Layer-by-layer assembled sulfonated-graphene/polyaniline nanocomposite films: enhanced electrical and ionic conductivities, and electrochromic properties†
Jinlin
Lu‡
,
Wanshuang
Liu‡
,
Han
Ling
,
Junhua
Kong
,
Guoqiang
Ding
,
Dan
Zhou
and
Xuehong
Lu
*
School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: asxhlu@ntu.edu.sg
First published on 11th September 2012
Abstract
In this article, we report the facile synthesis of sulfonic acid-grafted reduced graphene oxide (S-rGO) using a one-pot method under mild conditions, and layer-by-layer (LbL) assembly and electrochromic properties of S-rGO/polyaniline (S-rGO/PANI) nanocomposite thin films. It was found that the multilayer films of S-rGO/PANI exhibit much faster electrochromic switching kinetics than that of corresponding spin-coated PANI thin films. The enhancement can be attributed to the drastically increased electrical and ionic conductivities of the S-rGO/PANI films brought by the graphitic structure of the S-rGO sheets and the sulfonic acid groups attached to S-rGO, which lead to non-diffusion-controlled redox processes of PANI.
1. Introduction
Graphene is a single-atom-thick sheet of hexagonally arrayed sp2-bonded carbon atoms, which promises a great diversity of applications owing to its unique electronic, mechanical, thermal and chemical properties.1,2 In particular, its conjugated structure, excellent electron-transport property and versatile chemistry make it an ideal candidate for the fabrication of electronic, optoelectronic and electrochemical devices in conjunction with conjugated polymers. The interactions between graphene and conjugated polymers often lead to synergistic effects in improving performances of the devices such as field effect transistors, supercapacitors and sensors.3–5 A common way to combine graphene with conjugated polymers is through self assembly of graphene oxide (GO) with functionalized conjugated polymers followed by chemical reduction. For example, Zhang et al.6 synthesized a functionalized conjugated polyelectrolyte that could interact with reduced GO (rGO) via strong π–π interactions to improve the processability of the rGO. Kim et al.7 prepared an rGO/poly(3,4-ethylene dioxythiophene):poly(styrenesulfonate) (PSS) hybrid film where PSS acts as the dispersant in the reduction process. An alternative approach would be the self assembly of functionalized rGO with conjugated polymers, which is preferred for conjugated polymers that cannot survive harsh chemical reduction conditions or need to be re-oxidized to restore their functionalities.Electrochromism is a reversible change in optical properties due to electrochemically induced redox reactions.8,9 Many applications have been envisaged for systems based on electrochromic devices, e.g., light and overheating protection windows, mirrors, optical filters and display panels. Conjugated polymers have been widely used as electrochromic materials owing to their low cost, relatively high coloration efficiency and response speed.10 Conjugated polymer-based electrochromic thin films are commonly fabricated via spin-coating or electropolymerization. An attractive, alternative approach is to fabricate electrochromic thin films using a layer by layer (LbL) assembly technique, which offers several advantages over other thin film deposition methods, e.g. accurately controlled thickness and the formation of uniform, defect-free thin films that have better mechanical properties than conventional thin films of similar thickness, while retaining their flexibility.11,12 Furthermore, it allows a convenient incorporation of nanomaterials into polymers in a wide range of compositions without the issues of phase separation, providing huge interfacial areas between the polymers and nanomaterials.
Polyaniline (PANI) is one of the most widely used anodically coloring electrochromic polymers owing to its good environmental stability, low cost and multicolor capability.13 PANI/rGO nanocomposites have been prepared by reduction of PANI/GO prior to re-oxidation of PANI.14–16 Herein we report an innovative strategy to prepare electrochromic PANI/rGO thin films, i.e., one-pot synthesis of sulfonic acid (SO3H)-grafted reduced graphene oxide (S-rGO) and fabrication of S-rGO/PANI multilayer films via LbL assembly. The purpose of introducing SO3H groups is to facilitate the growth of the multilayer films via electrostatic interactions between S-rGO and PANI-EB, while the SO3H groups may also act as dopants for PANI during redox reactions. Furthermore, the SO3H groups may enhance ionic conduction while the graphene sheets may improve electron transport in the thin films. In this paper, the impressive enhancements in electrochemical and electrochromic properties provided by S-rGO/PANI are demonstrated, and the underlying mechanism for the enhancement is illustrated by the evidence from cyclic voltammetry and impedance analysis.
