one and two molecules in the triclinic unit cell respectively. When viewed from the rings C or B side of the compound, 11d displays the S configuration because of the presence of the chiral axis, whilst 17a displays the R configuration. The geometry around the spirane center C10 of 11d, is defined by the carbon atoms C9 and C11 of the five membered ring C and C19 and C23 belonging to six membered ring B. The two rings B and C are almost orthogonal with respect to each other having a dihedral angle of 82.11°. The dihedral angle between the rings A and C is 82.89°. The immediate geometry around the spirane center C1 of 17a is defined by the carbon atoms C2 and C12 belonging to five membered ring C and C13 and C21 belonging to six membered ring B. The two rings B and C are almost orthogonal with respect to each other having a dihedral angle of 77.44°. The dihedral angles between the rings A and C; C and E are 84.30° and 25.44°, respectively. In the solid state, 11d, displays intermolecular Br1⋯H21B interactions in antiparallel fashion with dimension 2.766 Å (symm. op.: −x, −y, 2 − z) and interaction angle 153.96° to generate a dimer configuration (Fig. 3).
space group having one and two molecules in the triclinic unit cell respectively. When viewed from the rings C or B side of the compound, 11d displays the S configuration because of the presence of the chiral axis, whilst 17a displays the R configuration. The geometry around the spirane center C10 of 11d, is defined by the carbon atoms C9 and C11 of the five membered ring C and C19 and C23 belonging to six membered ring B. The two rings B and C are almost orthogonal with respect to each other having a dihedral angle of 82.11°. The dihedral angle between the rings A and C is 82.89°. The immediate geometry around the spirane center C1 of 17a is defined by the carbon atoms C2 and C12 belonging to five membered ring C and C13 and C21 belonging to six membered ring B. The two rings B and C are almost orthogonal with respect to each other having a dihedral angle of 77.44°. The dihedral angles between the rings A and C; C and E are 84.30° and 25.44°, respectively. In the solid state, 11d, displays intermolecular Br1⋯H21B interactions in antiparallel fashion with dimension 2.766 Å (symm. op.: −x, −y, 2 − z) and interaction angle 153.96° to generate a dimer configuration (Fig. 3).
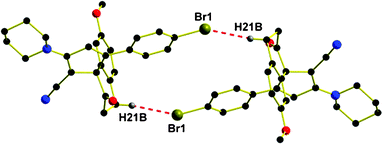 |
| | Fig. 3 Antiparallel dimeric structure for 11d displaying weak intermolecular C(Ar)–Br⋯H intermolecular halogen-hydrogen interaction. | |
This dimer in turn generates an elliptical cavity having a major axis of 11.20 Å and a minor axis of 4.46 Å. It is obvious that these interactions play an important role if the structure is to be rationalized in terms of interactions between the molecular fragments. However, the question as to what kind of intermolecular interaction(s) contribute to the binding energy between molecules and dimers in the structure need to be investigated. It is known that the covalent, H-bond, dipole–dipole, and van der Waals interaction energies are >1700, 70–50, 8–2, and <4 kJ mol−1, respectively.
In order to analyze the various interactions that lead to the crystal structure, interaction energies and electrostatic potentials (Fig. 4) have been calculated for dimer keeping Br1⋯H21B distance fixed at values obtained from the X-ray single-crystal structure analyses. This analysis of the interaction energy in the crystal structure of 11d by means of dimer unit at the MP2 level of theory yields interaction energy in the Br1⋯H21B dimer of −7 kJ mol−1. Hence this interaction can be regarded as the halogen-hydrogen bond.25
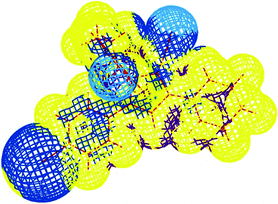 |
| | Fig. 4 Electrostatic potentials plotted at the van der Waals surfaces for 11d calculated at the MP2 level of theory, yellow/blue denote areas of low/high charge density. | |
Computational analysis.
In order to gain deeper insight into the stereochemical behaviour of 17a, the helimeric inversion barrier for this compound was calculated using quantum chemical methodology. The energy calculations reveal that the helimerization energy barrier for 17a is 11.70 kJ mol−1. The calculated low inversion barrier of the enantiomers is an indicator for rapid interconversion at room temperature.
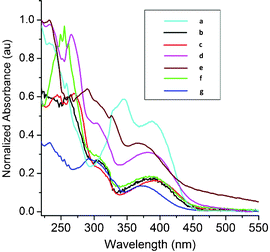
The electronic absorption recorded in a THF solution of 17a (Fig. 5) displays bands at ∼404, 374, 367, 322 and 278 nm. The observed electronic spectrum have been assigned with the help of time-dependent DFT (TD-DFT) calculations. Since each absorption line in a TD-DFT spectrum can arise from a several single orbital excitations, a description of the transition character is generally not straightforward. However, approximate assignments can be made, although they provide a simplified representation of the transitions. TD-DFT excitations were calculated both in the gas phase and in the solvent using dichloromethane (DCM). By comparing the calculated spectra it is evident that calculated transitions do not exhibit significant solvatochromic effects. In this view, only the DCM model results are presented in the Table 5. The first lower energy excitation calculated at 404 nm arises because of the π→π* transition within ring C. The next higher energy transition calculated at 374 nm occurs due to the π→π* transition between ring A and ring C. Also, in this case there is the involvement of the lone pair electron present on methoxy oxygen attached to ring A. The absorption calculated at 367 nm involves n→π* transition between the sulfur atom S1 of the ring D and π* orbitals of the ring C. The absorption calculated at 322 nm is ascribed to π→π* transitions from rings D and E to ring C; π→π* transition from ring C to ring B. The highest energy band calculated at 278 nm also arises because of the π→π* between ring C and rings B and E. Hence, from the TD-DFT calculations it can be concluded that the dominant electronic transition in 17a is π→π* with feeble contributions from the sulfur atom of the ring D. The morpholine ring does not play any role in the electronic absorption though it possesses lone pair of electrons at nitrogen and oxygen centers. This may also be due to inability of the morpholinyl moiety to undergo conjugation with the aromatic rings of the molecule.
Table 5 TD-DFT calculated excitations & approximate assignments, 17a
|
E
a
|
λ
b
|
F
c
|
Composition (%) |
Natured |
|
Excitation Energy (eV).
Wavelength (nm).
