DOI:
10.1039/C2RA21254D
(Paper)
RSC Adv., 2012,
2, 10529-10536
Simple, selective, and sensitive colorimetric and ratiometric fluorescence/phosphorescence probes for platinum(II) based on Salen-type Schiff bases†
Received
21st June 2012
, Accepted 2nd September 2012
First published on 4th September 2012
Abstract
Ratiometric fluorescence/phosphorescence probes for Pt2+ based on the fluorescence quenching from the ligand of Salen-type Schiff bases and phosphorescence enhancement from the resulting Pt(II) complexes have been demonstrated. For phosphorescence enhancement, a good linearity (correlation coefficient R2 = 0.996, n = 10) was established with detection limit of 11.6 ± 0.2 ppb. The detection limit would be decreased to 2.31 ± 0.03 ppb if degassing was adopted. To the best of our knowledge, this value is one of the most sensitive probes for Pt2+. This system also has good selectivity with little interference from Sr2+, Sn2+, Pb2+, Cr3+, Mn2+, Al3+, Fe3+, Ce3+, Ag+, Li+, Mg2+, Na+, Cd2+, Cu2+, Ca2+, Zn2+, K+, Co2+, Ni2+, Au3+, Ir3+, and Pd2+. Moreover, this system is suitable for detection of different Pt(II) sources, such as PtCl2, K2PtCl4, Pt(COD)Cl2 (COD = 1,5-cyclooctadiene), and cis-platin. It potentially provides a new and simple way to detect some useful transition metal ions, such as Cu+, Au+, Pd2+, and Ru2+.
Introduction
Platinum, one of the rarest elements in the Earth's crust, is widely used in various materials including dental crowns, catalysts, laboratory equipment, resistance thermometers, electrodes, fuel cells, optic devices, and jewellery. It is also used in pharmaceuticals; for example, cis-platin, carboplatin, and oxaliplatin are widely used as anticancer drugs.1 Highly-efficient phosphorescent organic light-emitting devices (OLEDs) based on Pt(II) complexes have became an area of topical interest in material science and display technology.2 Pt(II) complexes can be used as catalyst in carbophilic activation, oxidation, hydration, and so on.3 Despite the importance of platinum in these fields, a major problem is the subsequent pollution of the environment that comes with its frequent use, because heavy-metal platinum and its salts are considered to be potentially hazardous to human health.4 Accordingly, the recognition and detection of Pt2+ has also been an especially active research area. The traditional analytical methods used for the detection of platinum include various instrumental techniques such as flame atomic absorption spectrometry (FAAS) and inductively coupled plasma mass spectrometry (ICP-MS).5 However, these methods require large and expensive instruments and highly skilled individuals to operate them. Recently, fluorescence detection of Pt2+ has received growing attention due to its high sensitivity, easy visualization, and short response time. Up to now, two types of fluorescence probes have been designed to probe Pt2+.6 Koide et al. reported fluorescent methods for Pt2+ detection based on the Pt-catalyzed Tsuji–Trost type reaction.6a–c Tae et al. utilized a rhodamine derivative as a fluorescence probe for Pt2+ in aqueous solutions.6d
Phosphorescence probes, especially those based on phosphorescent heavy-metal complexes, such as Pt(II), Ru(II), Re(I), Ir(III), Cu(I), Au(I/III) and Os(II) complexes, offer several important advantages over the traditional fluorescent probe. They provides longer lifetimes of emission, so the interference from short-lived fluorescence and scattering light can be easily avoided using an appropriate delay time. They have larger Stokes' shifts, so simplify the spectral separation between the excitation light and the phosphorescent emission. They have richer excited states, such as triplet metal-to-ligand charge-transfer (3MLCT), intraligand charge-transfer (3ILCT), ligand-to-ligand charge-transfer (3LLCT), ligand-to-metal charge transfer (3LMCT), metal–metal-to-ligand charge-transfer (3MMLCT), ligand-to-metal–metal charge-transfer (3LMMCT) and metal-to-ligand–ligand charge-transfer (3MLLCT) states, which depend on the metal centers, chemical structures and triplet state energy levels of the ligands, local environment and intermolecular interaction.7 To date, several approaches have been reported for phosphorescent detection, as shown in Fig. 1a.7c They have a similar phenomenon. Firstly the phosphorescent heavy-metal complexes with signaling unit and receptor unit are prepared by the reaction between the heavy-metal ion and ligand. Then the receptor unit can selectively bind to the analytes. Finally the signaling unit can report the interaction between the receptor and analytes through changes in optical or other properties. Furthermore, ratiometric emission probes, widely used in biological and sensory materials chemistry,8 have multiple emissions with different colors induced by the analyte, which can be useful not only for the ratiometric method of detection but also for rapid visual sensing. Herein, we report novel, simple, selective, and sensitive colorimetric and ratiometric fluorescence/phosphorescence probes for Pt2+ based on the fluorescence from the ligand and phosphorescence from the resulting Pt(II) complex (Fig. 1b). Therefore, the complicated preparation of ligand containing receptor units would not be required.