2. Experimental
2.1 Materials
Natural graphite flake (43319, -10 mesh, 99.9%) and potassium permanganate were supplied by Alfa Aesar (USA). Polyaniline emeraldine base (PANI-EB, Mw ∼20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), N,N-dimethylformamide (DMF), (3-aminopropyl)triethoxysilane (APTES, 99%), sulfuric acid (>97.0%), sodium nitrate (>99.0%), hydrochloric acid (37%), hydrogen peroxide (30%), sodium borohydride (NaBH4 >98%) and 2-aminoethanesulfonic acid (taurine, ≥99.5%) were purchased from Sigma-Aldrich Chemicals Inc (USA) and used without further purification.
000), N,N-dimethylformamide (DMF), (3-aminopropyl)triethoxysilane (APTES, 99%), sulfuric acid (>97.0%), sodium nitrate (>99.0%), hydrochloric acid (37%), hydrogen peroxide (30%), sodium borohydride (NaBH4 >98%) and 2-aminoethanesulfonic acid (taurine, ≥99.5%) were purchased from Sigma-Aldrich Chemicals Inc (USA) and used without further purification.
2.2 Synthesis of S-rGO
GO was prepared from the natural graphite by a modified Hummers method.17 The details are provided in the ESI.† In a typical reaction to synthesize S-rGO, 600 mg GO was dispersed into 200 mL deionized (DI) water under ultrasonication. The suspension was centrifuged at 4000 rpm for 30 min to remove the small amount of unexfoliated particles. The obtained supernatant was transferred into a 500 ml flask purged by N2, followed by adding 12 g taurine. The mixture was then heated at 60 °C for 36 h under stirring. After cooling to room temperature, a 200 mL with 0.25 M NaBH4 aqueous solution was added to the mixture under stirring for 4 h. The mixture was then vacuum-filtered through 0.2 μm polycarbonate membrane and washed with a large amount of DI water to remove the residual ions and excessive raw materials. The obtained product, i.e. S-rGO was freeze-dried for 48 h and stored in a dry cabinet for further use.2.3 Preparation of dipping solutions and LbL assembly process
PANI-EB was dissolved in DMF at a concentration of 10 mg mL−1 by first stirring the solution overnight and then sonicating it for 10 h. The solution was passed through a 0.2 μm pore size filter to remove any undissolved PANI residues. The PANI dipping solution was prepared by slowly adding one part (by volume) of the filtered PANI solution to nine parts of Milli-Q water that has had its pH adjusted to 3.0 with HCl. PANI will change from its EB form to emeraldine salt (ES) form under this pH condition.18 The as-prepared PANI solution was used as the polycation dipping solution. The PANI dipping solution was typically used within two days after preparation. The 0.01 mg mL−1 S-rGO water solution with pH = 3.0 adjusted by 1 M HCl was used as the polyanion dipping solution.Indium tin oxide (ITO) (5–15 Ω □−1) coated glass substrates with the dimensions of 7 × 50 × 0.7 mm (Delta Technologies) were cleaned by ultrasonication in a series of solvents including acetone, ethanol and Milli-Q water for 30 min at each step. To provide a hydrophilic and positively charged surface, the cleaned substrates were then immersed in a 1 vol.% APTES aqueous solution for 30 min, followed by heating for 2 h at 120 °C in a vacuum oven. The LbL process was carried out by an automated dip coating equipment. The ITO-glass substrates were exposed first to polyanion solution for 15 min at room temperature, followed by three consecutive rinsing steps (2 min for each step) in Milli-Q water, then exposed to polycation solution for 15 min and another three rinsing steps in Milli-Q water. The cycle was repeated to create multilayer thin films of certain thickness.