Oscillator Strength.
Nature of the transition and approximate assignment of compound 17a (Fig. 2).
|
| 3.07 |
404 |
0.1773 |
HOMO→LUMO (45%) |
π (C)→π*(C) |
| 3.32 |
374 |
0.0025 |
HOMO-1→LUMO (46%) |
π(A)→π*(C) |
| 3.38 |
367 |
0.0175 |
HOMO-2→LUMO (43%) |
n(D)→π*(C) |
| 3.85 |
322 |
0.0453 |
HOMO-3→LUMO (19%) |
π(D, E & C)→π*(C) |
| HOMO→LUMO+1 (29%) |
| 4.45 |
278 |
0.0260 |
HOMO→LUMO+2 (41%) |
π(C)→π*(E) |
Conclusion
In conclusion, we have developed an unique, concise and economical one pot protocol for the construction of novel spiranes through base catalyzed ring contraction-cyclization reactions of suitably functionalized 2H-pyran-2-ones, benzo[h]chromene and thiochromeno[4,3-b]pyrans by 2-tetralones separately. This methodology provides a new avenue for the construction of highly functionalized spiranes and provides opportunity to transform functional groups present to newer heterocycles. The TD-DFT calculations indicated that the majority of electronic transitions are π→π* type but the presence of hetero atom in conjugation with aromatic ring may induce n→π* transition. Additionally, the compound 11d, displayed interesting halogen-hydrogen bonding to generate an elliptical cavity. This elliptical cavity may act as an excellent host for specific cations and may be relevant for the generation of host guest assembly. This will be our future target for investigation.
Experimental section
General
The reagents and the solvents used in this study were of analytical grade and were used without further purification. The melting points were determined on an electrically heated Townson Mercer melting point apparatus and are uncorrected. Commercial reagents were used without purification. 1H and 13C NMR spectra were recorded on a Bruker WM-300 (300 MHz)/Jeol-400 using CDCl3 and DMSO-d6 as the solvents. Chemical shifts are reported in parts per million (δ-value) from Me4Si (δ 0 ppm for 1H) or based on the middle peak of the solvent (CDCl3/DMSO-d6) (δ 100.00 ppm for 13C NMR) as the internal standard. Signal patterns are indicated as s, singlet; d, doublet; dd, double doublet; t, triplet; m, multiplet; brm, broad multiplet. Coupling constants (J) are given in Hertz. Infrared (IR) spectra were recorded on a Perkin-Elmer AX-1 spectrophotometer in KBr disc and reported in wave number (cm−1). JEOL MSRoute/ESIMS spectrometers were used for mass/HRMS spectra.
General procedure for the synthesis of 9,10-dihydrophenanthrenes (10) and 3′,4′-dihydro-2′H-spiro[cyclopenta[2,4]diene-1,1′-naphthalene]-2-carbonitrile (11)
2-Tetralone (9) (1.0 mmol) was added in 5 equivalent NaH (60%) in THF (8 mL). After stirring five minutes, 2-oxo-6-phenyl-4-sec-amino-2H-pyran-3-carbonitrile (8) (1.0 mmol) was added and stirred at 15-20 °C for 7 days, monitoring TLC at regular interval. The excess THF was evaporated or distilled at reduced pressure and reaction mixture was poured onto crushed ice with vigorous stirring and neutralized with 10% HCl. The crude product obtained was filtered, washed with water and purified by silica gel column chromatography using chloroform/DCM in hexane.
4-phenyl-2-(piperidin-1-yl)-9,10-dihydrophenanthrene-1-carbonitrile (10a).
White powder; Rf 0.78 (CHCl3); yield 10%; mp 184 °C; IR (KBr): 2213 cm−1; 1HNMR (400 MHz, CDCl3): δ 1.58–1.62 (m, 2H, CH2), 1.77–1.80 (m, 4H, CH2), 2.85 (t, J = 6.96 Hz, 2H, CH2), 3.04 (t, J = 6.96 Hz, 2H, CH2), 3.20 (t, J = 4.76 Hz, 4H, CH2), 6.64 (d, 1H, J = 7.28 Hz, Ar–H), 6.74–6.79 (m, 1H, Ar–H), 6.83 (s, 1H, Ar–H), 7.02 (dd, 1H, J = 6.96, Ar–H), 7.19 (d, 1H, J = 7.28 Hz, Ar–H), 7.24–7.27 (m, 2H, Ar–H), 7.31–7.35 (m, 3H, Ar–H); 13C NMR (100 MHz, CDCl3): δ 24.1, 26.1(2C), 28.9, 29.2, 53.2 (2C), 104.6, 120.1, 125.5, 126.5, 127.1, 127.3, 127.5, 128.6 (2C), 129.1(2C), 129.1(2C), 132.7, 138.3, 142.3, 144.7, 145.8, 155.5; MS: m/z = 364.22 (M+); HRMS (ESI): calc. for C26H24N2: 365.1973 (M++1); found: 365.1978.
2′-oxo-5-phenyl-3-(piperidin-1-yl)-3′,4′-dihydro-2′H-spiro [cyclopenta[2,4]diene-1,1′-naphthalene]-2-carbonitrile (11a).
Light yellow-greenish solid; Rf 0.40 (CHCl3); yield 40%; mp 182 °C; IR (KBr): 2162 (CN), 1708 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) cm−1; 1HNMR (400 MHz, CDCl3): δ 1.67–1.71 (brm, 6H, CH2), 2.81–2.85 (m, 1H, CH2), 3.20–3.32 (m, 2H, CH2), 3.38–3.43 (m, 1H, CH2), 3.63–3.67 (brm, 4H, CH2), 6.87 (d, J = 7.64 Hz, 1H, Ar–H), 6.97 (s, 1H, Ar–H), 7.04–7.11 (m, 4H, Ar–H), 7.15–7.24 (m, 4H, Ar–H); 13C NMR (100 MHz, CDCl3): δ 24.0, 25.7(2C), 29.1, 39.5, 49.6(2C), 70.7, 84.6, 120.7, 124.6, 126.7(2C), 127.3, 127.4, 127.5, 128.5(2C), 128.7(2C), 128.9, 132.4, 135.1, 135.4, 158.7, 160.8, 206.3; MS: m/z = 380.25 (M+); HRMS (ESI): calc. for C26H24N2O: 381.1922 (M++1); found: 381.1929.
