Facile synthesis of bifunctional silica-coated core–shell Y2O3:Eu3+,Co2+ composite particles for biomedical applications
Timur Sh.
Atabaev
*a,
Oh Seong
Jin
b,
Jong Ho
Lee
b,
Dong-Wook
Han
*b,
Hong Ha Thi
Vu
a,
Yoon-Hwae
Hwang
*a and
Hyung-Kook
Kim
*a
aDepartment of Nanomaterials Engineering and BK 21 Nano Fusion Technology Division, Pusan National University, Miryang, 627-706, Republic of Korea. E-mail: atabaev@pusan.ac.kr; yhwang@pusan.ac.kr; hkkim@pusan.ac.kr
bDepartment of Applied Nanoscience and BK 21 Nano Fusion Technology Division, Pusan National University, Miryang, 627-706, Republic of Korea. E-mail: nanohan@pusan.ac.kr
First published on 3rd August 2012
Abstract
In the present study, bifunctional silica-coated Y2O3:Eu3+,Co2+ spherical-shaped submicron composite particles were successfully prepared using a facile urea-assisted homogenous precipitation method. Synthesized Y2O3:Eu3+,Co2+ phosphor composites showed both ferromagnetic behaviour and strong visible red luminescence emission. The surface of the Y2O3:Eu3+,Co2+ phosphor composites was modified with a thin silica layer to enhance the luminescent properties and obtain higher biocompatibility of the samples. The composite particles with silica coatings resulted in a small decrease in the viability of L-929 fibroblastic cells at concentrations lower than 25 ppm and allowed cell imaging via internalization and wide distribution into the cells. Overall, these synthesized novel SiO2@Y2O3:Eu3+,Co2+ core–shell phosphor composites with both magnetic and luminescent properties have potential biomedical applications, including bioimaging and diagnosis, and can be also potentially used as MRI contrast agents.
1. Introduction
In recent years, there has been considerable interest in the development of novel multifunctional nano/submicron systems for biomedical applications. A combination of magnetic and fluorescent properties in a single nanostructure is expected to open up great prospects in both nano- and biotechnology. Therefore, considerable attention has been paid to the synthesis of nanocomposites with luminescent and magnetic properties, which can be used for real-time in vitro and in vivo imaging, as well as for potential drug delivery. Bifunctional nanocomposites normally consist of luminescent media (quantum dots or organic dyes) and magnetic iron oxide nanoparticles encapsulated in silica. Such a multifunctional system can easily be manipulated by an external magnetic field, which can bring particles to the site of interest, whereas the luminescent media can be used for real-time cellular tracking.1 For example, organic dyes (e.g. Cy5.5, FITC) are widely used as probes to visualize a range of biological processes, and magnetic nanoparticles can act as contrast agents for magnetic resonance imaging (MRI). Therefore, the conjugation of magnetic nanoparticles and dyes will offer bifunctional nanoprobes combining MRI and optical imaging.2 Compared to fluorescent dyes, quantum dots (QDs) do not suffer photobleaching and their emission colors can be tuned from the visible to the NIR region by varying their size or composition.3 QDs have been used extensively as cell labels, tissue imaging agents, sensing labels, etc. Although QDs have a wide range of applications in the biomedical field,4,5 they still exhibit chemical instability, size-dependent fluorescence and strong toxicity.Rare-earth-based phosphors are another class of luminescent material that can revolutionize biological labeling. In contrast to conventional dyes and QDs, a lanthanide-doped phosphor material exhibits superior photostability, with sharp emission lines and good chemical stability. The emission spectrum of phosphor materials, which depends only on the dopant type, shows size independent behavior, which can simplify the fabrication process and reduce the cost of synthesis. Furthermore, the facile urea-assisted homogenous precipitation method, which is a simple and green approach, allows the preparation of almost spherical rare-earth oxides of various sizes.6,7 Yttria has physical properties such as a high melting point (2400 °C), high refractive index (∼1.8), low cut-off phonon energy (380 cm−1), phase stability and a wide transparency range (0.2–8 μm) with a band gap of 5.6 eV. These attributes, as well as the similar chemical properties and ionic radius of rare-earth materials, make Y2O3 material one of the best hosts for rare-earth doping. One well-known dopant for the yttria host is trivalent europium Eu3+, the emission spectrum of which exhibits several main groups of emission lines with a strong dominant red emission (5D0→7F2 transition). Another important activator is divalent cobalt Co2+, which is a type of laser active ion that exhibits more stable magnetic behavior than any other element of iron group metals (Fe, Ni). Therefore, extensive research has been carried out on cobalt-based diluted magnetic semiconductors because of their potential applications in spintronics and optoelectronic devices.8 For example, Sharma et al. reported that Co2+-doped ZnO nanoparticles exhibit strong ferromagnetic properties with a broad green emission centered at 514 nm.9 On the other hand, to the best of the authors' knowledge, no research has been done on a combination of the luminescent properties of dielectric materials, particularly Y2O3:Eu3+ ceramic phosphors with the magnetic properties of cobalt ions in a single entity. In this study, spherical-shaped bifunctional ceramic Y2O3:Eu3+,Co2+ composite particles were synthesized using a facile urea-assisted homogenous precipitation method. The morphology and structural, optical and magnetic properties of the synthesized composite particles were investigated by using X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), Fourier transformed infrared (FT-IR) and photoluminescence (PL) spectroscopy. The magnetic measurements were performed using a quantum design vibrating sample magnetometer (QD-VSM). In the second step, the surface of the Y2O3:Eu3+,Co2+ composite particles was coated with a thin silica shell to achieve a higher biocompatibility and enhance the luminescent properties of the synthesized composites. Pilot studies examining the cytotoxicity and capability of Y2O3:Eu3+,Co2+ composite particles for fluorescent labeling of living cells were also performed using a murine fibroblast cell line (L-929 cells from subcutaneous connective tissue). Murine fibroblast L-929 cell lines were incubated with the samples, and intracellular particle distribution was visualized by confocal microscopy.