2.4 Characterization
Atomic force microscope (AFM) measurements were performed in the tapping mode using Digital Instruments Nanoscope DI 3100. Transmission electron microscopy (TEM) images were obtained using a JEOL Model JEM-2010 TEM system. Raman spectra of the materials were obtained using a Renishaw® RM1000 Raman spectroscope. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Kratos Analytical AXIS His spectrometer with a monochromatized Al Kα X-ray source (1486.6 eV photons). Elemental analysis was performed using Perkin Elmer Instruments CHNS-O Analyzer. Thermal gravimetric analysis (TGA) was performed on a TA Instruments TGA Q 500 under a nitrogen atmosphere over a temperature range of 25–800 °C at a heating rate of 10 °C min−1. Zeta potential was measured on a Zetasizer Nano Series 90 at 25 °C, using a DTS 1060C disposable capillary cell (Malvern Instruments, UK). The sheet resistances of the GO, rGO and S-rGO films were measured by four-point probe method (CMT-SR2000N). Thickness measurements were performed with an Alfa-step IQ surface profiler. The Fourier transform infrared spectroscopy-attenuated total reflectance (FTIR-ATR) measurements were carried out with a Perkin Elmer Model GX spectrometer. A diamond crystal was used as the ATR plate. All the electrochemical measurements were carried out in a 0.5 M H2SO4 aqueous solution without any salt. The cyclic voltammetry (CV) experiments were performed using a platinum wire (99.99%) as the counter electrode and an Ag/AgCl electrode as the reference electrode. In situ spectro-electrochemical properties of the thin films were recorded with a UV-vis spectrophotometer (Shimadzu UV-3600). The electrochemical impedance spectroscopy (EIS) was conducted in the frequency range of 1 MHz to 1 Hz with the signal amplitude of 10 mV under the open circuit potential using an Autolab PGSTAT30 potentiostat.3. Results and discussion
3.1 Preparation and structural verification of S-rGO and LbL-assembled S-rGO/PANI
It is well known that GO contains a large amount of oxygen-containing groups such as hydroxyl, epoxy, carboxyl and carbonyl groups, which open up a versatile avenue for a variety of chemical transformations.19 Among these groups, epoxy groups on the basal planes of GO can easily react with amines through a ring-opening reaction. In this work, taurine was used to functionalize the GO. The synthesis route for S-rGO is shown in Scheme 1a. Using this one-pot method, the whole process is carried out in aqueous solutions without adding any other organic solvents or surfactants that may generate undesirable byproducts or residues attaching on the surfaces of GO.20,21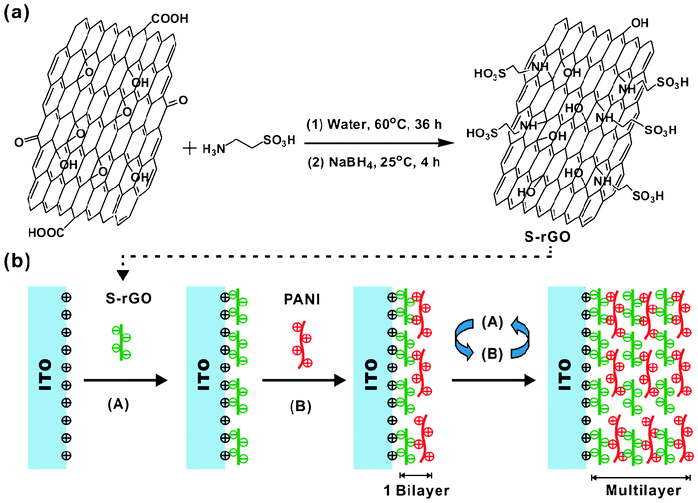 | ||
| Scheme 1 (a) Synthesis of S-rGO and (b) LBL self-assembly process. | ||
The morphologies of the GO and S-rGO were examined by AFM and TEM. The AFM images confirm that the dried dispersions of GO and S-rGO are comprised of isolated sheets (Fig. 1a and 1c). The GO sheets have lateral dimensions of several micrometers and a thickness of 0.8 nm, which is characteristic of a fully exfoliated GO sheet.22 The thickness of a single-layer S-rGO sheet is about 1.2 nm. This implies that the taurine groups have been grafted onto GO, giving rise to the thickness increase. The single-layer sheets can also be observed clearly for both GO and S-rGO by TEM (Fig. 1b and 1d). The corresponding selected-area electron diffraction (SAED) pattern of S-rGO coincides well with the typical SAED pattern of a single layer of rGO (inset in Fig. 1d).23
 | ||
| Fig. 1 (a) AFM and (b) TEM images of GO; (c) AFM and (d) TEM images of S-rGO. The inset picture in the top-right part of (d) is the SAED image of S-rGO. | ||