O) cm−1; 1HNMR (400 MHz, CDCl3): δ 1.67–1.71 (brm, 6H, CH2), 2.81–2.85 (m, 1H, CH2), 3.20–3.32 (m, 2H, CH2), 3.38–3.43 (m, 1H, CH2), 3.63–3.67 (brm, 4H, CH2), 6.87 (d, J = 7.64 Hz, 1H, Ar–H), 6.97 (s, 1H, Ar–H), 7.04–7.11 (m, 4H, Ar–H), 7.15–7.24 (m, 4H, Ar–H); 13C NMR (100 MHz, CDCl3): δ 24.0, 25.7(2C), 29.1, 39.5, 49.6(2C), 70.7, 84.6, 120.7, 124.6, 126.7(2C), 127.3, 127.4, 127.5, 128.5(2C), 128.7(2C), 128.9, 132.4, 135.1, 135.4, 158.7, 160.8, 206.3; MS: m/z = 380.25 (M+); HRMS (ESI): calc. for C26H24N2O: 381.1922 (M++1); found: 381.1929.
2-(piperidin-1-yl)-4-p-tolyl-9,10-dihydrophenanthrene-1-carbonitrile (10b).
White powder; Rf 0.25 (CHCl3:Hexane; 1:1); yield 11%; mp 144 °C; IR (KBr): 2203 cm−1; 1HNMR (400 MHz, CDCl3): δ 1.58–1.61 (m, 2H, CH2), 1.75–1.80 (m, 4H, CH2), 2.37 (s, 3H, CH3), 2.85 (t, J = 6.60 Hz, 2H, CH2), 3.03 (t, J = 6.60 Hz, 2H, CH2), 3.18 (t, J = 5.52 Hz, 4H, CH2), 6.69 (d, 1H, J = 7.32 Hz, Ar–H), 6.77–6.85 (m, 2H, Ar–H), 6.99–7.05 (m, 1H, Ar–H), 7.11–7.16 (m, 4H, Ar–H), 7.18 (d, 1H, J = 7.32 Hz, Ar–H); 13C NMR (100 MHz, CDCl3): δ 21.2, 24.1, 26.1(2C), 28.9, 29.2, 53.2(2C), 104.4, 118.9, 120.2, 125.5, 126.5, 127.3, 128.9(2C), 129.1(2C), 129.3(2C), 132.8, 137.4, 138.2, 139.4, 144.8, 145.8, 155.4; MS: m/z = 379.40 (M++1); HRMS (ESI): calc. for C27H26N2: 379.2130 (M++1); found: 379.2136.
2′-oxo-3-(piperidin-1-yl)-5-p-tolyl-3′,4′-dihydro-2′H-spiro[cyclopenta[2,4]diene-1,1′-naphthalene]-2-carbonitrile (11b).
Light yellow-greenish solid; Rf 0.47 (DCM); yield 34%; mp 204 °C; IR (KBr): 2152 (CN), 1705 (CO) cm−1; 1HNMR (400 MHz, CDCl3): δ 1.65–1.69 (brm, 6H, CH2), 2.22 (s, 3H, CH3), 2.80 (td, J = 5.12 and 13.9 Hz, 1H, CH2), 3.15–3.32 (m, 2H, CH2), 3.39 (td, J = 5.12 and 13.9 Hz, 1H, CH2), 3.60–3.67 (brm, 4H, CH2), 6.83 (d, J = 8.76 Hz, 1H, Ar–H), 6.91 (s, 1H, Ar–H), 6.94–6.98 (m, 4H, Ar–H), 7.05–7.09 (m, 1H, Ar–H), 7.14–7.22 (m, 2H, Ar–H); 13C NMR (100 MHz, CDCl3): δ 21.2, 24.0, 25.7(2C), 29.2, 39.5, 49.7(2C), 70.6, 84.2, 120.9, 123.6, 126.6(2C), 127.3(2C), 127.4, 128.7, 129.3, 129.6(2C), 135.0, 135.6, 139.1, 158.7, 160.9, 206.3; MS: m/z = 395.10 (M++1); HRMS (ESI): calc. for C27H26N2O: 395.2079 (M++1); found: 395.2082.
6-methoxy-2-(piperidin-1-yl)-4-p-tolyl-9,10-dihydrophenanthrene-1-carbonitrile (10c).
White powder, Rf 0.21 (CHCl3:Hexane; 1:1); yield 19%; mp 170 °C; IR (KBr): 2201 cm−1; 1HNMR (400 MHz, CDCl3): δ 1.64–1.70 (m, 2H, CH2), 1.83–1.87 (m, 4H, CH2), 2.43 (s, 3H, CH3), 2.85 (t, J = 6.56 Hz, 2H, CH2), 3.09 (t, J = 6.56 Hz, 2H, CH2), 3.25–3.30 (m, 7H, OCH3 and CH2), 6.35 (d, 1H, J = 2.20 Hz, Ar–H), 6.67 (dd, 1H, J = 2.20 and 8.76 Hz, Ar–H), 6.90 (s, 1H, Ar–H), 7.15 (d, 1H, J = 8.76 Hz, Ar–H), 7.20–7.26 (m, 4H, Ar–H); 13C NMR (100 MHz, CDCl3): δ 21.1, 24.1, 26.1(2C), 27.9, 29.5, 53.2(2C), 54.4, 104.6, 113.3, 113.9, 117.6, 120.0, 127.2, 128.0(2C), 129.0(2C), 129.4, 130.4, 133.5, 137.3, 139.6, 144.7, 145.8, 155.5, 157.0; MS: m/z = 408.90 (M++1); HRMS (ESI): calc. for C28H28N2O: 409.2235 (M++1); found: 409.2238.
7′-methoxy-2′-oxo-3-(piperidin-1-yl)-5-p-tolyl-3′,4′-dihydro-2′H-spiro[cyclopenta[2,4]diene-1,1′-naphthalene]-2-carbonitrile (11c).