 |
| | Fig. 1 The reported approaches (a) and new approach in this work (b) for phosphorescence probes. | |
It is well-known that d8 square planar Pt(II) complexes with N⁁N or C⁁N bidentate, N⁁N⁁N, C⁁N⁁N or N⁁C⁁N terdentate, or N⁁N⁁N⁁N or O⁁N⁁N⁁O tetradentate conjugated aromatic ligands are highly phosphorescent at room temperature and widely used in OLEDs2b and O2 sensing.7c,d,9 The presence of conjugated aromatic ligands around a heavy-metal ion of Pt2+ introduces 3ILCT (π–π*) and/or 3MLCT (d–π*) excited states. In many Pt(II) complexes, either the MLCT or the ILCT states, or indeed both, may lie at lower energies than the d–d states and have intrinsically larger radiative decay rate constant kr values than the d–d transition. In order to achieve ratiometric fluorescence from the ligand and phosphorescence from its Pt(II) complex, the ligand system was carefully chosen, according to the following factors. (1) Not only the ligands should be highly fluorescent but also their Pt(II) complexes should be highly phosphorescent at room temperature. (2) The chemical reaction between the ligand and Pt2+ should be complete and prompt under moderate conditions, so tetradentate chelating ligands might be better. (3) The ligands and their Pt(II) complexes should be stable enough for the application. (4) The wavelength difference between the maximum fluorescence (λF,max) of ligand and the maximum phosphorescence (λP,max) of its Pt(II) complex should be large. Therefore, the tetradentate chelating ligands of porphyrin and Salen-type Schiff base (Fig. 2) were firstly chosen, due to the fact that these ligands and their Pt(II) complexes usually are highly emissive and stable.2a,10 Unfortunately, porphyrins are not suitable for applications in ratiometric fluorescence/phosphorescence sensing, because the phosphorescence of Pt(II) porphyrin complexes, usually originated in 3ILCT, is similar to the fluorescence of free porphyrin (Fig. S1 in the ESI†). Especially, salen, a particular class of tetradentate O⁁N⁁N⁁O chelating bis-Schiff base, can be synthesized by the condensation of salicylaldehyde or its derivatives with 1,2-diamines. Due to their stabilities, biological activities, and photophysical properties, salen-type Schiff bases and their metal complexes have exhibited many potential applications in many fields, such as catalysts, DNA cleavage, optical materials, magnetic materials, and probes.11 Furthermore, the recent study demonstrated that PtL3 (Fig. 2), prepared by the reaction between equivalent molecular Pt2+ and L3 at 70 °C with a good isolated yield of 82%, showed good thermal stability with the decomposition temperature of 415 °C and strong red phosphorescence (3MLCT mixed with 3ILCT) with a quantum efficiency (ΦP) of 0.26 and long emission decay lifetime (τ) of 3.1 μs in degassed benzene and was used in the application of OLEDs.10f Thus, salen-type Schiff bases (L1–L4, Fig. 2) were finally adopted. To the best of our knowledge, this is the first example of detection of Pt2+ based on ratiometric dual fluorescent/phosphorescent emission.
 |
| | Fig. 2 The chemical structures of L1–L4 and Pt(II) complex of PtL3. | |
Results and discussion
Synthesis and features of the ligands and Pt(II) complexes
L1–L4 were simply synthesized by a one-pot reaction of diamine and 5-sulfonatosalicylaldehyde in ethanol/H2O according to a modified literature method.12 The presence of sulfonate groups in L4 can improve the solubility of the ligand and its metal complex in the water.11h,13 All the ligands in DMF have relatively intense absorption bands centred at λabs,max = 315–331 nm (molar extinction coefficient, ε = 1.4–2.5 × 104 dm3 mol−1 cm−1), which are assigned to the π–π* transition involving molecular orbitals essentially localized on the C=N group and the benzene ring (Fig. 3). The lower intensity absorption bands (ε = 102–103 dm3 mol−1 cm−1) in the λabs,max = 380–420 nm region are assigned to the n–π* transition involving molecular orbitals of the C=N chromophore and the benzene ring.14 As shown in Fig. 3, L1–L4 exhibit strong fluorescence in DMF with a ΦF of 0.057, 0.097, 0.19, and 0.56, respectively, attributed to the 1ILCT (π–π*) transition. It is well known that the π-conjugation enhancement should lead to a considerable decrease in the energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), resulting in a red shift in emission spectra, which is confirmed by the fact that the emission peak of L3 and L4 with the π-conjugation of 1,2-phenylenediamine bridge is at λF,max = 497 and 457 nm, respectively, and that of L1 and L2 with non-π-conjugation of 1,2-ethylenediamine bridge is at λF,max = 420 and 428 nm, respectively (Fig. 3). Sulfonate groups are often believed not to affect the spectroscopic properties of organic dyes, which have been discussed in detail in our recently published paper.11h
At room temperature, the reaction between 1 equiv. of L3 and Pt2+ in DMF with 2 equiv. of sodium acetate was a bit slow and needed 12 h to complete, but the reaction could be promoted by a moderate temperature of 70 °C and finished fully in 3 h with an excellent isolated yield of up to 86%. The resulting PtL3 complex, identified by 1H NMR, mass spectrum, and elemental analysis (see the Experimental Section), gives a strong red phosphorescence (λP,max = 608) in degassed DMF with ΦP of 0.20 and long τ of 2.8 μs, similar with the reported data.10f Both solid state and solution of the L3 and PtL3 complex are stable in air for several months. The combined results reveal that L3 might have potential applications in ratiometric fluorescence/phosphorescence probe for Pt2+.