Fig. 2 shows the XPS spectra of GO, rGO and S-rGO. There are four types of carbons: C–O–C (286.7 eV), C–OH (286.0 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.7 eV) and OC–O (289.5 eV) in the spectra of the GO (Fig. 2a), which are in good agreement with previous reports.24,25 After the reduction, the peak intensities from oxygen-related groups decrease significantly (Fig. 2b and 2c). In addition, the peaks related to C–N at 286.5 eV (Fig. 2c), S2p at 168.5 eV (Fig. 2d) and N1s at 399.8 eV (Fig. 2e) can be observed clearly in the spectra of S-rGO. The results indicate that taurine molecules have been successfully grafted on the GO. In TGA tests, S-rGO shows an obvious weight loss between 200 °C and 400 °C, compared to rGO (Fig. S1, ESI†). This is caused by the decomposition of the grafted taurine molecules. Elemental analysis data (Table S1, ESI†) indicate that the C, O and H contents in the GO and rGO are within the range of what has been reported in the literature.26,27 The N and S elements found for S-rGO evidently come from the grafted taurine molecules and account for 2.3 wt.% and 5.2 wt.% of the total mass of S-rGO, respectively. Based on this, the weight percent of the grafted taurine molecules is about 21.0 wt.%, which is in good agreement with the TGA results.
O (287.7 eV) and OC–O (289.5 eV) in the spectra of the GO (Fig. 2a), which are in good agreement with previous reports.24,25 After the reduction, the peak intensities from oxygen-related groups decrease significantly (Fig. 2b and 2c). In addition, the peaks related to C–N at 286.5 eV (Fig. 2c), S2p at 168.5 eV (Fig. 2d) and N1s at 399.8 eV (Fig. 2e) can be observed clearly in the spectra of S-rGO. The results indicate that taurine molecules have been successfully grafted on the GO. In TGA tests, S-rGO shows an obvious weight loss between 200 °C and 400 °C, compared to rGO (Fig. S1, ESI†). This is caused by the decomposition of the grafted taurine molecules. Elemental analysis data (Table S1, ESI†) indicate that the C, O and H contents in the GO and rGO are within the range of what has been reported in the literature.26,27 The N and S elements found for S-rGO evidently come from the grafted taurine molecules and account for 2.3 wt.% and 5.2 wt.% of the total mass of S-rGO, respectively. Based on this, the weight percent of the grafted taurine molecules is about 21.0 wt.%, which is in good agreement with the TGA results.
 | ||
| Fig. 2 C1s XPS spectra of (a) GO, (b) rGO and (c) S-rGO; (d) S2p XPS spectra, (e) N1s XPS spectra and (f) XPS survey scan of S-rGO. | ||
Raman spectroscopy for the graphitic modes of carbon provided additional evidence for the structural changes caused by the reactions (Fig. S2, ESI†). GO, rGO and S-rGO samples all exhibit a D band at around 1332 cm−1 and a G band at around 1586 cm−1. However, S-rGO shows a slightly higher D/G intensity ratio than that of rGO, implying that more small domains of sp2 structure were created, which is consistent with previous reports on functionalized graphene.26,27 To further probe the conjugated structures of the samples, the electrical conductivities (σe) of the GO, rGO and S-rGO films were measured by a four-point probe method and the data are listed in Table 1. The pristine GO shows a conductivity of around 2.4 × 10−4 S cm−1. The electrical conductivity of S-rGO is four-orders of magnitude greater than that of GO, although it is slightly lower than that of rGO. The results further verify that the π-conjugated system has been effectively restored in S-rGO after the reduction.