Light yellow-greenish solid; Rf 0.20 (CHCl3); yield 31%; mp 234 °C; IR (KBr): 2150 (CN), 1701 (CO) cm−1; 1HNMR (400 MHz, CDCl3): δ 1.65–1.68 (brm, 6H, CH2), 2.23 (s, 3H, CH3), 2.75–2.80 (m, 1H, CH2), 3.08–3.16 (m, 1H, CH2), 3.21–3.26 (m, 1H, CH2), 3.28–3.36 (m, 1H, CH2), 3.62–3.68 (brm, 7H, CH3 and CH2), 6.37 (d, 1H, J = 2.20 Hz, Ar–H), 6.73 (dd, 1H, J = 2.20 and 8.76 Hz, Ar–H),6.88 (s, 1H, Ar–H), 6.95–6.99 (m, 4H, Ar–H), 7.12 (d, 1H, J = 8.76 Hz, Ar–H); 13C NMR (100 MHz, CDCl3): δ 21.2, 24.0, 25.8(2C), 28.4, 39.7, 49.6(2C), 55.1, 70.6, 76.7, 84.0, 112.5, 113.0, 120.9, 123.5, 126.6(2C), 127.4, 129.3(2C), 129.5, 129.6, 136.9, 139.1, 158.7, 160.9, 206.3; HRMS (ESI): calc. for C28H28N2O2: 425.2184 (M++1); found: 425.2187.
4-(4-bromophenyl)-6-methoxy-2-(piperidin-1-yl)-9,10-dihydrophenanthrene-1-carbonitrile (10d).
White (light yellowish) rod shaped crystal from hexane:CHCl3 (1:1); Rf 0.31 (CHCl3:hexane; 1:1); yield 17%; mp 178 °C; IR (KBr): 2209 cm−1; 1HNMR (400 MHz, CDCl3): δ 1.57–1.62 (m, 2H, CH2), 1.77–1.80 (m, 4H, CH2), 2.77 (t, J = 7.32 Hz, 2H, CH2), 3.01 (t, J = 7.32 Hz, 2H, CH2), 3.20 (t, J = 5.12 Hz, 4H, CH2), 3.26 (s, 3H, OCH3), 6.20 (d, 1H, J = 2.92 Hz, Ar–H), 6.61 (dd, 1H, J = 2.92 and 8.0 Hz, Ar–H), 6.77 (s, 1H, Ar–H), 7.08 (d, 1H, J = 8.0 Hz, Ar–H), 7.14 (d, 2H, J = 6.6 Hz, Ar–H), 7.48 (d, 2H, J = 6.6 Hz, Ar–H); 13C NMR (100 MHz, CDCl3): δ 24.0, 26.1(2C), 27.8, 29.5, 53.1(2C), 54.2, 54.6, 105.0, 113.6, 113.9, 117.4, 119.6, 127.0, 128.2, 130.4, 130.8(2C), 131.8(2C), 133.0, 141.3, 143.2, 146.1, 155.5, 157.2; MS: m/z = 473.20 (M++1); HRMS (ESI): calc. for C27H25BrN2O: 473.1184 (M++1); found: 473.1193.
5-(4-bromophenyl)-7′-methoxy-2′-oxo-3-(piperidin-1-yl)-3′,4′-dihydro-2′H-spiro[cyclopenta[2,4]diene-1,1′-naphthalene]-2-carbonitrile (11d).
Light yellow-greenish crystalline solid crystallized from EtOAc and hexane (1:25); Rf 0.20 (CHCl3); yield 29%; mp 262 °C; IR (KBr): 2152 (CN), 1708 (CO) cm−1; 1HNMR (400 MHz, CDCl3): δ 1.65–1.68 (brm, 6H, CH2), 2.74–2.80 (m, 1H, CH2), 3.08–3.13 (m, 1H, CH2), 3.21–3.30 (m, 2H, CH2), 3.61–3.65 (brm, 4H, CH2), 3.68 (s, 3H, OCH3), 6.34 (d, 1H, J = 2.8 Hz, Ar–H), 6.74 (dd, 1H, J = 2.8 and 8.8 Hz, Ar–H), 6.88–6.96 (m, 3H, Ar–H), 7.13 (d, 1H, J = 8.8 Hz, Ar–H), 7.29 (d, 2H, J = 8.8 Hz, Ar–H); 13C NMR (100 MHz, CDCl3): δ 23.9, 25.8(2C), 28.4, 39.7, 49.7(2C), 55.2, 70.7, 84.5, 112.5, 113.1, 120.5, 123.2, 125.1, 127.3, 128.2(2C), 129.9, 131.4(2C), 131.8, 136.3, 157.4, 158.8, 160.4, 206.0; MS: m/z = 489.30 (M++1); HRMS (ESI): calc. for C27H25BrN2O2: 489.1133 (M++1); found: 489.1139.
General procedure for the synthesis of spiranes (17)
2-Tetralone (9) (1.0 mmol) was added to a suspension of 5 equivalent NaH (60%) in THF (8 mL). After stirring five minutes 4-sec-amino-2-oxo-5,6-dihydro-2H-benzo[h]chromene-3-carbonitrile/4-sec-amino-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitrile (14) (1.0 mmol) was added and reaction mixture was stirred at 15–20 °C for 7 days by monitoring TLC at regular interval. The excess THF was distilled at reduced pressure and thereafter, reaction mixture was poured onto crushed ice with vigorous stirring and neutralized with 10% HCl. The crude product obtained was filtered, washed with water, purified by silica gel column chromatography using pure chloroform/DCM as eluent and finally crystallized with DCM:THF (10:3).
7′-methoxy-3-morpholino-2′-oxo-3′,4′-dihydro-2′H,4H-spiro[cyclopenta[c]thiochromene-1,1′-naphthalene]-2-carbonitrile (17a).
Light yellow-greenish tiny prism shaped crystal crystallized from THF:DCM (3:10); Rf 0.83 (DCM); yield 45%; mp 270 °C; IR (KBr): 2182 (CN), 1708 (CO) cm−1; 1HNMR (300 MHz, DMSO-d6): δ 2.82 (m, 2H, CH2), 2.99 (m, 2H, CH2), 3.33 (m, 4H, CH2), 3.61 (s, 3H, OCH3), 3.72 (brm, 4H, OCH2), 3.9 (d, J = 15.3 Hz, 1H, SCH2), 4.0 (d, J = 15.3 Hz, 1H, SCH2), 6.24 (d, J = 2.1 Hz, 1H, Ar–H), 6.43 (d, J = 8.1 Hz, 1H, Ar–H), 6.89 (m, 2H, Ar–H), 7.11 (t, J = 7.5 Hz, 1H, Ar–H), 7.30 (d, J = 6.6 Hz, 2H, Ar–H); 13CNMR (100 MHz, CDCl3): δ 22.9, 27.1, 39.0, 39.2, 39.4, 49.6(2C), 54.5, 65.6(2C), 68.8, 94.5, 111.0, 112.8, 117.6, 124.6, 125.1, 126.5, 128.2, 129.3, 132.1, 132.9, 134.1, 150.7, 158.0, 162.3, 204.2; MS: m/z = 457.3 (M++1); HRMS (ESI): calc. for C27H24N2O3S: 457.1541 (M++1); found: 457.1545.