Colorimetric analysis of L3
The colorimetric analysis of L3 toward various metal ions, such as Pt2+, Sr2+, Sn2+, Pb2+, Cr3+, Mn2+, Al3+, Fe3+, Ce3+, Ag+, Li+, Mg2+, Na+, Cd2+, Cu2+, Ca2+, Zn2+, K+, Co2+, Ni2+, Pd2+, Au3+, and Ir3+ was studied. After incubating at 70 °C for 3 h, the color change of solutions was visualized by naked eye analysis and optical response was studied by UV/Vis spectroscopy, as shown in Fig. 4, S2†, and S3†. Except Pt2+ (PtCl2 as source), the addition of 2 equiv. of any other metal ion to the slight yellow solution of L3 did not show any drastic change in color monitored by the naked eye. However, drastic changes were found by UV/Vis absorption spectroscopy, indicating the complexation of the metal ion and L3 (Fig. 4a). Upon addition of Pt2+ to the L3 solution, there is a remarkable change, not only by the naked eye (Fig. 4b), but also by UV/Vis absorption spectroscopy. The absorption band of L3 at 375 nm gradually increased following the formation of new bands centered at 450 and 525 nm upon the addition of Pt2+, resulting in a color change from a slight yellow color to a red color (Fig. S4†).
 |
| | Fig. 4 Absorption spectra (a) and photographs (b) top: under sunlight; bottom: under 360 nm UV light of L3 (1.0 × 10−5 mol dm−3 in air-saturated DMF) upon addition 2 equiv. of different metal ions. | |
Fluorescence and phosphorescence sensing for Pt2+ based on L3
The fluorescence and phosphorescence titration of L3 (1.0 × 10−5 mol dm−3 in air-saturated DMF) in the presence of Pt2+ was recorded at room temperature after the reaction fully finished (incubating at 70 °C for 3 h). Notably, upon addition of Pt2+ to the L3 solution, the green-yellow fluorescence (λF,max = 497 nm) of L3 reduced gradually and red phosphorescence (λP,max = 608 nm) subsequently appeared when excited at 400 nm in air (Fig. 5). The green-yellow fluorescence of L3 is highly sensitive to Pt2+, which will be reduced 87% and 97% upon the addition of 1 and 2 equiv. of Pt2+, respectively. When excited at 525 nm, where not L3 but PtL3 had strong absorption (Fig. 4a), the interference from the fluorescence (λF,max = 497 nm) was eliminated and the linear function between phosphorescence intensity at 608 nm and Pt2+ concentration was found (inset image of Fig. 5a). In air, the phosphorescence intensity is relative weak compared with fluorescence intensity using a single excitation wavelength of 400 nm, so the phosphorescence intensity needs to be enhanced for the purpose of ratiometric dual fluorescence/phosphorescence sensing. It is well known that the triplet phosphorescence can be quenched by O2 because the ground state of O2 is triplet state.15 For example, the ΦP of PtL3 in degassed DMF is 0.20, much higher than that (0.031) in air-saturated DMF. Therefore, degassing is an alternative way to achieve ratiometric dual fluorescence/phosphorescence sensing, as shown in Fig. 5b (a good linearity was found in the Pt2+ concentration between 0 and 0.5 × 10−5 mol dm−3).
 |
| | Fig. 5 (a) Emission spectra (excited at 400 nm) of L3 (1.0 × 10−5 mol dm−3 in air-saturated DMF) upon addition different equiv. of Pt2+ (0, 0.10, 0.20, 0.30, 0.40, 0.50, 0.60, 0.70, 0.80, 0.90, 1.0, 1.2, 1.5, and 2.0). Inset image: excited at 525 nm; (b) ratiometric emission spectra (excited at 400 nm) of L3 (1.0 × 10−5 mol dm−3 in degassed DMF) upon addition different equiv. of Pt2+. Inset image: emission titration profile of I497/I608versus the equivalents of Pt2+ ions added. | |
The plots of emission intensity as a function of the concentration of Pt2+ are shown in Fig. 6. Both the fluorescence quenching (λF,max = 497 nm) and phosphorescence enhancement (λP,max = 608 nm) of L3 in air-saturated DMF are suitable for detection of Pt2+, which also reveal that the stoichiometry of the PtL3 complex formed between L3 and Pt2+ ions is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. For fluorescence quenching, a good linearity (correlation coefficient R2 = 0.994, n = 10) of the fluorescence intensity as the function of the Pt2+ concentration between 0 and 1.0 × 10−5 mol dm−3 was established. The detection limit, based on the definition by IUPAC (CDL = 3 Sd/m) [Sd = standard deviation, m = sensitivity of the method (a change in the inhibition percent divided by the change in concentration near the detection limit)],16 was found to be 2.57 ± 0.03 μmol dm−3 (500 ± 6 ppb) from 10 blank solutions. For phosphorescence enhancement, a good linearity (correlation coefficient R2 = 0.996, n = 10) was also established with detection limit of 59.5 ± 1.0 nmol dm−3 (11.6 ± 0.2 ppb). The detection limit could be further decreased to 11.9 ± 1.3 nmol dm−3 (2.31 ± 0.03 ppb) by degassing. To the best of our knowledge, this value is one of the most sensitive probes for Pt2+.6