| Sample name | GO | rGO | S-rGO | PANI |
|---|---|---|---|---|
| σ e (S cm−1) | 2.4 × 10−4 | 5.2 | 4.3 | 2.0 × 10−2 |
| Zeta potential (mV) | −27.6 | −10.4 | −62.4 | 21.8 |
The zeta potentials of the GO, rGO and S-rGO suspensions are listed in Table 1. The S-rGO aqueous suspension at a concentration around 0.01 mg mL−1 under neutral conditions has a zeta potential of −62.4 mV, which is much lower than −40 mV, indicating that the suspension has a very good stability.22,28 Moreover, there is no sign of coagulation of S-rGO sheets after more than 3 months (Fig. S3, ESI†), making it an ideal candidate for LbL assembly.
The S-rGO and PANI dipping solutions were used to fabricate nanocomposite thin films using the LbL self-assembly technique as illustrated in Scheme 1b. Fig. 3a shows representative optical images of LbL assembled (S-rGO/PANI)n films on ITO-coated glass slides with different numbers of bilayers. The films appear darker with increasing number of bilayers, and the thickness of the films is a linear function of the number of bilayers (Fig. 3b).
 | ||
| Fig. 3 (a) A photograph of the LbL assembled (S-rGO/PANI)n films on the ITO-coated glass slides. (b) Thickness of the thin films as a function of the number of bilayers. | ||
Fig. 4 shows the FTIR spectra of S-rGO, PANI-EB and a 25-bilayer S-rGO/PANI film ((S-rGO/PANI)25). The FTIR spectra of GO and rGO can be found in Fig. S4, ESI.† For S-rGO, the absorbance bands at 1044 and 1196 cm−1 can be attributed to the symmetric and asymmetric stretching band of –SO3 groups,29,30 which evidently demonstrate the successful grafting of taurine molecules on the GO and are in good agreement with the AFM and XPS results. For PANI-EB, the characteristic bands at 1536 and 1622 cm−1 are attributed to the CC stretching deformation modes of the benzenoid and quinoid rings in PANI molecules.31 In comparison with that of the spin-coated PANI-EB film, new bands appear at 1044 and 1196 cm−1 for the (S-rGO/PANI)25 film, which can be ascribed to the effective incorporation of S-rGO by LbL assembly. The clear shifts of relatively sharp r(CH2) band from 738 cm−1 to 719 cm−1 and v(C2H4) band from 1340 cm−1 to 1322 cm−1 signify the interactions between S-rGO and PANI.32
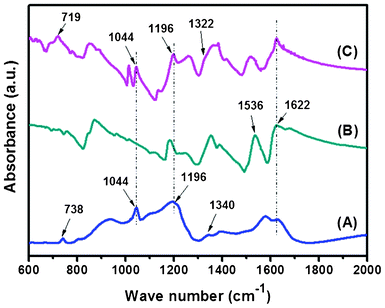 | ||
| Fig. 4 FTIR curves of the different samples (A) S-rGO, (B) PANI, (C) (S-rGO/PANI)25. | ||
3.2 Electrochemical and electrochromic properties of S-rGO/PANI
To identify unique electrochemical and electrochromic properties of the S-rGO/PANI nanocomposite films, a spin-coated PANI film with a thickness of ∼90 nm was fabricated as a reference because the thickness of the (S-rGO/PANI)25 film is ∼120 nm, while the thickness of 25 layers of S-rGO is ∼30 nm. The CV curves of the spin-coated PANI and (S-rGO/PANI)25 films at different scan rates are shown in Fig. 5a and 5c. There are two oxidation and reduction peaks for both films. The first oxidation peak at lower potentials can be assigned to the leucoemeraldine base (LEB) to ES transition, and the second oxidation peak at higher potentials is due to the transition from the ES to pernigraniline state.33 It can be seen clearly that the oxidation peak potentials of the spin-coated PANI film shift positively and the corresponding reduction peak potentials shift negatively with increasing