7′-methoxy-2′-oxo-3-(piperidin-1-yl)-3′,4′-dihydro-2′H,4H-spiro[cyclopenta[c]thiochromene-1,1′-naphthalene]-2-carbonitrile (17b)
Greenish-yellow solid; Rf 0.22 (CHCl3); yield 39%; mp 290 °C; IR (KBr): 2175 (CN), 1712 (CO) cm−1; 1HNMR (300 MHz, CDCl3): δ 1.70 (brm, 6H, , CH2), 2.81–2.87 (m, 1H, CH2), 3.16–3.31 (m, 2H, CH2), 3.37 (brm, 5H, CH2), 3.68 (s, 3H, OCH3), 3.77 (d, J = 6.7 Hz, 2H, SCH2), 6.33 (d, J = 2.4, 1H, Ar–H), 6.50 (d, J = 7.7, 1H, Ar–H), 6.78 (dd, J = 2.5, 8.4, 1H, Ar–H), 6.86 (t, J = 7.2, 1H, Ar–H), 7.05 (t, J = 7.0, 1H, Ar–H), 7.18 (d, J = 8.5, 1H, Ar–H), 7.28 (m, 1H, Ar–H); 13CNMR (100 MHz, CDCl3): δ 23.8, 23.9, 25.8(2C), 28.1, 29.6, 39.7, 51.3(2C), 55.1, 69.4, 94.2, 112.0, 113.3, 119.9, 125.4, 125.8, 127.3, 127.4, 128.7, 129.8, 133.9, 134.1, 135.9, 151.3, 158.7, 164.0, 205.4; HRMS (ESI): calc. for C28H26N2O2S: 455.1749 (M++1); found: 455.1754.
6′-methoxy-2′-oxo-3-(piperidin-1-yl)-3′,4′-dihydro-2′H,4H-spiro[cyclopenta[c]thiochromene-1,1′-naphthalene]-2-carbonitrile (17c).
Greenish-yellow solid; Rf 0.44 (CHCl3); yield 40%; mp 256 °C; IR (KBr): 2180 (CN), 1714 (CO) cm−1; 1HNMR (300 MHz, CDCl3): δ 1.70 (brm, 6H, CH2), 2.81–2.87 (m, 1H, CH2), 3.19–3.28 (m, 2H, CH2), 3.37–3.47 (m, 5H, CH2), 3.71–3.83 (m, 5H, CH2), 6.49 (d, J = 7.9, 1H, Ar–H), 6.70 (m, 2H, Ar–H), 6.76 (s, 1H, Ar–H), 6.86 (t, J = 7.7, 1H, Ar–H), 7.05 (t, J = 7.5, 1H, Ar–H), 7.30 (s, 1H, Ar–H); HRMS (ESI): calc. for C28H26N2O2S: 455.1749 (M++1); found: 455.1758.
8-chloro-6′-methoxy-2′-oxo-3-(piperidin-1-yl)-3′,4′-dihydro-2′H,4H-spiro[cyclopenta[c]thiochromene-1,1′-naphthalene]-2-carbonitrile (17d).
Greenish-yellow solid; Rf 0.69 (CHCl3); yield 42%; mp 258 °C; IR (KBr): 2176 (CN), 1714 (CO) cm−1; 1HNMR (300 MHz, CDCl3): δ 1.69 (brm, 6H, CH2), 2.80–2.88 (m, 1H, CH2), 3.23–3.44 (m, 7H, CH2), 3.75 (m, 2H, CH2), 3.80 (s, 3H, OCH3), 6.44 (d, J = 3.2, 1H, Ar–H), 6.68 (d, J = 2.3, 2H, Ar–H), 6.77 (s, 1H, Ar–H), 7.00 (dd, J = 3.3, 12.5, 1H, Ar–H), 7.27 (s, 1H, Ar–H); HRMS (ESI): calc. for C28H25ClN2O2S: 489.1359 (M++1); found: 489.1368.
6′-methoxy-2′-oxo-3-(piperidin-1-yl)-3′,4,4′,5-tetrahydro-2′H-spiro[cyclopenta[a]naphthalene-1,1′-naphthalene]-2-carbonitrile (17e).
Greenish-yellow solid; Rf 0.48 (CHCl3); yield 37%; mp 246 °C; IR (KBr): 2175 (CN), 1714 (CO) cm−1; 1HNMR (400 MHz, CDCl3): δ 1.65 (brm, 6H, CH2), 2.65 (m, 2H, CH2), 2.79 (m, 1H, CH2), 2.86 (m, 1H, CH2), 2.98 (m, 1H, CH2), 3.19 (m, 2H, CH2), 3.40 (m, 5H, CH2), 3.77 (s, 3H, OCH3), 6.39 (d, J = 8.0, 1H, Ar–H), 6.65–6.74 (m, 3H, Ar–H), 6.90 (m, 1H, Ar–H), 7.05–7.11 (m, 2H, Ar–H); HRMS (ESI): calc. for C29H28N2O2: 437.2184 (M++1); found: 437.2188.
2′-oxo-3-(piperidin-1-yl)-3′,4′-dihydro-2′H,4H-spiro[cyclopenta[c]thiochromene-1,1′-naphthalene]-2-carbonitrile (17f).
Greenish-yellow solid; Rf 0.48 (DCM); yield 35%; mp 278 °C; IR (KBr): 2176 (CN), 1714 (CO) cm−1; 1HNMR (300 MHz, CDCl3): δ 1.69 (brm, 6H, CH2), 3.24 (m, 2H, CH2), 3.34 (m, 4H, CH2), 3.44 (m, 2H, CH2), 3.74 (m, 2H, CH2), 6.43 (d, J = 8.0, 1H, Ar–H), 6.78–6.82 (m, 2H, Ar–H), 7.02–7.12 (m, 2H, Ar–H), 7.24 (m, 3H, Ar–H); 13CNMR (100 MHz, CDCl3): δ 23.7, 23.9, 25.9(2C), 29.0, 39.5, 51.4(2C), 69.4, 94.5, 119.9, 125.4, 125.8, 127.0, 127.4, 127.7, 127.8, 128.8, 129.8, 131.8, 133.8, 135.0, 135.5, 151.4, 164.1, 166.2, 205.5; HRMS (ESI): calc. for C27H24N2OS: 425.1643 (M++1); found: 425.1646.