:1. For fluorescence quenching, a good linearity (correlation coefficient R2 = 0.994, n = 10) of the fluorescence intensity as the function of the Pt2+ concentration between 0 and 1.0 × 10−5 mol dm−3 was established. The detection limit, based on the definition by IUPAC (CDL = 3 Sd/m) [Sd = standard deviation, m = sensitivity of the method (a change in the inhibition percent divided by the change in concentration near the detection limit)],16 was found to be 2.57 ± 0.03 μmol dm−3 (500 ± 6 ppb) from 10 blank solutions. For phosphorescence enhancement, a good linearity (correlation coefficient R2 = 0.996, n = 10) was also established with detection limit of 59.5 ± 1.0 nmol dm−3 (11.6 ± 0.2 ppb). The detection limit could be further decreased to 11.9 ± 1.3 nmol dm−3 (2.31 ± 0.03 ppb) by degassing. To the best of our knowledge, this value is one of the most sensitive probes for Pt2+.6
 |
| | Fig. 6 Titration curves (a: fluorescence was excited at 400 nm and measured at 497 nm; b: phosphorescence was excited at 525 nm and measured at 608 nm) of L3 (1.0 × 10−5 mol dm−3 in air-saturated DMF) with addition of Pt2+. I0 and I are the emission intensities before and after the addition of Pt2+, respectively. | |
The selectivity behavior is obviously one of the most important characteristics of a probe, that is, the relative probe response for the primary ion over other ions present in solution. In order to evaluate the selectivity of L3, other 20 different kinds of metal ions (Sr2+, Sn2+, Pb2+, Cr3+, Mn2+, Al3+, Fe3+, Ce3+, Ag+, Li+, Mg2+, Na+, Cd2+, Cu2+, Ca2+, Zn2+, K+, Co2+, Ni2+, and Pd2+), which probably interfere with the detection of Pt2+, were performed under the similar conditions: the concentrations of L3 were all kept at 1.0 × 10−5 mol dm−3 and 2 equiv. of metal ion were added and heated at 70 °C for 3 h. When excited by UV light at 400 nm, as depicted in Fig. 4b and S5†, several metal ions, such as Pt2+, Ni2+, Pd2+, and Cu2+, gave almost 100% quenching of the fluorescence intensity at 497 nm, on the other hand, Mg2+, Cd2+, Sr2+, and so on, showed fluorescence enhancement, indicating bad selectivity of fluorescence quenching. However, red phosphorescence at 608 nm was only observed after adding Pt2+, revealing that L3 had excellent selectivity for Pt2+. Furthermore, competition experiments were also performed in the presence 2 equiv. of L3 and 1 equiv. of Pt2+ mixed with 1 equiv. of the other metal ions (Fig. 7) to explore practical applicability of L3 as a Pt2+-selective phosphorescence probe. The red phosphorescence (excited at 525 nm) intensity at 608 nm of solutions containing both Pt2+ and the other metal ions showed no obvious variation (± 10%) compared with those containing only Pt2+. The phosphorescence selectivity between Pt2+ and its similar metal ions, such as Pd2+, Ni2+, Cd2+, and Co2+, is especially promising because it is typically difficult to discriminate these similar metal ions.6 Moreover, it is well known that Au(III) and Ir(III) complexes usually are highly phosphorescent, but Au3+ and Ir3+ have little interference with phosphorescence detection of Pt2+ (Fig. S3 and S6†).
 |
| | Fig. 7 Phosphorescence (excited at 525 nm and measured at 608 nm) selectivity of 2 equiv. of L3 (1.0 × 10−5 mol dm−3 in air-saturated DMF) toward 1 equiv. of Pt2+ and 1 equiv. of other metal ions (0, L3; 1, Pt2+; 2, Sr2+; 3, Sn2+; 4, Pb2+; 5, Cr3+; 6, Mn2+; 7, Al3+; 8, Fe3+; 9, Ce3+; 10, Ag+; 11, Li+; 12, Mg2+; 13, Na+; 14, Cd2+; 15, Cu2+; 16, Ca2+; 17, Zn2+; 18, K+; 19, Co2+; 20, Ni2+; and 21, Pd2+). | |
In order to investigated the application scope of L3, different Pt(II) sources, such as PtCl2, K2PtCl4, Pt(COD)Cl2 (COD = 1,5-cyclooctadiene), and cis-platin, were examined (Fig. 8 and 9). Cis-platin and Pt(COD)Cl2 showed relatively smaller phosphorescence enhancements compared with PtCl2 and K2PtCl4, presumably because the ligand exchange reactions of the cis-platin and Pt(COD)Cl2 took place slowly.6d It is possible for cis-platin to enhance phosphorescence intensity and sensitivity by increasing reaction time. The above combined results reveal that L3 can be used in the application of ratiometric fluorescence/phosphorescence sensing for the detection of Pt2+ with minimum interference from other metal ions.
 |
| | Fig. 8 Phosphorescence (excited at 525 nm) intensities at 608 nm of L3 (1.0 × 10−5 mol dm−3 in air-saturated DMF) in the presence of 2 equiv. of different Pt(II) sources. | |
 |
| | Fig. 9 Phosphorescence (excited at 525 nm) intensities at 608 nm of L3 (1.0 × 10−5 mol dm−3 in air-saturated DMF) in the presence of different equiv. of cis-platin (0, 0.30, 0.60, 1.0, 1.5, and 2.0). | |
Fluorescence and phosphorescence sensing for Pt2+ based on L1, L2, and L4
L1, L2, and L4 were also used to probe Pt2+, but the results (Table 1 and Fig. S7†) were not as good as L3. Excited at 350 nm, a good linearity of the fluorescence intensity of L1 (λF,max = 420 nm) and L2 (λF,max = 430 nm) as the function of the Pt2+ concentration between 0 and 1.0 × 10−5 mol dm−3 was also found, but the linear relationship between the phosphorescence intensity at λP,max = 560 nm of L1/L2 and Pt2+ concentration was not observed, even excited at the longer wavelength of 440 nm. The failures to detect Pt2+ by phosphorescence enhancement might be caused by the incomplete reaction between the ligand and Pt2+. A moderate yield of 50–60% of the reaction between the L1/L2 and Pt2+ in DMF was obtained, even after heating at 70 °C for 10 h. This might be explained by their single-crystal X-ray diffraction structures (Fig. 10). The two N atoms and two H atoms in –OH of L1 or L2 are far apart and not in one plane. On the contrary, these four atoms in L3 are almost close in one plane, which facilitates formation of square planar Pt(II) complex between the ligand and Pt2+, resulting in a high yield of PtL3 of up to 86%. L4, with the help of two sulfonate groups, has good solubility in water, so it can be used to detect Pt2+ in aqueous solution (Fig. S8 and S9†). Since the ΦF of L4 in water is high, up to 0.33, L4 has the lowest detection limit of 906 nmol dm−3 (180 ppb) for Pt2+ based on fluorescence (excited at 365 nm) quenching at 435 nm. Based on phosphorescence (excited at 480 nm) enhancement, a linearity of the phosphorescence intensity at 585 nm of L4 as the function of Pt2+ concentration was not found. We failed to identify the chemical structure of the resulting complex of the reaction between L4 and Pt2+, because the pure resulting complex was not obtainable by either column chromatography or recrystallization.
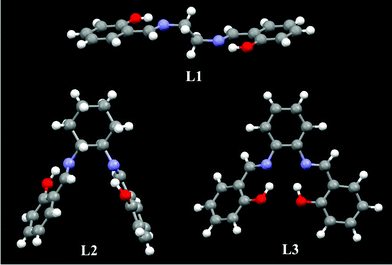 |
| | Fig. 10 Single-crystal X-ray diffraction structures of L1, L2, and L3. | |
Table 1 The performances of L1–L4 as Pt2+ probe
| Ligand (λEm/nm)a |
Ab |
Bb |
R2 |
C
DL
|
|
wavelength of emission was measured.
in the job plot: Y = A + B × CPt (10−5 mol dm−3).
in air-saturated DMF.
in degassed DMF.
in air-saturated water.
|
| L1 (420)c |
3.50 × 10−2 |
0.964 |
0.994 |
2.28 ± 0.03 ppm |
| L2 (430)c |
7.13 × 10−2 |
0.880 |
0.988 |
2.22 ± 0.03 ppm |
| L3 (497)c |
1.06 × 10−1 |
0.776 |
0.994 |
500 ± 7 ppb |
| L3 (608)c |
8.17 |
80.95 |
0.996 |
11.6 ± 0.2 ppb |
| L3 (608)d |
3.54 × 101 |
398 |
0.995 |
2.31 ± 0.03 ppb |
| L4 (435)e |
1.67 × 10−1 |
0.799 |
0.985 |
180 ± 2 ppb |
Experimental
Materials and instrumentation
All reagents were purchased from commercial suppliers and used without further purification. 1HNMR (400 MHz) spectra were recorded in [D6]DMSO. Chemical shifts are reported in ppm using tetramethylsilane as internal standard. UV/vis absorption spectra were recorded using a UV 765 spectrophotometer with quartz cuvettes of 1 cm pathlength. Emission spectra were obtained using FluoroMax-4 Spectrofluorophotometer (HORIBA Jobin Yvon) linked to a Pentium PC running SpectraCalc software package at 298 K. The slit width was 5.0 nm for both excitation and emission. The photon multiplier voltage was 400 V. Samples in solution and powder were contained in 10.0 mm path length quartz cuvettes (3.5 mL volume) and quartz tube, respectively. Emission decay lifetime measurements were performed with a Quanta Ray DCR-3 pulsed Nd:YAG laser system (pulse output 355 nm, 8 ns). Decay times were registered using a FAST Comtec multichannel scaler PCI card with a time resolution of 250 ps.
Synthesis of N,N-bis(salicylidene)-1,2-ethylenediamine (L1)12
The compound salicylaldehyde (1.22 g, 10 mmol) was poured in a round-bottomed flask which containing 50 mL ethanol. To the reaction mixture, the ethanolic solution (10 mL) of 1,2-ethylenediamine (0.30 g, 5.0 mmol) was added and the mixture was stirred at 80 °C for 2 h. Then, the reaction mixture was filtered and the residue was washed with ethanol and diethyl ether to obtain L1 as a yellow crystalline solid. Yield: 1.41 g (93%); 1HNMR (400 MHz, CDCl3): δ = 13.23 (s, 2H), 8.39 (s, 2H), 7.34 (m, 2H), 7.30 (m, 2H), 6.98 (d, J = 8.0 Hz, 2H), 6.90 (m, 2H), 3.97 (s, 4H). FAB-MS: m/z: [269 M+]; elemental analysis calcd (%) for C16H16N2O2: C 71.62, H 6.01, N 10.44; found: C 71.43, H 6.06, N 10.50.
Synthesis of N,N-bis(salicylidene)-1,2-cyclohexanediamine (L2)12
L2 was prepared by a similar procedure as that used for L1, but with salicylaldehyde (1.22 g, 10 mmol) and 1,2-cyclohexanediamine (0.57 g, 5.0 mmol) to give pale yellow crystalline solid. Yield: 1.65 g (92%); 1HNMR (400 MHz, CDCl3): δ = 13.36 (s, 2H), 8.28 (s, 2H), 7.28 (m, 2H), 7.19 (d, J = 7.6 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 6.84(t, J = 7.6 Hz, 2H), 3.37 (m, 2H), 1.99 (m, 4H), 1.77 (m, 4H); FAB-MS: m/z: [323 M+]; elemental analysis calcd (%) for C20H22N2O2: C 74.51, H 6.88, N 8.69; found: C 74.88, H 6.82, N 8.62.
Synthesis of N,N-bis(salicylidene)-1,2-phenylenediamine (L3)12
L3 was prepared by a similar procedure as that used for L1, but with salicylaldehyde (1.22 g, 10 mmol) and 1,2-phenylenediamine (0.54 g, 5.0 mmol) to give an orange crystalline solid. Yield: 1.67 g (95%); 1HNMR (400 MHz, CDCl3): δ = 13.10 (s, 2H), 8.67 (s, 2H), 7.42 (dd, J = 1.6,7.6 Hz, 4H), 7.53 (dd, J = 3.6,2.0 Hz, 2H), 7.29 (m, 2H), 7.09 (d, J = 8.4 Hz, 2H), 6.97 (t, J = 7.2 Hz, 2H); FAB-MS: m/z: [317 M+]; elemental analysis calcd (%) for C20H16N2O2: C 75.93, H 5.10, N 8.86; found: C 75.61, H 5.14, N 8.79.
Synthesis of N,N′-bis(5-sulfonatosalicylidene)-1,2-phenylenediamine disodium salt (L4)12
The compound salicylaldehyde-5-sulfonate sodium salt (1.12 g, 5 mmol) was poured in a round-bottomed flask containing ethanol/water 50 mL (4:1, v/v). To the reaction mixture, the ethanol solution of 1,2-phenylenediamine (0.29 g, 2.7 mmol) was added and the mixture was stirred at 80 °C for 2 h. Then, the reaction mixture was filtered and the residue was washed with ethanol and diethyl ether to obtain L4 as a yellow powder. Yield: 1.19 g (85%); 1HNMR (400 MHz, [D6]DMSO): δ = 13.21 (s, 2H), 9.00 (s, 2H), 7.96 (d, J = 2.0 Hz, 2H), 7.65 (m, 2H), 7.53 (m, 2H), 7.43 (m, 2H), 6.93 (d, J = 8.4 Hz, 2H); ESI-MS (negative): m/z = [474 M–2Na+]; elemental analysis calcd (%) for C20H14N2 Na2O8S2·6H2O: C 38.22, H 4.17, N 4.46; found: C 38.43, H 4.20, N 4.42.
Synthesis of N,N-bis(salicylidene)-1,2-phenylenediamine Pt(II) Complex (PtL1)10f
Complex PtL1 was prepared by modification of the literature procedures.10f Sodium acetate (0.082 g, 1.0 mmol) was suspended in a solution of L1 (0.13 g, 0.5 mmol) in DMF (15 mL). K2PtCl4 (0.21 g, 0.5 mmol) dissolved in DMSO (3 mL) was added dropwise to the suspension at 70 °C and the resulting mixture was stirred at the same temperature for 10 h. Upon cooling, diethyl ether (50 mL) was added to the mixture to afford an orange precipitate. The solid product was filtered and washed with Et2O. Recrystallization from hot DMF afforded the product as yellow crystals. Yield: 0.11 g (50%); 1HNMR [D6]DMSO): δ = 8.55 (s, 2H), 7.51 (dd, J = 7.9, 1.7 Hz, 2H), 7.44 (td, J = 7.2, 1.8 Hz, 2H), 6.91 (d, J = 8.4 Hz, 2H), 6.62 (td, J = 7.4, 1.0 Hz, 2H), 3.83 (s, 4H). FAB-MS: m/z: [461 M+]. elemental analysis calcd (%) for C16H14N2O2Pt: C 41.65, H 3.06, N 6.07; Found: C 41.67, H 3.09, N 6.11.
Synthesis of N,N-bis(salicylidene)-1,2-cyclohexanediamine Pt(II) Complex (PtL2)
PtL2 was prepared by a similar procedure as that used for PtL2 but with L2 (0.16 g, 0.5 mmol) to give yellow crystals. Yield: 0.15 g (60%); 1HNMR (400 MHz CDCl3): δ = 7.80 (s, 2 H), 7.47 (t, J = 4.6, 2.4 Hz, 2 H), 7.21 (d, J = 8.8 Hz, 2H); 7.13 (d, J = 7.6 Hz, 2 H), 6.60 (m, 2 H),3.68 (s, 2 H), 1.98 (m, 4H), 1.58 (m, 4H); FAB-MS: m/z: [516 M+]; elemental analysis calcd (%) for C20H20N2O2Pt: C 46.60, H 3.91, N 5.43; found: C 46.44, H 3.87, N 5.46.
Synthesis of N,N-bis(salicylidene)-1,2-phenylenediamine Pt(II) Complex (PtL3)
PtL3 was prepared by a similar procedure as that used for PtL1 but with L3 (0.16 g, 0.5 mmol). Removal of solvent from the reaction mixture followed by purification through column chromatography using dichloromethane/ethyl acetate (3:1, v/v) as eluent afforded a red solid. Yield: 0.22 g (86%); 1HNMR (400 MHz [D6]DMSO): δ = 9.51 (s, H), 8.44 (dd, J = 4.6, 2.4 Hz, 2H), 7.86 (m, 2 H), 7.57 (m, 2H), 7.44 (m, 2H), 7.11 (d, J = 6.4 Hz, 2H), 6.78 (m, 2H); FAB-MS: m/z: [510 M+]; elemental analysis calcd (%) for C20H14 N2O2Pt: C 47.15, H 2.77, N 5.50; found: C 46.91, H 2.74, N 5.45.
Measurement of fluorescence quantum yield (Φ)
Φ was measured by the optical dilute method of Demas and Crosby17 with a standard of quinine sulfate (Φr = 0.55, quinine in 0.05 mol dm−3 in sulfuric acid) calculated by: Φs = Φr(Br/Bs)(ns/nr)2(Ds/Dr), where the subscripts s and r refer to the sample and reference standard solution respectively, n is the refractive index of the solvents, D is the integrated intensity under the emission spectrum. The excitation intensity B is calculated by: B = 1 − 10−AL, where A is the absorbance at the excitation wavelength and L is the optical path length (L = 1 cm in all cases). The refractive indices of the solvents at room temperature are taken from standard source. Errors for Φ values (± 10%) are estimated.
Measurement of metal ion sensing
Each metal ion titration experiment was started with ligand (3 mL) of known concentration (1.0 × 10−5 mol dm−3 in DMF or water) in presence of 2 equiv. of sodium acetate. Pt(II) salt and other various metal salts (nitrate in H2O) were used for the titration. All types of emission measurement were monitored after addition of the metal salt to the ligand solutions and incubating 70 °C for 3 h in air. Solution degassing was carried out three times with freeze-pump-thaw cycles.
Conclusions
In conclusion, we have described novel, simple, highly selective and sensitive colorimetric and ratiometric dual fluorescence/phosphorescence probes for Pt2+ in DMF or water based on Salen-type Schiff bases. We believe that it potentially provides a new and simple way to detect some useful transition metal ions, such as Cu+, Au+, Pt2+, Pd2+, and Ru2+. Further studies on the detection of Cu+ utilizing this kind of method are currently underway in our laboratory.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21172160) and Ministry of Education of China (no. 20100181120039).
References
-
(a) J. Reedijk, Pure Appl. Chem., 1987, 59, 181 CrossRef CAS;
(b) L. Kelland, Nat. Rev. Cancer, 2007, 7, 573 CrossRef CAS.
-
(a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS;
(b) J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205 CrossRef CAS;
(c)
Highly efficient OLEDs with phosphorescent materials, ed. H. Yersin, WILEY-VCH, Weinheim, 2007 Search PubMed;
(d)
WOLEDs and Organic Photovoltaics, ed. V. W. W. Yam, Springer-VerlagBerlin Heidelberg, 2010 Search PubMed.
-
(a) A. Furstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef;
(b) A. Leyva-Perez and A. Corma, Angew. Chem., Int. Ed., 2012, 51, 614 CrossRef CAS.
-
(a) J. H. Schiller and G. Bittner, Clin. Cancer Res., 1999, 5, 4287 CAS;
(b) E. Rudolph, S. Hann, G. Stingeder and C. Reiter, Anal. Bioanal. Chem., 2005, 382, 1500 CrossRef CAS;
(c) A. J. Danford, D. Wang, Q. Wang, T. D. Tullius and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12311 CrossRef CAS.
-
(a) G. S. Reddi and C. R. M. Rao, Analyst, 1999, 124, 1531 RSC;
(b) J. G. Morrison, P. White, S. McDougall, J. W. Firth, S. G. Woolfrey, M. A. Graham and D. Greenslade, J. Pharm. Biomed. Anal., 2000, 24, 1 CrossRef CAS;
(c) A. R. Ghezzi, M. Aceto, C. Cassino, E. Gabano and D. Osella, J. Inorg. Biochem., 2004, 98, 73 CrossRef CAS;
(d) A. V. Klein and T. W. Hambley, Chem. Rev., 2009, 109, 4911 CrossRef CAS.
-
(a) A. L. Garner and K. Koide, J. Am. Chem. Soc., 2008, 130, 16472 CrossRef CAS;
(b) A. L. Garner and K. Koide, Chem. Commun., 2009, 83 RSC;
(c) A. L. Garner and K. Koide, Chem. Commun., 2009, 86 RSC;
(d) H. Kim, S. Lee, J. Lee and J. Tae, Org. Lett., 2010, 12, 5342 CrossRef CAS;
(e) J. F. Zhang, Y. Zhou, J. Yoon and J. S. Kim, Chem. Soc. Rev., 2011, 40, 3416 RSC.
-
(a) Z. M. Hudson and S. N. Wang, Acc. Chem. Res., 2009, 42, 1584 CrossRef CAS;
(b) C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418 CrossRef CAS;
(c) Q. Zhao, F. Y. Li and C. H. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC;
(d) Q. Zhao, C. H. Huang and F. Y. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC.
-
(a) Y. Kubo, M. Yamamoto, M. Ikeda, M. Takeuchi, S. Shinkai, S. Yamaguchi and K. Tamao, Angew. Chem., 2003, 115, 2082 CrossRef;
(b) W. J. Xu, S. J. Liu, X. Y. Zhao, S. Sun, S. Cheng, T. C. Ma, H. B. Sun, Q. Zhao and W. Huang, Chem. Eur. J., 2010, 16, 7125 CAS;
(c) Z. M. Hudson, S. B. Zhao, R. Y. Wang and S. N. Wang, Chem.–Eur. J., 2009, 15, 6131 CrossRef CAS;
(d) P. H. Lanoe, J. L. Fillaut, L. Toupet, J. A. G. Williams, H. L. Bozec and V. Guerchais, Chem. Commun., 2008, 4333 RSC.
-
(a) A. Mills, Platinum Metals Rev., 1997, 41, 115 CAS;
(b) Y. Amao, Microchim. Acta, 2003, 143, 1 CrossRef CAS.
-
(a) H. F. Xiang, S. C. Yu, C. M. Che and P. T. Lai, Appl. Phys. Lett., 2003, 83, 1518 CrossRef CAS;
(b) C. M. Che, Y. J. Hou, M. C. W. Chan, J. H. Guo, Y. Liu and Y. Wang, J. Mater. Chem., 2003, 13, 1362 RSC;
(c) C. M. Che, S. C. Chan, H. F. Xiang, M. C. W. Chan, Y. Liu and Y. Wang, Chem. Commun., 2004, 1484 RSC;
(d) H. F. Xiang, S. C. Chan, K. K. Y. Wu, C. M. Che and P. T. Lai, Chem. Commun., 2005, 1408 RSC;
(e) F. Galbrecht, X. H. Yang, B. S. Nehls, D. Neher, T. Farrell and U. Scherf, Chem. Commun., 2005, 2378 RSC;
(f) C. M. Che, C. C. Kwok, S. W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Y. Zhu and H. Yersin, Chem.–Eur. J., 2010, 16, 233 CrossRef CAS.
-
(a) E. Lamour, S. Routier, J. L. Bernier, J. P. Catteau, C. Bailly and H. Vezin, J. Am. Chem. Soc., 1999, 121, 1862 CrossRef CAS;
(b) P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410 RSC;
(c) H. Miyasaka, A. Saitoh and S. Abe, Coord. Chem. Rev., 2007, 251, 2622 CrossRef CAS;
(d) F. Faridbod, M. R. Ganjali, R. Dinarvand, P. Norouzi and S. Riahi, Sensors, 2008, 8, 1645 CrossRef CAS;
(e) A. W. Kleij, Eur. J. Inorg. Chem., 2009, 193 CrossRef CAS;
(f) P. Wu, D. L. Ma, C. H. Leung, S. C. Yan, N. Y. Zhu, R. Abagyan and C. M. Che, Chem.–Eur. J., 2009, 15, 13008 CrossRef CAS;
(g) M. Andruh, Chem. Commun., 2011, 47, 3025 RSC;
(h) L. Zhou, P. Y. Cai, Y. Feng, J. H. Cheng, H. F. Xiang, J. Liu, D. Wu and X. G. Zhou, Anal. Chim. Acta, 2012, 735, 96 CrossRef CAS.
-
(a) K. J. Berry, F. Moya, K. S. Murray, A. B. van der Bergen and B. O. West, J. Chem. Soc., Dalton Trans., 1982, 109 RSC;
(b) M. Botsivali, D. F. Evans, P. H. Missen and M. W. Upton, J. Chem. Soc., Dalton Trans., 1985, 1147 RSC;
(c) D. A. Atwood and M. J. Harvey, Chem. Rev., 2001, 101, 37 CrossRef CAS;
(d) P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410 RSC.
-
(a) Z. Q. Wu, Z. Q. Jiang, D. Wu, H. F. Xiang and X. G. Zhou, Eur. J. Org. Chem., 2010, 1854 CrossRef CAS;
(b) Y. Y. Qu, F. Ke, L. Zhou, Z. K. Li, H. F. Xiang, D. Wu and X. G. Zhou, Chem. Commun., 2011, 47, 3912 RSC.
-
(a) S. Di Bella, I. Fragala, I. Ledoux, M. A. Diaz-Garcia and T. J. Marks, J. Am. Chem. Soc., 1997, 119, 9550 CrossRef CAS;
(b) S. C. Wang, G. W. Men, L. Y. Zhao, Q. F. Hou and S. M. Jiang, Sens. Actuators, B, 2010, 145, 826 CrossRef;
(c) M. Hosseini, Z. Vaezi, M. R. Ganjali, F. Faridbod, S. D. Abkenar, K. Alizadeh and M. Salavati-Niasari, Spectrochim. Acta, Part A, 2010, 75, 978 CrossRef;
(d) C. Gou, S. H. Qin, H. Q. Wu, Y. Wang, J. Luo and X. Y. Liu, Inorg. Chem. Commun., 2011, 14, 1622 CrossRef CAS.
- Y. Feng, J. H. Cheng, L. Zhou, X. G. Zhou and H. F. Xiang, Analyst 10.1039/c2an35907c.
- L. A. Currie, Anal. Chim. Acta, 1999, 391, 105 CrossRef CAS.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21254d |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 