scan rate, especially for the peaks between the LEB and ES transition. This is mainly caused by the rather high resistance of the spin-coated PANI film. However, for the (S-rGO/PANI)25 film, the peak potentials for the LEB to ES transition obtained at different scan rates are all around 0.22 V for oxidation and 0.12 V for reduction. The anodic and cathodic peak current densities at the lower potentials are extracted and plotted against the scan rate. As can be seen from Fig. 5d, it is a good linear relationship for both the oxidation and reduction processes of the (S-rGO/PANI)25 film, whereas the spin-coated PANI film displays a pair of semiparabolic curves (Fig. 5b). These results indicate that the redox reactions in the (S-rGO/PANI)25 film are close to a non-diffusion-controlled process in the studied scan rate range, whereas the redox reactions in the spin-coated PANI film are most likely controlled by the ionic diffusion process,34,35 which will be discussed with further evidence later. | ||
| Fig. 5 CV curves of (a) spin-coated PANI film and (c) (S-rGO/PANI)25 film in 0.5 mol L−1 H2SO4 aqueous solutions at different scan rates: 10, 20, 30, 40, 50, 60, 100 mV s−1. Plots of peak current densities of (b) spin-coated PANI film and (d) (S-rGO/PANI)25 film vs. scan rate. | ||
Spectroelectrochemistry plays a key role in examining the optical changes that occur upon doping or dedoping processes of an electrochromic film. The UV-vis absorbance spectra of the spin-coated PANI and (S-rGO/PANI)25 films under −0.6, 0 and +0.8 V are shown in Fig. 6a and 6b, and the full spectra under various applied potentials in the range of −0.6 V to +0.8 V are shown in Fig. S5, ESI.† The absorbance increases gradually for the spin-coated PANI and (S-rGO/PANI)25 films with increasing potential. Both the spin-coated PANI and (S-rGO/PANI)25 films can be switched between a reduced state (relatively-transparent with a yellow color) and oxidized state (opaque with a blue-green color). The maximum change in absorbance (ΔA) occurs at ∼620 nm between +0.8 V and −0.6 V. Although the (S-rGO/PANI)25 film shows a slightly higher absorbance than the spin-coated PANI film at the reduced state due to the incorporation of S-rGO, the ΔA of the (S-rGO/PANI)25 film shows a more than 30% higher absorbance than that of the spin-coated PANI film at 620 nm. These results indicate that the PANI units in the (S-rGO/PANI)25 film are more redox-active than those in the spin-coated PANI film. The optical switching behaviors of the films are investigated at a wavelength of 620 nm with the applied potential stepped between −0.6 V and +0.8 V. Each cycle consists of an oxidation step with 20 s and a reduction step with 20 s. As can be seen clearly in Fig. 6c, under the dynamic conditions the ΔA of the (S-rGO/PANI)25 film is significantly higher than that of the spin-coated PANI film. More importantly, the coloration time, that is the time for achieving 90% of their total absorbance change, is 6 s for the LbL assembled (S-rGO/PANI)25 film, but 15 s for the spin-coated PANI film. It is well known that the redox process of PANI involves both electron and proton exchange. The great enhancement in switching kinetics is due to the drastically improved electron and hydrogen ion transport brought by S-rGO, as discussed further below.