6′-methoxy-2′-oxo-3-(4-phenylpiperazin-1-yl)-3′,4′-dihydro-2′H,4H-spiro[cyclopenta[c]thiochromene-1,1′-naphthalene]-2-carbonitrile (17g).
Orange solid; Rf 0.40 (DCM); yield 31%; mp 216 °C; IR (KBr): 2180 (CN), 1713 (CO) cm−1; 1HNMR (300 MHz, CDCl3): δ 2.83–2.90 (m, 1H, CH2), 3.21–3.29 (m, 2H, CH2), 3.33 (t, J = 4.6, 4H, CH2), 3.43–3.50 (m, 1H, CH2), 3.60 (d, J = 3.8, 4H, CH2), 3.81 (s, 3H, OCH3), 3.84 (s, 2H, SCH2), 6.51 (d, J = 7.7, 1H, Ar–H), 6.71 (m, 2H, Ar–H), 6.79 (s, 1H, Ar–H), 6.88 (m, 1H, Ar–H), 6.96 (t, J = 7.6, 3H, Ar–H), 7.07 (t, J = 7.4, 1H, Ar–H), 7.29–7.34 (m, 3H, Ar–H); 13CNMR (100 MHz, CDCl3): 23.94, 29.07, 39.41, 49.36(2C), 50.08(2C), 55.21, 69.44, 93.08, 113.27, 114.50, 116.58(2C), 119.50, 120.63, 125.52, 125.89, 128.13, 128.21, 128.90, 129.19, 129.26(2C), 131.89, 131.96, 132.01, 133.60, 150.74, 151.77, 159.02, 161.70, 205.38; HRMS (ESI): calc. for C33H29N3O2S: 532.2014 (M++1); found: 532.2019.
Crystallography
Intensity data for the colorless crystals of 11d was collected at 298(2) K on Sapphire2-CCD, OXFORD diffractometer system and for 17a was collected at 132(2) K on a Bruker APEX-II CCD diffractometer system, both equipped with graphite monochromated Mo-Kα radiation λ = 0.71073 Å. The final unit cell determination, scaling of the data, and corrections for Lorentz and polarization effects were performed with CrysAlis RED26 and Bruker SAINT.27 Symmetry-related multi-scan absorption corrections have been applied. The structures were solved by direct methods (SHELXS-97)28 and refined by a full-matrix least-squares procedure based on F2.29 All non-hydrogen atoms were refined anisotropically; hydrogen atoms were located at calculated positions and refined using a riding model with isotropic thermal parameters fixed at 1.2 times the Ueq value of the appropriate carrier atom.
Crystal data for 11d (CCDC No. 841513)30.
C27H25BrN2O2, formula mass 489.40, Triclinic space group P, a = 7.883(5), b = 11.024(5), c = 14.736(5) Å, α = 110.363(5), β = 96.456(5) γ = 104.041(5)° V = 1137.1(10) Å3, Z = 2, dcalcd = 1.429 mg m−3, linear absorption coefficient 1.835 mm−1, F(000) = 504, crystal size 0.25 × 0.20 × 0.15 mm, reflections collected 10971, independent reflections 5316 [Rint = 0.0334], Final indices [I > 2σ(I)] R1 = 0.0595 wR2 = 0.1195, R indices (all data) R1 = 0.1148, wR2 = 0.1409, gof 0.970, Largest difference peak and hole 0.377 and −0.434 e Å−3.
Crystal data for 17a (CCDC No. 831581)30.
C27H24N2O3S, formula mass 456.54, Triclinic space group P, a = 7.7227(6), b = 11.4586(9), c = 13.0552(11) Å, α = 78.782(4), β = 81.895(4) γ = 80.857(4)° V = 1111.55(15) Å3, Z = 2, dcalcd = 1.364 mg m−3, linear absorption coefficient 0.179 mm−1, F(000) = 480, crystal size 0.54 × 0.08 × 0.05 mm, reflections collected 15809, independent reflections 4535 [Rint = 0.0949], Final indices [I > 2σ(I)] R1 = 0.0653 wR2 = 0.1539, R indices (all data) R1 = 0.0929, wR2 = 0.1666, gof 1.181, Largest difference peak and hole 0.448 and −0.455 e Å−3.
Computational details
Density functional theory (DFT) calculations have been performed using the Gaussian 03 program.31 The optimized ground and transition state geometries were calculated using the B3LYP exchange–correlation functional32 and using GDIIS algorithm.33 The triple zeta 6-311+G* basis set for all atoms and tight SCF convergence criteria were used for the geometry optimization. For ground state optimization, wave function stability calculations were performed to confirm that the calculated wave functions corresponded to the ground state. The presence of one negative frequency was observed in the case of the transitions state geometries. The optimized ground state structures of the compounds were used for molecular orbital analyses and time-dependent DFT (TD-DFT) calculations at the B3LYP/6-311G** level of theory with the polarized continuum model (PCM).34 The solvent parameters were those of the chloroform. The energies and intensities of the 30 lowest energy spin-allowed electronic excitations were calculated using TD-DFT. The intermolecular interaction energies have been estimated at the MP2 level of theory. For the interaction energy calculations, the Br⋯H distance has been fixed for the dimer while all other degrees of freedom were relaxed in the geometry optimization. The magnitude of energy corresponding to this dimer was substracted from twice the energy of monomer. The intermolecular interaction strengths are significantly weaker than either ionic or covalent bonding, therefore it was essential to do basis set superposition error (BSSE) corrections. The BSSE corrections in the interaction energies were done using Boys-Bernardi Scheme.35 In this paper all interaction energies have been reported after BSSE correction.
Acknowledgements
HKM is thankful to DST, New Delhi, India for DST Fast Track Young Scientist Project.VJR is also thankful to UGC, New Delhi, India for Emeritus fellowship.
References
-
(a) T. R. Kelly, N. Ohashi, R. J. Armstrong-Chong and S. H. Bell, J. Am. Chem. Soc., 1986, 108, 7100 CrossRef CAS;
(b) G. Mehta and D. Subrahmanyam, Tetrahedron Lett., 1987, 28, 479 CrossRef CAS;
(c) A. V. Rama Rao, A. K. Singh, B. Venkateswara Rao and K. M. Reddy, Tetrahedron Lett., 1993, 34, 2665 CrossRef;
(d) Y. Kita, K. Higuchi, Y. Yoshida, K. Iio, S. Kitagaki, S. Akai and H. Fujioka, Angew. Chem., Int. Ed., 1999, 38, 683 CrossRef CAS;
(e) Y. Kita, K. Higuchi, Y. Yoshida, K. Iio, S. Kitagaki, K. Ueda, S. Akai and H. Fujioka, J. Am. Chem. Soc., 2001, 123, 3214 CrossRef CAS.
-
(a) J. A. Marshall, Prog. Chem. Nat. Prod., 1974, 31, 283 CAS;
(b)
C. K. Heathcock, S. L. Graham, F. P. Palvac, C. T. White, Total Synthesis of Natural Products, Vol. 11.; ApSimon, J. Ed. John Wiley: New York, 1983, 264–314 Search PubMed;
(c)
Atta-ur-Rahman, Studies in Natural Products Chemistry; Elsevier: Oxford, 1989 Search PubMed;
(d) H. Spreitzer, I. Piringer, W. Holzer and M. Widhalm, Helv. Chim. Acta, 1998, 81, 2292 CrossRef CAS;
(e) J. D. White, J. F. Ruppert, M. A. Avery, S. Torii and J. Nokami, J. Am. Chem. Soc., 1981, 103, 1813 CrossRef CAS;
(f) L. C. Pati, A. Roy and D. Mukherjee, Tetrahedron, 2002, 58, 1773 CrossRef.
-
(a) A. Longeon, M. Guyot and J. Vacelet, Experientia, 1990, 46, 548 CrossRef CAS;
(b) J. Kobayashi, M. Tsuda, K. Agemi, H. Shigemori, M. Ishibashi, T. Sasaki and Y. Mikami, Tetrahedron, 1991, 47, 6617 CrossRef CAS;
(c) D. M. James, H. B. Kunze and D. J. Faulkner, J. Nat. Prod., 1991, 54, 1137 CrossRef CAS.
-
(a) J. A. Wendt, P. J. Gauvreau and R. D. Bach, J. Am. Chem. Soc., 1994, 116, 9921 CrossRef CAS;
(b) G. Mehta and A. Srikrishna, Chem. Rev., 1997, 97, 671 CrossRef CAS.
- V. Fajardo, M. Araya, P. Cuadra, A. Oyarzun, A. Gallardo, M. Cueto, A. R. Diaz-Marrero, J. Darias, L. Villarroel, C. Lvarez, Y. Mora-Pérez and P. Joseph-Nathan, J. Nat. Prod., 2009, 73, 1355 CrossRef.
- C. Fourneau, R. Hocquemiller, D. Guédon and A. Cavé, Tetrahedron Lett., 1997, 38, 2965 CrossRef CAS.
- R. Pradhan, M. Patra, A. K. Behera, B. K. Mishra and R. K. Behera, Tetrahedron, 2006, 62, 779 CrossRef CAS.
- N. Watanabe, A. Ikeno, H. Minato, H. Nakagawa, C. Kohayakawa and J.-I. Tsuji, J. Med. Chem., 2003, 46, 3961 CrossRef CAS.
- E. Fisher and Y. Hirshberg, J. Chem. Soc., 1952, 4522 Search PubMed.
- N. Feuerbacher, F. Vögtle, J. Windscheidt, E. Poetsch and M. Nieger, Synthesis, 1999, 117 CrossRef CAS.
-
(a) G. H. Posner and T. G. Hamil, J. Org. Chem., 1988, 53, 6031 CrossRef CAS;
(b) S. N. Naik, B. Pandey and N. R. Ayyangar, Synth. Commun., 1988, 18, 633 CrossRef CAS;
(c) Y. Sakata, S. Nakashima, Y. Goto, H. Tatemitsu and S. Misumi, J. Am. Chem. Soc., 1989, 111, 8979 CrossRef CAS;
(d) S. Kotha, A. C. Deb and S. Chattopadhya, Lett. Org. Chem., 2006, 3, 39 CrossRef;
(e) H. Huang and C. J. Forsyth, J. Org. Chem., 1995, 60, 2773 CrossRef CAS;
(f) V. Bjornstad and K. Undheim, Synthesis, 2008, 962 Search PubMed;
(g) D. R. Goldberg, J. A. Hansen and R. J. Giguere, Tetrahedron Lett., 1993, 34, 8003 CrossRef CAS.
-
(a) B. M. Trost, D. C. Lee and F. Rise, Tetrahedron Lett., 1989, 30, 651 CrossRef CAS;
(b) B. M. Trost and Y. Shi, J. Am. Chem. Soc., 1991, 113, 701 CrossRef CAS;
(c) A. J. Pearson and X. Wang, Tetrahedron Lett., 2002, 43, 7513 CrossRef CAS;
(d) J. J. Gonzalez, N. Garcia, B. Gómez-Lor and A. M. Echavarren, J. Org. Chem., 1997, 62, 1286 CrossRef CAS;
(e) R. Shintani, S. Park and T. Hayshi, J. Am. Chem. Soc., 2007, 129, 14866 CrossRef CAS;
(f) T. J. Katz, A. M. Gilbert, M. E. Huttenloch, G. Min-Min and H. H. Brintzinger, Tetrahedron Lett., 1993, 34, 3551 CrossRef CAS.
-
(a) C. Kuroda and Y. Hirono, Tetrahedron Lett., 1993, 35, 6895 CrossRef;
(b) C. Kuroda and H. Koshio, Chem. Lett., 2000, 962 CrossRef CAS;
(c) D. B. England, G. Merey and A. Padwa, Org. Lett., 2007, 9, 3805 CrossRef CAS;
(d) J. Huang and A. J. Frontier, J. Am. Chem. Soc., 2007, 129, 8060 CrossRef CAS;
(e) T. Hundlicky, L. Radesca-Kwart, T.-Q. Li and T. Bryant, Tetrahedron Lett., 1988, 29, 3283 CrossRef.