2. Experimental details
2.1 Synthesis of Y2O3:Eu3+,Co2+ composite particles
Analytical grade yttrium oxide Y2O3 (99.9%), europium oxide Eu2O3 (99.9%), cobalt(II) oxide CoO (99.9%), nitric acid HNO3 (70%), tetraethyl orthosilicate TEOS (99%), ethanol C2H5OH (99.5%), cyclohexane C6H12 (99.5%), ammonium hydroxide NH4OH (28%) and urea (99–100.5%) were purchased from Sigma-Aldrich and used as received. Spherical Y2O3:Eu3+,Co2+ composite particles were synthesized using urea homogeneous precipitation method according to the reported protocols. 10–12 In a typical synthesis method, an appropriate amount of yttrium oxide and dopant oxides (a total of 0.001 mol for each sample) were diluted with nitric acid and stirred vigorously until they dissolved completely. In each case, the total concentration of Eu3+ was kept constant at 1 mol%, whereas the concentration of Co2+ varied in the range 0–3 mol%. Each dried sample was then mixed with 40 ml deionized water and 0.5 g urea, and stirred vigorously for 30 min to form a clear solution. Sealed beakers with freshly prepared solutions were then placed into an electrical furnace and heated to 90 °C for 1 h. The resulting colloidal solutions were centrifuged at 5000 rpm for 30 min. The precipitates were washed 3 times with ethanol and DI water and dried at 70 °C for 24 h under vacuum. The dried precipitates were then calcined in air at 900 °C for 1 h to produce the oxide particles.2.2 Synthesis of core–shell SiO2@Y2O3:Eu3+,Co2+ composite particles
Silica coated Y2O3:Eu3+,Co2+ composite particles were synthesized using a reverse microemulsion system according to the reported protocol with some minor modifications.13 0.2 g Y2O3:Eu3+,Co2+ composite particles was dispersed in 45 mL cyclohexane and sonicated for 30 min. Subsequently, 5 mL pure ethanol was added. The resulting mixture was stirred and 0.4 mL ammonium hydroxide (28–30%) was added to form a transparent solution. Finally, 0.1 mL tetraethyl orthosilicate (TEOS) was added and the reaction was continued for 4 h with vigorous stirring. The resulting SiO2@Y2O3:Eu3+,Co2+ core–shell composites were collected by centrifugation and washed several times in DI water.2.3 Physical characterization
The structure of the prepared powders was examined by XRD (Bruker D8 Discover) using Cu-Kα radiation (λ = 0.15405 nm) at a 2θ scan range 20–60°. The structural properties were also analyzed by FTIR (Jasco FT-IR6300). The morphology of the particles was characterized by FESEM (Hitachi S-4700) and TEM (JEOL JEM-2100F). Elemental analysis was carried out using EDX (Horiba, 6853-H). The PL measurements were performed with a Hitachi F-7000 spectrophotometer equipped with a 150 W Xenon lamp as an excitation source. The magnetization measurements were performed using a quantum design vibrating sample magnetometer (QD-VSM on a PPMS 6000 machine). All measurements were performed at room temperature.2.4 Cell culture and cytotoxicity assay
A murine fibroblast cell line (L-929 cells from subcutaneous connective tissue) was obtained from the American Type Culture Collection (ATCC CCL-1™, Rockville, MD). The cells were routinely maintained in Dulbecco's modified Eagle's medium (Sigma-Aldrich), supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% antibiotic antimycotic solution (including 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units penicillin, 10 mg streptomycin and 25 μg amphotericin B per ml, Sigma-Aldrich) at 37 °C in 95% humidity and 5% CO2. The number of viable cells was indirectly quantified using highly water-soluble tetrazolium salt [WST-8,2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] (Dojindo Lab., Kumamoto, Japan), reduced to a water-soluble formazan dye by mitochondrial dehydrogenases. The cell viability was found to be directly proportional to the metabolic reaction products obtained in WST-8. Briefly, the WST-8 assay was conducted as follows. L-929 cells were treated with increasing concentrations (0–100 ppm) of Y2O3:Eu3+,Co2+ composite particles with or without silica coating and then incubated with WST-8 for the last 4 h of the culture periods (24 h) at 37 °C in the dark. Parallel sets of wells containing freshly cultured nontreated cells were regarded as negative controls. The absorbance was determined to be 450 nm using an ELISA reader (SpectraMax® 340, Molecular Device Co., Sunnyvale, CA). The relative cell viability was determined as the percentage ratio of the optical densities in the medium (containing the composite particles with or without silica at each concentration) to that of the fresh control medium. The IC50, the concentration (%) inhibiting the growth of cells by 50%, was estimated from the relative cell viability profiles.