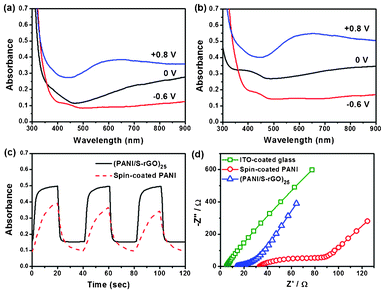 | ||
| Fig. 6 UV-vis absorbance spectra of (a) spin-coated PANI film and (b) (S-rGO/PANI)25 film under varying potentials at −0.6, 0 and 0.8 V; (c) the absorbance of spin-coated PANI and (S-rGO/PANI)25 films at 620 nm under switched potentials between −0.6 V and +0.8 V with 20 s each step; (d) EIS results of the different samples. | ||
The charge transport behaviors in the electrochromic thin films in the acid solution were investigated by electrochemical impedance spectroscopy (EIS). Fig. 6d shows the EIS results for the spin-coated PANI and (S-rGO/PANI)25 films. Based on a widely used model for conducting polymer films, the electrical and ionic conductivities (σe and σion) of the films can be calculated from the EIS results.36–39 The calculated conductivity data are tabulated in Table 2. The detailed descriptions for the conductivity calculation can be found in the ESI.† The spin-coated PANI film shows reasonable σion and σe, the latter is close to the value obtained from the four-point probe measurement. In contrast, the (S-rGO/PANI)25 film shows an increase in electrical conductivity by one order of magnitude and an increase in ionic conductivity by two orders, as compared with that of the spin-coated PANI film. The drastic increase in electrical and ionic conductivities can be attributed to the incorporation of S-rGO, which not only has high electrical conductivity but also bears sulfonic acid groups that are good ionic conductors. The enhanced electron and ion movements in the films during the redox processes improve the electrochromic switching of PANI.40
| Samples | σ e (S cm−1) | σ ion (S cm−1) |
|---|---|---|
| Spin-coated PANI | 2.2 × 10−2 | 3.6 × 10−6 |
| (S-rGO/PANI)25 | 5.3 × 10−1 | 4.5 × 10−4 |
4. Conclusion
S-rGO has been readily synthesized using a one-pot method under mild conditions. The obtained S-rGO exhibits reasonably high electrical conductivity and can form highly stable colloidal suspensions in aqueous media. The (S-rGO/PANI)25 nanocomposite film fabricated via LbL assembly shows significantly increased electrical and ionic conductivities owing to the incorporation of S-rGO. Consequently, the redox reactions in the (S-rGO/PANI)25 film are a non-diffusion-controlled process in the studied scan rate range. This gives the (S-rGO/PANI)25 film a slightly higher contrast and much faster switching kinetics as compared with those of the corresponding spin-coated PANI film. We believe that S-rGO is also highly promising for other technological applications, such as for fabrication of flexible transparent electrodes with conjugated polymers.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- Z. Yan, J. Yao, Z. Sun, Y. Zhu and J. M. Tour, Small, 2012, 8, 59–62 CrossRef CAS.
- S. Biswas and L. T. Drzal, Chem. Mater., 2010, 22, 5667–5671 CrossRef CAS.
- V. Eswaraiah, K. Balasubramaniam and S. Ramaprabhu, J. Mater. Chem., 2011, 21, 12626–12628 RSC.
- X. Qi, K. Y. Pu, X. Zhou, H. Li, B. Liu, F. Boey, W. Huang and H. Zhang, Small, 2010, 6, 663–669 CrossRef CAS.
- K. Jo, T. Lee, H. J. Choi, J. H. Park, D. J. Lee, D. W. Lee and B. S. Kim, Langmuir, 2011, 27, 2014–2018 CrossRef CAS.
- V. Kumar, G. Ding, J. Ma, P. S. Lee and X. Lu, Adv. Mater., 2012, 24, 4071–4096 CrossRef.
- S. Xiong, J. Ma and X. Lu, Sol. Energy Mater. Sol. Cells, 2009, 93, 2113–2117 CrossRef CAS.
- A. A. Argun, P. H. Aubert, B. C. Thompson, I. Schwendeman, C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G. MacDiarmid and J. R. Reynolds, Chem. Mater., 2004, 16, 4401–4412 CrossRef CAS.
- A. L. Becker, A. P. R. Johnston and F. Caruso, Small, 2010, 6, 1836–1852 CAS.
- J. F. Quinn and F. Caruso, Macromolecules, 2005, 38, 3414–3419 CrossRef CAS.
- C. Bechinger, S. Ferrere, A. Zaban, J. Sprague and B. A. Gregg, Nature, 1996, 383, 608–610 CrossRef CAS.