-
(a) R. D. Bach and R. C. Klix, Tetrahedron Lett., 1986, 27, 1983 CrossRef CAS;
(b) R. D. Bach and R. C. Klix, J. Org. Chem., 1985, 50, 5434 CrossRef;
(c) J. R. Hwu and J. M. Wetzel, J. Org. Chem., 1992, 57, 922 CrossRef CAS.
-
(a) P. A. Wender and R. J. Ternansky, Tetrahedron Lett., 1985, 26, 2625 CrossRef CAS;
(b) E. R. Koft and A. B. III Smith, J. Org. Chem., 1984, 49, 832 CrossRef CAS;
(c) D. Caine, A. A. Boucugnani, S. T. Chao, J. B. Dawson and P. F. Ingwalson, J. Org. Chem., 1976, 41, 1539 CrossRef CAS.
-
(a) J. N. Marx and L. R. Norman, J. Org. Chem., 1975, 40, 1602 CrossRef CAS;
(b) T. G. Back and J. E. Payne, Org. Lett., 1999, 1, 663 CrossRef CAS;
(c) D. R. Goldberg, J. A. Hansen and R. J. Giguere, Tetrahedron Lett., 1993, 34, 8003 CrossRef CAS;
(d) H.-J. Knölker, P. G. Jones and R. Graf, Synlett, 1996, 1155 CrossRef;
(e) H.-J. Knölker, E. Baum, R. Graf, P. G. Jones and O. Spiess, Angew. Chem., Int. Ed., 1999, 38, 2583 CrossRef;
(f) S. Kotha and E. Manivannan, J. Chem. Soc., Perkin Trans. 1, 2001, 2543 RSC.
-
(a) D. P. Curran and S. C. Kuo, J. Am. Chem. Soc., 1986, 108, 1106 CrossRef CAS;
(b) P. Shanmugam, R. Srinivasan and K. Rajagopalan, Tetrahedron, 1997, 53, 1185 Search PubMed;
(c) M. Inui, A. Nakazaki and S. Kobayashi, Org. Lett., 2007, 9, 467 CrossRef;
(d) T. Taniguchi, G. Tanabe, O. Muraoka and H. Ishibashi, Org. Lett., 2008, 10, 197 CrossRef CAS.
-
(a) K. Hammer and K. Undheim, Tetrahedron, 1997, 53, 2309 CrossRef CAS;
(b) M. J. Bassindale, P. Hamley, A. Leitner and J. P. A. Harrity, Tetrahedron Lett., 1999, 40, 3247 CrossRef CAS;
(c) S. Kotha, E. Manivannan, T. Ganesh, N. Sreenivasachary and A. Deb, Synlett, 1999, 1618 CrossRef CAS;
(d) D. L. Wright, J. P. II Schulte and M. A. Page, Org. Lett., 2000, 2, 1847 CrossRef CAS;
(e) S. Kotha, K. Mandal, A. Tiwari and S. M. Mobin, Chem.–Eur. J., 2006, 12, 8024 CrossRef CAS.
-
(a) Y. Tominaga, A. Ushirogouchi and Y. Matsuda, J. Heterocycl. Chem., 1987, 24, 1557 CrossRef CAS;
(b) V. J. Ram, M. Verma, F. A. Hussaini and A. Shoeb, J. Chem. Res. (S), 1991, 98 CAS;
(c) V. J. Ram, M. Verma, F. A. Hussaini and A. Shoeb, Liebigs Ann. Chem., 1991, 1229 CrossRef CAS;
(d) V. J. Ram, M. Nath, P. Srivastava, S. Sarkhel and P. R. Maulik, J. Chem. Soc., Perkin Trans. 1, 2000, 3719 RSC.
- R. Pratap and V. J. Ram, Tetrahedron Lett., 2007, 48, 1715 CrossRef CAS.
- R. Gomper and W. Töpfl, Chem. Ber., 1962, 95, 2861 CrossRef.
-
(a) A. Goel, D. Verma, R. Pratap, G. Taneja, Y. Hemberger, M. Knauer, R. Raghunandan, P. R. Maulik, V. J. Ram and G. Bringmann, Eur. J. Org. Chem., 2011, 2940–2947 CrossRef CAS;
(b) R. Pratap, A. Kumar, R. Pick, V. Huch and V. J. Ram, RSC Adv., 2012, 2, 1557–1564 RSC.
-
(a) H. K. Maurya, V. K. Tandon, B. Kumar, A. Kumar, V. Huch and V. J. Ram, Org. Biomol. Chem., 2012, 10, 605–613 RSC;
(b) H. K. Maurya, S. K. Gautam, R. Pratap, V. K. Tandon, A. Kumar, V. Bajpai, B. Kumar and V. J. Ram, Org. Biomol. Chem., 2012, 10, 4977 RSC.
- For X-ray structures 11d and 17a also see supporting data..
-
P. Metrangolo and H. D. Arman, Halogen Bonding: Fundamentals and Applications; Springer-VerlagBerlin Heidelberg, 2008 Search PubMed.
- CrysAlis CCD, RED version 1.711.13, copyright 1995-2003, Oxford Diffraction Poland Sp..
-
SAINT+ 6.02 ed.; Bruker AXS; Madison, WI, 1999 Search PubMed.
-
G. M. Sheldrick, SHELXS97, Program for Crystal Structure Solution, University of Göttingen, Göttingen, 1997 Search PubMed.
-
G. M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, 1997 Search PubMed.
- The supplementary crystallographic data of 11d [CCDC No. 841513] and 17a [CCDC No. 831581] can be obtained free of charge from www.ccdc.cam.uk/conts/retrieving.html or from the Cambridge
Crystallographic Data Center, 12 Union Road, Cambridge, CB2 1EZ, U. K; Fax: (internet) + 44-1223/336-033; E-mail: deposit@ccdc.cam.ac.uk.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision D.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS;
(b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 1133 Search PubMed.
-
(a) D. Farkas and H. B. Schlegal, J. Chem. Phys., 1998, 109, 7100 CrossRef;
(b) D. Farkas and H. B. Schlegal, J. Chem. Phys., 1999, 111, 10806 CrossRef.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Perspective view of 11d and 17a showing absolute configuration, crystallographic information files (CIF) of spiranes 11d and 17a, Optimized cartesian coordinates for dimer, monomer for 11d and transition state coordinates for 17a and copies of HRMS and NMRs Spectra of all new compounds. CCDC reference numbers 831581 and 841513. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21587j |
| ‡ Responsbile for Quantum Chemical Study. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.