000 units penicillin, 10 mg streptomycin and 25 μg amphotericin B per ml, Sigma-Aldrich) at 37 °C in 95% humidity and 5% CO2. The number of viable cells was indirectly quantified using highly water-soluble tetrazolium salt [WST-8,2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] (Dojindo Lab., Kumamoto, Japan), reduced to a water-soluble formazan dye by mitochondrial dehydrogenases. The cell viability was found to be directly proportional to the metabolic reaction products obtained in WST-8. Briefly, the WST-8 assay was conducted as follows. L-929 cells were treated with increasing concentrations (0–100 ppm) of Y2O3:Eu3+,Co2+ composite particles with or without silica coating and then incubated with WST-8 for the last 4 h of the culture periods (24 h) at 37 °C in the dark. Parallel sets of wells containing freshly cultured nontreated cells were regarded as negative controls. The absorbance was determined to be 450 nm using an ELISA reader (SpectraMax® 340, Molecular Device Co., Sunnyvale, CA). The relative cell viability was determined as the percentage ratio of the optical densities in the medium (containing the composite particles with or without silica at each concentration) to that of the fresh control medium. The IC50, the concentration (%) inhibiting the growth of cells by 50%, was estimated from the relative cell viability profiles.
2.5 Fluorescence microscopy
To examine the cellular uptake and distribution of Y2O3:Eu3+,Co2+ composite particles with or without silica into L-929 cells and subsequent cell imaging, the cells were treated with 10 ppm of composite particles for 1 h. After treatment, the cells were fixed with 3.5% paraformaldehyde (Sigma-Aldrich) in 0.1 M phosphate buffer (pH 7) for 5 min at room temperature and immediately observed under a fluorescence microscope (IX81-F72, Olympus Optical, Osaka, Japan). Cell nuclei were counterstained with 10 μmol L−1 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich) before 20 min of observation.2.6 Statistical analysis
All variables were tested in three independent cultures for cytotoxicity assay, which was repeated twice (n = 6). The cytotoxicity results are reported as the mean ± standard deviation (SD), compared with the nontreated controls. A one-way analysis of the variance (ANOVA, SAS Institute Inc, Cary, NC), which was followed by a Tukey honestly significant difference (HSD) test for the multiple comparisons, was used to detect the dose-dependent effects of the composite particles with or without silica on L-929 cells. A P-value <0.05 was considered statistically significant.3. Results and discussion
3.1 Synthesis, structural measurements and morphology
Samples with different Co2+ concentrations were synthesized to allow a comparative study of the physical, magnetic and optical properties. The Eu3+ concentration was kept constant at 1 mol%, whereas the Co2+ concentration was varied in the range 0–3 mol%. A total of four samples (Y2O3:1%Eu3+; Y2O3:1%Eu3+-1%Co2+; Y2O3:1%Eu3+-2%Co2+ and Y2O3:1%Eu3+-3%Co2+) were synthesized and characterized using a range of microscopy and analysis techniques. The morphology and elemental composition of the synthesized Y2O3:Eu3+,Co2+ composites were examined by FESEM and EDX, respectively. Fig. 1(a) shows a FESEM image of pure Y2O3:1%Eu3+ particles without Co2+ ions. The sample consists of relatively spherical particles with a size distribution in the range 70–90 nm. After codoping with Co2+, the spherical morphology of the composite particles still remained unchanged except for a slightly larger particle size (80–110 nm), as shown in Fig. 1(b–d), which was attributed mainly to the effect of codoping with Co2+ ions. The pure Y2O3:1%Eu3+ and Y2O3:1% Eu3+-1% Co2+ particles were used as an example to determine the element composition by EDX. EDX analysis clearly indicated the presence of specific dopant ions (Eu in Y2O3:1%Eu3+, Fig. 1(e)) or (Eu and Co in Y2O3:1%Eu3+-1%Co2+, Fig. 1(f)) in the synthesized composite particles.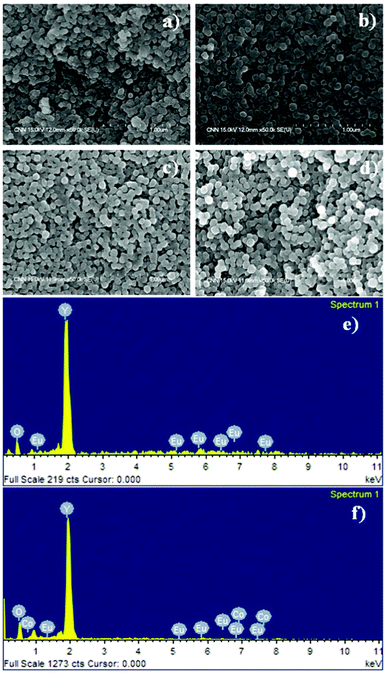 | ||
| Fig. 1 FESEM images of (a) Y2O3:1%Eu3+, (b) Y2O3:1%Eu3+-1%Co2+, (c) Y2O3:1%Eu3+-2%Co2+, (d) Y2O3:1%Eu3+-3%Co2+, and EDX analysis spectra of (e) Y2O3:1%Eu3+, (f) Y2O3:1%Eu3+-1%Co2+ composite particles. | ||
The structural properties of the prepared composite particles were examined by XRD. Fig. 2(a–d) show the XRD patterns of the synthesized composite particles after calcination at 900 °C for 1 h, and the standard peak positions of cubic Y2O3 (JCPDS no. 86-1107) as a reference. No additional peaks from the doped components could be detected due to the low concentration of codopant ions, and all XRD peaks could be indexed to the single-phase body-centered cubic Y2O3 structure with the space group Ia3 (206).14,15 Moreover, all the diffraction peaks of the samples were very strong and sharp, indicating that the final product with high crystallinity can be obtained using this method, which is very important for the luminescence properties of the phosphor composites.10,12 The structure of Y2O3 belongs to c-RE (rare-earths), where six tetrahedral gaps are occupied by O ions and other tetrahedral gaps without O ions. The calculated lattice parameters of the prepared composites for the Y2O3:1%Eu3+, Y2O3:1%Eu3+-1%Co2+, Y2O3:1%Eu3+-2%Co2+ and Y2O3:1%Eu3+-3%Co2+ samples were 10.62, 10.69, 10.73 and 10.76 Å, respectively, which is larger than the value of 10.59 Å reported for a pure Y2O3 phase.16 Since the ionic radius of O2− (0.132 nm) is larger than that of Co2+ (0.072 nm); the lattice expansion suggests that some cobalt ions may probably enter into the remnant tetrahedral gaps without oxygen ions in the body-centered cubic Y2O3 lattice.16 Furthermore, the composite particles obtained using the urea homogeneous precipitation method might allow a homogeneous distribution of codopant ions in the particles, which is also favorable for strong luminescence properties.6
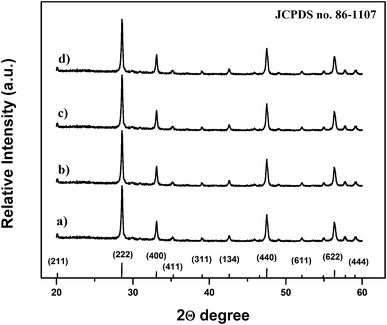 | ||
| Fig. 2 X-Ray diffraction patterns of (a) Y2O3:1%Eu3+, (b) Y2O3:1%Eu3+-1%Co2+, (c) Y2O3:1%Eu3+-2%Co2+ and (d) Y2O3:1%Eu3+-3%Co2+ composite particles. | ||
Fig. 3 shows the excitation (a) and emission (b) spectra of the synthesized particles. The samples exhibited strong red emission under ultraviolet UV (λexc = 255 nm) light irradiation, and the spectral properties are well-known typical Y2O3:Eu3+ phosphors. In the excitation spectrum (Fig. 3(a)) monitored by the Eu3+ 5D0→7F2 hypersensitive transition at 612 nm, the broad band with a maximum at 258 nm originates from the oxygen-to-europium charge-transfer band (CTB), with some weak peaks in the longer wavelength region assigned to the f–f transitions of Eu3+ ions. 6,11 Upon excitation to CTB of the Eu3+ at 255 nm, the emission spectrum (Fig. 3(b)) of the synthesized composite particles consists of the characteristic transition lines between Eu3+ levels. The location and their assignments are also labeled.
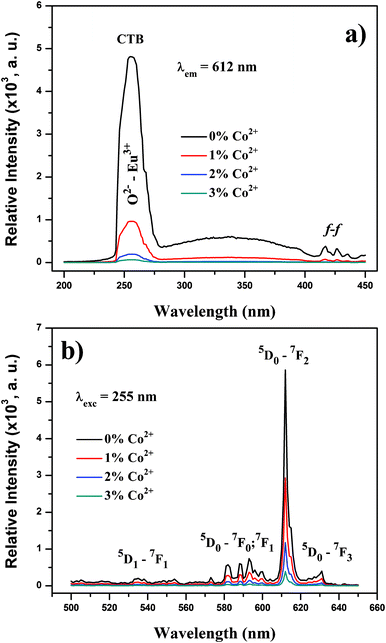 | ||
| Fig. 3 (a) PL excitation and (b) PL emission spectra of synthesized composite particles. | ||
The emission spectrum exhibits five groups of emission lines, which were assigned to the 5D1→7F1 and 5D0→7Fj (j = 0, 1, 2, 3) transitions of Eu3+. The emission spectrum is dominated by the red 5D0→7F2 (612 nm) transition, which is an electric-dipole allowed transition.17,18 Codoping with trace amounts of Co2+ does not change the Eu3+ peak positions, but strongly affects the intensity of these peaks. The luminescence intensity of the Eu3+ peaks deteriorates with increasing amounts of codoped Co2+ ions. The same situation was also observed with the O2−–Eu3+ CTB. The absolute QY values of Y2O3:1%Eu3+, Y2O3:1%Eu3+-1%Co2+, Y2O3:1%Eu3+-2%Co2+ and Y2O3:1%Eu3+-3%Co2+ are measured to be 13.3%, 5.4%, 2.1% and 0.7%, respectively. Therefore, it is reasonable to assume that an increased Co2+ content can give rise to deeper levels in the forbidden band gap, which act as non-radiative recombination centers. Thereafter, the excitation energy absorbed by the luminescent centers is transferred to these deep levels without emitting radiation.
In this study we examined the magnetic properties of Y2O3:1%Eu3+-x%Co2+ (x = 0, 1, 2 and 3) composite particles using a QD-VSM. Fig. 4(a) and (b) present the room-temperature magnetization data of the samples. With the incorporation of Co2+ ions into the Y2O3:1%Eu3+ structure, composite particles began showing a distinct ferromagnetic behavior (hysteresis with the coercive field Hc of 63 ± 8.1 Oe at 300 K) compared to the pure Y2O3:1%Eu3+, which was found to be diamagnetic. As the doping concentration increases, the strength of magnetization also increases, which appears to be needed to increase the magnetic properties of composites. On the other hand, as demonstrated previously, the increased Co2+ content can quench the luminescence intensity. Furthermore, as the XRD measurements (Fig. 2) did not reveal the peaks and presence of cobalt oxides in any form of valence, the observed ferromagnetic properties of Y2O3:1%Eu3+-x%Co2+ composites also confirmed the incorporation of Co2+ ions into the Y2O3:1%Eu3+ structure.
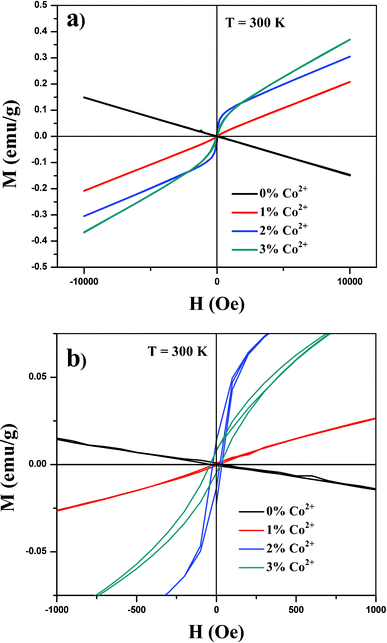 | ||
| Fig. 4 (a) Magnetic hysteresis loop at 300 K of synthesized composite particles and (b) magnified portion of the curves. | ||
The Y2O3:1%Eu3+-2%Co2+ sample, which showed both good magnetic and optical properties, was used in further experiments. The surface chemistry, dose and composition of the particles are critical parameters that affect the particle–cell interactions and cytotoxicity. The Y2O3:1%Eu3+-2%Co2+ composite particles were coated with a thin silica-shell layer, with the main objective being to develop bifunctional composite particles with low toxicity.
There are several reasons for choosing silica as a coating for composite particles. First, a silica coating can reduce any potential toxic effects of the bare composite particles. Second, the silica shell is relatively inert and can be surface-functionalized easily. This also helps prevent particle aggregation and increases their stability in solution. Finally, a silica coating can enhance the luminescence properties and prevent interaction between the surfaces of phosphor materials and OH groups (known as luminescence killers).19 During the experiment, the composite particles were redispersed in cyclohexane for 30 min before coating with a thin silica-shell (see experimental section). After the experiment, the core–shell composites were collected by centrifugation and dried. Fig. 5(a–d) show, respectively, the TEM images of the as-coated Y2O3:1%Eu3+-2%Co2+, FESEM images of the core–shell SiO2@Y2O3:1%Eu3+-2%Co2+, the size distribution pattern of the core–shell SiO2@Y2O3:1%Eu3+-2%Co2+, and the EDX spectrum. After coating with a thin silica shell layer, the resulting core–shell SiO2@Y2O3:1%Eu3+-2%Co2+ structure consists of well-separated spherical particles with a mean size of ∼120 nm (Fig. 5(a) and (b)). The core–shell structure can be observed clearly in Fig. 5(a), due to the different electron penetrability of the cores and shells. The cores are black rough spheres, whereas the shells have a grey color with a mean thickness of ∼15 nm. EDX analysis confirmed the presence of Y, O, Co and Eu (from the Y2O3:1%Eu3+-2%Co2+ core) and Si (from the SiO2 shell), as shown in Fig. 5(d). It should be mentioned that the SiO2 shell layers on the surface of Y2O3:1%Eu3+-2%Co2+ are amorphous.
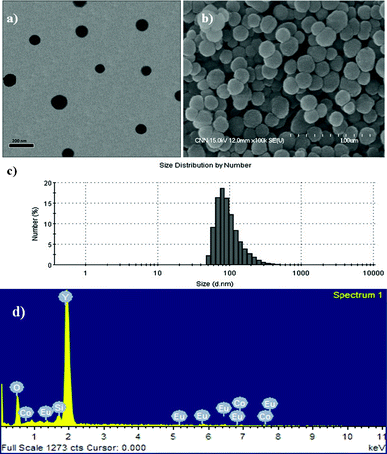 | ||
| Fig. 5 (a) TEM, (b) FESEM, (c) size measurements and (d) EDX analysis of core–shell SiO2@Y2O3:1%Eu3+-2%Co2+ composite particles. | ||
Fig. 6 shows the FT-IR spectra of the pure Y2O3:1%Eu3+-2%Co2+ and SiO2@Y2O3:1%Eu3+-2%Co2+ composite particles. Both samples exhibit the characteristic absorption peaks of Y–O bonds (∼560 cm−1), which appear in the host material.10 Furthermore, both samples exhibit angular deformation of water molecules (band at around 1660 cm−1) and stretch vibrations of the OH group (band at around 3600 cm−1). The characteristic Si–O–Si bond (1109 cm−1) for amorphous SiO2 was only clearly observed in the silica coated sample, which further confirms the formation of an amorphous silica coating on the surface of the Y2O3:1%Eu3+-2%Co2+ composite particles.20
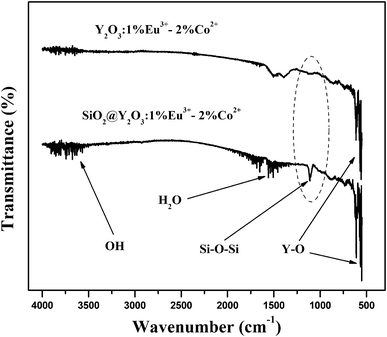 | ||
| Fig. 6 FT-IR spectra of pure Y2O3:1%Eu3+-2%Co2+ and core–shell SiO2@Y2O3:1%Eu3+-2%Co2+ composite particles. | ||
3.2 Cytotoxicity profiles
To provide the comprehensive results about the potential applications of synthesized composite particles for non-invasive imaging purposes, it is essential to measure the cytotoxicity of the synthesized particles. As shown in Fig. 7, the cytotoxicity profiles of composite particles were determined in L-929 fibroblastic cells using a WST-8 assay. The cells exposed to increasing concentrations (0–100 ppm) of composite particles for 24 h showed a noticeable dose-dependent decrease in their relative cell viability. The composite particles without a SiO2 coating began to induce a significant (P < 0.05) decrease in cell viability from 3.13 ppm. On the contrary, the composite particles with a SiO2 coating resulted in no significant decrease in cell viability at relatively lower concentrations (<6.25 ppm) and slightly decreased cell viability even at 25 ppm. The IC50 value of the particles without a SiO2 coating was calculated to be approximately 63 ppm, and they caused more than 55% loss of cell viability at 100 ppm. Considering the in vitro cytotoxicity only, it can be concluded that silica-coated core–shell SiO2@Y2O3:1%Eu3+-2%Co2+ composite particles can be safely applied for bio-imaging including live-cell imaging and MRI at concentrations <10 ppm.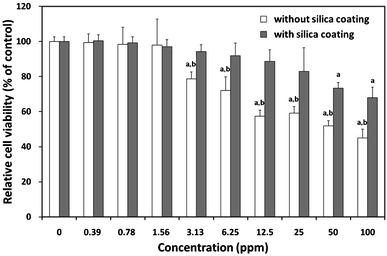 | ||
| Fig. 7 Effect of composite particles with or without silica coating on mitochondrial activity. Relative cell viability of L-929 cells exposed for 24 h to increasing concentrations (0–100 ppm) of composite particles with or without SiO2 coating was evaluated using the WST-8 assay. P < 0.05 vs. nontreated control (labelled a), P < 0.05 vs. cells treated with silica-coated particles (labelled b). | ||
On the other hand, the cytotoxicity against cells exposed to particles should be also determined by other viability end-point measurements, because a WST-8 assay is based only on the activity of mitochondrial dehydrogenases. A recent study demonstrated that the cellular response of human foreskin fibroblasts was different for the different morphologies of Y2O3, with spherical particles exhibiting no cytotoxicity, rod-like particles increasing cell proliferation, and platelet particles being markedly cytotoxic.21 Once a metal-containing particle has penetrated the cells, metal ions can leach from the particle and generate reactive oxygen species in the cell interior leading to oxidative stress to cells—in what is called a “Trojan horse” mechanism.22
3.3 Cellular uptake
In order to investigate the cellular incorporation of Y2O3:Eu3+,Co2+ composite particles with and without silica and compare their potentials for cell imaging, L-929 cells treated with 10 ppm composite particles for 1 h were observed by fluorescence microscopy. As shown in Fig. 8, the cells grew with normal fibroblast-like morphologies after co-labeling with the particles and DAPI for cell nuclei counterstaining.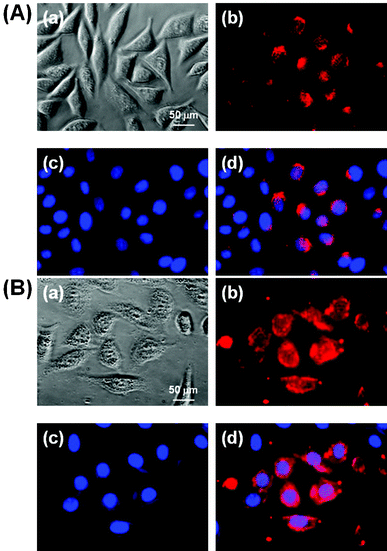 | ||
| Fig. 8 Fluorescence micrographs (×400) of L-929 cells treated with 10 ppm composite particles without silica coating (A) or with silica coating (B) for 1 h, followed by cell nuclei counterstaining with 10 μmol L−1 DAPI. (a) Phase contrast image of the cells co-labeled with the particles and DAPI. (b) and (c) fluorescence images of the cells collected at (b) λexc = 255 nm (red from the particles) and (c) λexc = 350 nm (blue, from DAPI), respectively. (d) Merged image of (b) and (c). All photographs shown in this figure are representative of six independent experiments with similar results. | ||
The fluorescence images demonstrated that the particles without a silica coating were clearly adsorbed onto the membrane and internalized into the cytoplasm of the cells, particularly with relatively weak red (Fig. 8(A)) luminescence. In contrast, the particles with a silica coating fluoresced more strongly, since the SiO2 shell layer can protect the phosphors from luminescence killers like water (Fig. 8(B)). This result suggests that the particles with a SiO2 coating can make cell imaging, tracking and targeting possible through internalization and wide distribution inside the cells, including the cytoplasm and membrane, but excluding the nucleus. A recent study reported that non-functionalized NaYF4:Er3+,Yb3+ NPs incorporated into HeLa cells by direct endocytosis were redistributed inside the cells as the incubation time was increased.23 The mechanisms for the cellular uptake of nanoparticles are very complex and considered as endocytotic pathways such as phagocytosis, pinocytosis, nonspecific endocytosis and receptor-mediated endocytosis.24,25 Although the further detailed study for the cellular uptake mechanism was not examined here, previous seminal studies have shown that larger particles (100–200 nm) would be incorporated into the cells more easily by endocytosis than smaller ones (∼10 nm), which are generally subject to pinocytosis.26,27 Therefore, the composite particles are considered to permeate into the cell membrane by nonspecific endocytosis, since the particles are about 100 nm in diameter and not covered with specific ligands for receptors on the cell membrane.
Conclusions
Highly uniform, bifunctional silica-coated, core–shell SiO2@Y2O3:1%Eu3+-2%Co2+ ceramic composite particles were prepared for the first time. The synthesized composite particles showed both magnetic and luminescent properties, which originate principally from the codoped Eu3+ and Co2+ ions. The synthesized composite particles showed potential as fluorescent contrast agents in cell imaging. Owing to the presence of magnetic properties, the synthesized composite particles can also be used as potential MRI contrast agents. The presence of a SiO2-shell layer on the surface of the composites is highly favorable, because it can reduce the toxic effect and enhance the luminescent properties of the composite particles. The advantages of the bifunctional composite particles prepared by such a route are the easy availability of a spherical morphology and their wide applicability.Acknowledgements
This work was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science, and Technology (Grant No: 2010-0010575 and 2012010181).References
- S. A. Corr, Y. P. Rakovich and Y. K. Gun'ko., Nanoscale Res. Lett., 2008, 3, 87–104 CrossRef CAS.
- J. Cheon and J. H. Lee, Acc. Chem. Res., 2008, 41, 1630–1640 CrossRef CAS.
- S. T. Selvan, T. T. Y Tan, D. K. Yi and N. R. Jana, Langmuir, 2010, 26(14), 11631–11641 CrossRef CAS.
- G. Liu, M. Swierczewska, S. Lee and X. Chen, Nanotoday, 2010, 5, 524–539 CrossRef CAS.
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS.
- T. S. Atabaev, H. K. Kim and Y. H. Hwang, J. Colloid Interface Sci., 2012, 373, 14–19 CrossRef CAS.
- J. G. Li, X. Li, X. Sun, T. Ikegami and T. Ishigaki, Chem. Mater., 2008, 20, 2274–2281 CrossRef CAS.
- T. Busgen, M. Hilgendorff, S. Irsen, F. Wilhelm, A. Rogalev, D. Goll and M. Giersig, J. Phys. Chem. C, 2008, 112, 2412–2417 Search PubMed.
- P. K. Sharma, R. K. Dutta and A. C. Pandey, J. Colloid Interface Sci., 2010, 345, 149–153 CrossRef CAS.
- T. S. Atabaev, H. H. V. Thi, Y. D. Kim, J. H. Lee, H.-K. Kim and Y.-H. Hwang, J. Phys. Chem. Solids., 2012, 73, 176–181 CAS.
- T. S. Atabaev, H.-H. T. Vu, H.-K. Kim and Y.-H. Hwang, J. Alloys Compd., 2012, 525, 8–13 CrossRef CAS.
- T. S. Atabaev, H.-H. T. Vu, H.-K. Kim and Y.-H. Hwang, J. Korean Phys. Soc., 2012, 60, 244–248 CrossRef CAS.
- Y. Zhang, S. Pan, X. Teng, Y. Luo and G. Li, J. Phys. Chem. C, 2008, 112, 9623–9626 CAS.
- H. Guo and Y. M. Qiao, Opt. Mater., 2009, 31, 583–589 CrossRef CAS.
- F. Zhang, G. B Braun, Y. Shi, Y. Zhang, X. Sun, N. O. Reich, D. Zhao and G. Stucky, J. Am. Chem. Soc., 2010, 132(9), 2850 CrossRef CAS.
- X.G. Liu, J. Du, D. Y. Geng, S. Ma, J. M. Liang, M. Tong and Z. D. Zhang, J. Alloys Compd., 2008, 457, 517–521 CrossRef CAS.
- J. L. Ferrari, A. M. Pires and M. R. Davolos, Mater. Chem. Phys., 2009, 113, 587–590 CrossRef CAS.
- J. L. Ferrari, M. A. Cebim, A. M. Pires, M. A. Couto dos Santos and M. R. Davolos, J. Solid State Chem., 2010, 183, 2110–2115 CrossRef CAS.
- Q. Lü, A. H. Li, F. Y. Guo, L. Sun and L. C. Zhao, Nanotechnology, 2008, 19, 205704 CrossRef.
- Q. Zhang, C. Chen, M. Wang, J. Cai, J. Xu and C. Xia, Nanoscale Res. Lett., 2011, 6, 586 CrossRef.
- T. Andelman, S. Gordonov, G. Busto, P. V. Moghe and R. E. Riman, Nanoscale Res. Lett., 2010, 5, 263–273 CrossRef CAS.
- L. Thrall, Environ Sci. Technol., 2007, 41, 3791–3792 CAS.
- F. Vetrone, R. Naccache, A. Juarranz de la Fuente, F. Sanz-Rodríguez, A. Blazquez-Castro, E. M. Rodriguez, D. Jaque, J. G Solé and J. A. Capobianco, Nanoscale, 2010, 2, 495–498 RSC.
- J. Lee, H. Y. Kim, H. Zhou, S. Hwang, K. Koh, D.-W. Han and J. Lee, J. Mater. Chem., 2011, 21(35), 13316–13326 RSC.
- R. Vácha, F. J. Martinez-Veracoechea and D. Frenkel, Nano Lett., 2011, 11(12), 5391 CrossRef.
- W. Jiang, B. Y. Kim, J. T. Rutka and W. C. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS.
- T. S. Atabaev, J. H. Lee, D.-W. Han, Y. H. Hwang and H. K. Kim, J Biomed Mater Res A., 2012, 100A, 2287–2294 CAS.
| This journal is © The Royal Society of Chemistry 2012 |