- K. Sheng, H. Bai, Y. Sun, C. Li and G. Shi, Polymer, 2011, 52, 5567–5572 CrossRef CAS.
- L. Zhao, Y. Xu, T. Qiu, L. Zhi and G. Shi, Electrochim. Acta, 2009, 55, 491–497 CrossRef CAS.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- M. K. Ram, M. Salerno, M. Adami, P. Faraci and C. Nicolini, Langmuir, 1999, 15, 1252–1259 CrossRef CAS.
- S. Wang, P. J. Chia, L. L. Chua, L. H. Zhao, R. Q. Png, S. Sivaramakrishnan, M. Zhou, R. G. S. Goh, R. H. Friend, A. T. S. Wee and P. K. H. Ho, Adv. Mater., 2008, 20, 3440–3446 CrossRef CAS.
- Z. J. Fan, W. Kai, J. Yan, T. Wei, L. J. Zhi, J. Feng, Y. M. Ren, L. P. Song and F. Wei, ACS Nano, 2011, 5, 191–198 CrossRef CAS.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Carbon, 2012, 50, 914–921 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60–63 CrossRef CAS.
- H. Yang, F. Li, C. Shan, D. Han, Q. Zhang, L. Niu and A. Ivaska, J. Mater. Chem., 2009, 19, 4632–4638 RSC.
- O. C. Compton, D. A. Dikin, K. W. Putz, L. C. Brinson and S. T. Nguyen, Adv. Mater., 2010, 22, 892–896 CrossRef CAS.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- F. Yang, Y. Liu, L. Gao and J. Sun, J. Phys. Chem. C, 2010, 114, 22085–22091 CAS.
- D. D. Jiang, Q. Yao, M. A. McKinney and C. A. Wilkie, Polym. Degrad. Stab., 1999, 63, 423–434 CrossRef CAS.
- W. Yin and E. Ruckenstein, Synth. Met., 2000, 108, 39–46 CrossRef CAS.
- S. Xiong, F. Yang, G. Ding, K. Y. Mya, J. Ma and X. Lu, Electrochim. Acta, 2012, 67, 194–200 CrossRef CAS.
- P. T. C. Freire, F. E. A. Melo and J. Mendes Filho, J. Raman Spectrosc., 1996, 27, 507–512 CrossRef CAS.
- P. Jia, A. A. Argun, J. Xu, S. Xiong, J. Ma, P. T. Hammond and X. Lu, Chem. Mater., 2009, 21, 4434–4441 CrossRef CAS.
- S. Xiong, S. L. Phua, B. S. Dunn, J. Ma and X. Lu, Chem. Mater., 2010, 22, 255–260 CrossRef CAS.
- P. Jia, A. A. Argun, J. Xu, S. Xiong, J. Ma, P. T. Hammond and X. Lu, Chem. Mater., 2010, 22, 6085–6091 CrossRef CAS.
- X. Ren and P. G. Pickup, J. Electroanal. Chem., 1997, 420, 251–257 CrossRef CAS.
- P. G. Pickup, J. Chem. Soc., Faraday Trans., 1990, 86, 3631–3636 RSC.
- P. G. Pickup and R. A. Osteryoung, J. Am. Chem. Soc., 1984, 106, 2294–2299 CrossRef CAS.
- X. Ren and P. G. Pickup, J. Phys. Chem., 1993, 97, 3941–3943 CrossRef CAS.
- S. Xiong, Y. Xiao, J. Ma, L. Zhang and X. Lu, Macromol. Rapid Commun., 2007, 28, 281–285 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of GO and rGO, calculation for the electrical and ionic conductivities of the conducting polymer films, TGA curves of GO, rGO and S-rGO; Raman spectra of GO, rGO and S-rGO; a photograph of the different dispersions after storing for 3 months; FTIR spectra of GO and rGO; the full spectra of UV-vis absorbance for spin-coated PANI film and (S-rGO/PANI)25 film under different potentials and elemental analysis of of GO, rGO and S-rGO. See DOI: 10.1039/c2ra21579a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |