Crystallization induced porosity control and photocatalytic activity of ordered mesoporous TiO2
Chang Woo
Kim
a,
Umapada
Pal
b,
Sangji
Park
c,
Young Hwan
Kim
d,
Jinheung
Kim
c and
Young Soo
Kang
*a
aKorea Center for Artificial Photosynthesis, Department of Chemistry, Sogang University, Seoul 121-741, Korea. E-mail: yskang@sogang.ac.kr; Fax: 82 2 701 0967; Tel: 82 2 701 6379
bInstituto de Física, Universidad Autónoma de Puebla, Apdo. Postal J-48, Puebla 72570, Mexico
cDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul, 120-750, Korea
dDepartment of Functional Layers, GMBU e. V., P. O. Box 52 01 65, D-01317 Dresden, Germany
First published on 3rd October 2012
Abstract
The evolution of the porous network in mesoporous TiO2 structures during the calcination induced crystallization process has been studied by transmission electron microscopy, small angle X-ray diffraction and N2 adsorption/desorption techniques. It has been observed that on increasing the calcination temperature, the mesoporous structures undergo phase transition from amorphous to crystalline and their porosity changes from the ordered to the disordered state, generating crystalline fragments or aggregated porous particles. The optimum calcination temperature required for fabricating ordered metal oxide meso-structures using a polymer template has been discussed. The ordered mesoporous TiO2 with a crystalline framework showed prolonged photocatalytic activity and repeated usage as an industrial photodegradation catalyst, advantageous to the commercial nanocrystalline TiO2 Degussa P25. Mechanisms of formation and deformation of mesoporous metal oxide crystalline frameworks are proposed.
Introduction
Since the first report on mesoporous silica derived from layered polysilicate kanemite by Kuroda and co-workers,1 porous materials with a narrow pore size distribution, controlled specific surface area, and ordered pore networks have attracted much attention due to their potential applications in the fields of catalysis, separation, and chemical sensing.2 Use of a structure-directing agent (SDA), first introduced by the researchers of Mobil Company, allowed us to generate mesoporous silica of the M41S family by engineering their pore connectivity and channel networks.3 Based on this approach, many research groups have fabricated various porous materials with uniform porosity and interconnected channels useful as nanocomposites, nano-reactors, effective adsorbents, and drug releasing agents, through various synthesis strategies.3b The SDA approach allows us to fabricate periodic two dimensional meso-silica structures that could be extended to three dimensional networks. Since the fabrication of M41S members using linear alkyl chain surfactants, several SDAs were successfully applied to synthesize mesoporous silica of SBA (Santa Barbara Amorphous series), HMS (Hexagonal Mesoporous Silica), MSU (Michigan State University series), KIT (Korea Advanced Institute of Science and Technology series), and FDU (Fudan University series) families.4Mesoporous silica frameworks have also been utilized to incorporate various crystalline materials into their well-arranged pores to fabricate crystalline mesoporous metal oxides.5 In particular, the crystalline mesoporous transition-metal oxides have great potential for applications as effective catalysts, photocatalysts, and electrode materials, due to their high thermal and mechanical stability, and characteristic catalytic, optical and electronic properties.6 In particular, titanium dioxide (TiO2) is a low-cost, promising semiconducting transition-metal oxide with low toxicity and high stability.7 As the photocatalytic activity of TiO2 is closely related to its crystallinity and specific surface area, in mesoporous form, it is expected to have outstanding photocatalytic activity for the degradation of organic compounds.
Most applications of transition-metal oxide meso-structures are related to framework crystallinity. Although a tremendous effort has been devoted to transform the amorphous meso-structures to crystalline frameworks through several methods, in most cases the final product includes only crystalline fragments or possesses inter-particle spaces.8 Therefore, the synthesis of highly crystalline ordered mesoporous transition-metal oxide structures remained challenging. To obtain highly crystalline and ordered porous metal oxide structures, it is necessary to understand the transition process from their initial amorphous to ‘highly’ crystalline framework during the crystallization process. Until now, several reports on the synthesis of crystalline mesoporous metal oxide structures and the mechanisms of formation of such geometries or morphologies have been proposed.8 However, there exists no reliable description of the detailed process and clear-cut mechanism of the production of those porous frameworks during their crystallization process.
In the present work, the process of phase transition in the mesoporous TiO2 is studied during its calcination process. Variations in pore size, pore-wall thickness and pore ordering during crystallization are monitored. Possible mechanisms of formation and deformation of mesoporous structures are proposed. For better understanding of the mechanisms, the crystallization process of mesoporous SiO2 synthesized through similar methods has been studied. Finally, the photocatalytic behavior of the highly crystalline mesoporous TiO2 in methylene blue degradation is studied and compared with the commercial nanocrystalline TiO2, Degussa P25.
Experimental section
Reagent and chemicals
Titanium(IV) butoxide ((Ti(OC4H9)4), 97.0%, Aldrich) used as a titanium precursor, tri-block copolymer EO106PO70EO106 (Pluronic F127, Sigma Aldrich) used as SDA, methylene blue (MB) used as a dye, ethyl alcohol ((C2H5OH), 94.0%, Samchun Chemicals), and hydrochloric acid (35∼37%, Samchun Chemicals) used as a solvent and a pH controlling agent, respectively, were used as received, without further purification. Ultrahigh purity deionized water (>18 MΩ, Millipore) was used throughout the experiment.Synthesis of mesoporous TiO2
For preparing Ti-sol, 2.7 mL (0.38 M) titanium(IV) butoxide was dissolved and stabilized in 2.9 mL HCl solution, under vigorous magnetic stirring for 3 h. The prepared yellowish Ti-sol was then added to the Pluronic F-127 solution (0.001 M in ethanol) under vigorous magnetic stirring. The stirring process was continued for 12 h until the mixture become colorless and transparent. The resulting mixture was transferred to glass petri-dishes and the solvent was allowed to evaporate under atmospheric conditions. After a week, the white films formed over the petri-dishes were removed and calcinated for 4 h using a 1 °C min−1 heating ramp of a muffle furnace. The crystallization process was monitored by calcinating the samples at different temperatures i.e. 260 °C, 315 °C, 350 °C, 400 °C, 500 °C, and 700 °C.Photocatalytic measurement
The photocatalytic activity of mesoporous TiO2 was monitored by photodegradation of MB at room temperature, and compared with that of commercial titania, Degussa P25. Photocatalytic degradation was carried out in a 50 mL glass vial. About 30 mL aqueous slurry containing 15 mg mesoporous TiO2/Degussa P25 and MB (1.0 × 10−4 M) was aged in the dark for 30 min for the dye to be adsorbed onto the surface of TiO2. A 300 W Xe lamp (ARC research sce MODEL 66984, USA) with a cut-on 299 nm filter was used as a light source. The UV degradation of MB as a function of time was monitored by a UV/VIS (Model: Jasco V-600 series) spectrophotometer.Characterizations
Thermogravimetric analysis (TGA) of the samples was carried out using a Thermal Advantage, TGA-2050 analyzer from 50 to 600 °C under nitrogen or air with a heating rate of 10 °C min−1. For transmission electron microscopic (TEM) observations, the samples were prepared by placing one drop of the colloidal solution onto a carbon coated copper grid of 200 mesh size and drying for a few minutes. The porosity of the samples was evaluated using their TEM micrographs. To determine the crystallinity and structure of the synthesized samples, a Rigaku D'Max 2200 V (Cu-Kα radiation, λ = 1.5406 Å) wide angle X-ray diffraction system, and a Rigaku D'Max 2500 18 K small angle X-ray scattering system were utilized. Nitrogen adsorption/desorption isotherms of the samples were measured at 77 K with an automated QUADRASORB ‘SI’ analyzer of Quantachrome Instruments. Prior to their adsorption/desorption measurement; the samples were degassed in a vacuum at 300 °C for 10 h.Results and discussion
The formation of micelles of each block segment in commercial tri-block copolymers, especially of the Pluronic series in selective solvents, has been widely demonstrated by several researchers.4c,9 The interaction of inorganic species with tri-block copolymer micelles can be understood from the schematic diagram presented in Fig. 1 associated with meso-silica formation. It has been demonstrated that the micelle structures of PEO-b-PPO-b-PEO in a hydrophilic solvent consist of individual micelles, in which hydrophilic polymer segments remain outside of the hydrophobic polymer block core. After the formation of polymer micelles in a selective solvent, incorporated inorganic species such as Si and Ti interact with the oxygen of PEO polymer segments through hydrogen bonding. At iso-electric conditions of the inorganic species, the micelles can form a stable self-assembly.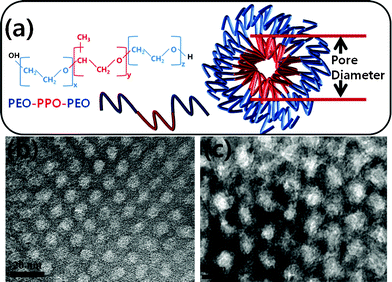 | ||
| Fig. 1 Schematic presentation of the formation of SDA polymer micelles (a), hexagonally close- packed porous silica (b), and titania (c) meso-structures prepared using SDA micelles and subsequent calcinations at 600 and 350 °C, respectively. | ||
It is well-known that low temperature sol–gel methods produce amorphous solids, as the formed inorganic molecules cannot get sufficient thermal energy to arrange their atoms at specific positions on the solid lattice.5e,10 Therefore, a posterior thermal treatment is necessary to convert the amorphous structures to their crystalline phase, as reported by Kondo and Domen.10 Herein, we must take into account the dissociation temperature (Td) of the polymer template and the crystallization temperature (Tc) of the inorganic species, to identify the adequate temperature for thermal treatment or calcination. As indicated by the TGA traces presented in Fig. 2, pure mesoporous frameworks can be obtained from the SiO2–polymer/SDA and TiO2–polymer composites, only after calcinating them in between 500 and 600 °C. As the temperatures in this range are well below the Tc of silica (∼1200 °C) and above the Td of the polymer template, the formed mesoporous silica frameworks are expected to be in the amorphous phase and free from carbon residue. The obtained results indicate that SiO2 samples with stable mesoporous frameworks can be formed by calcinating the inorganic–polymer composite in between Tc and Td. Fig. 1b and c show typical TEM images of hexagonally close packed mesoporous SiO2 and TiO2 samples obtained after calcinating their polymer composites at 600 and 350 °C, respectively. After obtaining the inorganic–polymer composites by sol–gel process, the mesoporous framework can be obtained by removing the polymer template through calcination at a suitable temperature, determined through the TGA analysis of the polymer and the composite in consideration (Fig. 2).
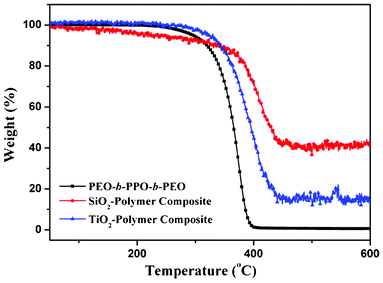
As stated earlier, the calcination temperature for obtaining ordered mesoporous structures could be fixed considering their Td and Tc. However, though the Tc in the case of SiO2 is much higher than Td, for most of the transition-metal oxides the Tc is lower than Td. Therefore, obtaining a crystalline mesoporous framework of transition-metal oxides is practically difficult. Fig. 3 shows typical TEM images of TiO2 frameworks obtained after calcination at 260 °C for 12 h, at 315 °C for 12 h, at 350 °C for 4 h, at 350 °C for 8 h, at 400 °C for 4 h, and at 500 °C for 4 h. As can be seen from the TGA curve of F127 in Fig. 2, the polymer template cannot be removed completely below 350 °C. Therefore, due to the presence of the carbon residue, there appeared to be no clear porous framework for the TiO2 sample calcinated at 260 °C (Fig. 3a), even after 12 h. Even after calcination at 315 °C for 12 h, the porous framework of the sample was not revealed clearly (Fig. 3b). The clear, continuous, and well-ordered porous network of the samples could be revealed when they are calcinated at 350 °C, for 4 h (Fig. 3c and d). Fig. 3c reveals even the lattice image of the porous framework. Fig. 3e presents a typical TEM image of the TiO2 sample calcinated at 350 °C for 8 h. However, a prolonged calcination treatment (Fig. 3e) at 350 °C generated porous fragments of the sample. On prolonged calcination at 350 °C (Fig. 3e) or at higher temperatures (Fig. 3f and g), though the porous frameworks crystallize well (Fig. 3f), due to the brittle nature of the material the pore walls begin to collapse (Fig. 3e). The process is more pronounced for higher calcination temperatures. Finally, on calcinating at about 500 °C, the collapsed structures form aggregated crystalline particles in which the porous network gets destroyed completely (Fig. 3g).
 | ||
| Fig. 3 Typical TEM images of porous TiO2 frameworks obtained after calcination of the inorganic–polymer composite at 260 °C for 12 h (a), at 315 °C for 12 h (b), at 350 °C for 4 h (c, d), at 350 °C for 8 h (e), at 400 °C for 4 h (f), and at 500 °C for 4 h (g). | ||
Fig. 4 shows the wide angle X-ray diffraction (WAXD) patterns of the TiO2 samples calcinated under different conditions. As can be observed, the inorganic–polymer composite starts to crystallize at temperatures as low as 260 °C (Fig. 4a), even when the polymer template does not start to dissociate. On increasing the calcination temperature, along with the dissociation and removal of the polymer template, the amorphous TiO2 gets crystallized in the anatase phase (Fig. 4b, JCPDS # 861157). While the anatase phase was maintained up to 350 °C (Fig. 4c), on increasing the calcination temperature further the rutile phase begin to appear along with the anatase. The fraction of the rutile phase increases on increasing the calcination temperature (Fig. 4d and e). Considering the Debye–Scherrer equation, the crystallized grain size varied from 8 nm to 24 nm as the calcination temperature increased. It is noted that the TEM estimated (Fig. 3g) size of particles formed by collapsing the regular meso-structure calcinated at 500 °C matches well with the grain size estimated from XRD analysis. Considering both the TEM images and XRD results, it is apparent that the crystallites were initially nucleated in the amorphous TiO2 framework derived from the inorganic–polymer composite as the calcination temperature increased. At high calcination temperatures, the crystalline meso-TiO2 framework collapses because of its brittle nature and aggregates to form bigger crystallites at higher temperatures Fig. 4e.
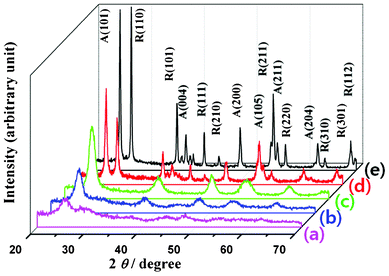 | ||
| Fig. 4 XRD spectra of the TiO2 meso-structures obtained by calcinating the samples at 260 °C for 12 h (a), at 315 °C for 12 h (b), at 350 °C for 4 h (c), at 500 °C for 4 h (d), and at 700 °C for 4 h (e). A and R correspond to the anatase and rutile phases of TiO2, respectively. | ||
To understand the pore formation and crystallinity, the SAXS patterns of the TiO2 frameworks calcinated under different conditions were measured (Fig. 5), which clearly revealed the mesoporous nature of the TiO2 frameworks. For a sample calcinated at 260 °C (Fig. 5a), the SAXS pattern revealed a first order peak corresponding to the inter-planar spacing (d) of 8.18 nm and a0 of 9.45 nm at 2θ = 1.1 (2d = a0√3), which corresponds to the (100) plane (* see Fig. 5) of the hexagonal mesoporous structure. As evident from the TEM image of Fig. 3a, pore walls of the sample annealed at 260 °C were not crystalline due to incomplete removal of the polymer micelle. However, for the samples calcinated at higher temperatures, an interesting feature was observed regarding the change in inter-planar spacing (d) as a function of annealing temperature. For the sample calcinated at 315 °C (12 h), the d value was found to be 10.77 nm, and it was 14.72 nm for the 350 °C (4 h) annealed sample (Fig. 5c). Such a gradual increase in d value (shift of the diffraction peak towards lower angles) with increased pore diameter indicates a gradual removal of carbon residue from the framework on increasing the calcination temperature. The intensity of the diffraction peak increased on increasing the calcination temperature up to 350 °C, due to the removal of residual carbon in the porous network. However, the peak intensity decreased again on calcinating the sample at 400 °C or higher temperatures due to the breakdown of the porous network, leading to the formation of aggregated particles. The d value decreased to 12.9 nm on calcinating the sample at 500 °C, and the SAXS pattern did not show any peak on annealing the sample at 700 °C (Fig. 5e), due to a total collapse of the crystalline framework. The observation is in accordance with the TEM and XRD results.
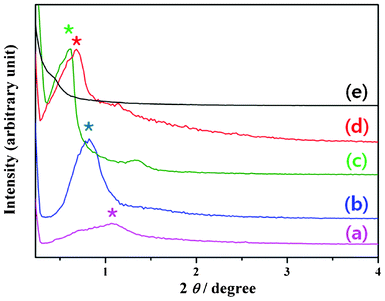 | ||
| Fig. 5 Typical SAXS patterns of the TiO2 meso-structures calcinated at 260 °C for 12 h (a), at 315 °C for 12 h (b), at 350 °C for 4 h (c), at 500 °C for 4 h (d), and at 700 °C for 4 h (e). The (100) plane of each of the samples is indicated by “*”. | ||
Fig. 6 shows the N2 adsorption/desorption isotherms of the mesoporous TiO2 samples calcinated under different conditions. All samples revealed type-IV isotherms according to the IUPAC classification.11 The abrupt increase in N2 adsorption from STP (Standard Temperature and Pressure) to a relative pressure of 0.4 is a typical characteristic of mesoporous materials.11 Considering the five types of hysteresis loops defined by de Boer, which correspond to various pore shapes, the revealed adsorption/desorption isotherms of titania samples can be attributed to their pores.12 For the adsorption/desorption isotherm of the sample calcinated at 350 °C for 4 h (Fig. 6c), the area under the hysteresis loop is much higher than for the other samples. The samples calcinated at lower and higher temperatures contain either residual carbon (Fig. 6a and b), or a collapsed crystalline framework (Fig. 6d). Therefore, the calcination process at 350 °C for 4 h (Fig. 6c) is found to be the most adequate for removing the residual carbon from the porous TiO2 networks without collapsing. The average pore size in the sample calcinated at 350 °C for 4 h, as determined from the BJH method, was 3.31 nm, and the calculated pore wall thickness was about 7.0 nm. The BET surface area and BJH cumulative desorption pore volume for the sample were 184.2 m2 g−1 and 0.78 cc g−1, respectively. It should be noted that the TiO2 samples calcinated at 400 °C and above did not reveal a characteristic adsorption/desorption loop corresponding to mesoporous material, indicating a collapse of mesoporous framework in them, as also revealed from their TEM and XRD analysis.
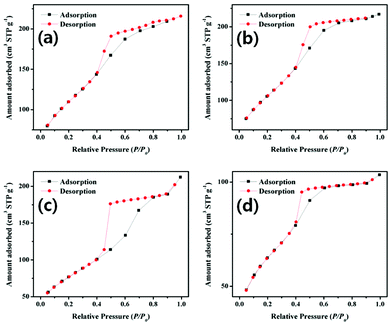 | ||
| Fig. 6 N2 adsorption/desorption isotherms of TiO2 meso-structures calcinated at 260 °C for 12 h (a), at 315 °C for 12 h (b), at 350 °C for 4 h (c), and at 350 °C for 8 h (d). | ||
Fig. 7 presents the absorption spectra of the mesoporous TiO2–MB solution and P25–MB solution exposed to UV light for different intervals. As the UV irradiation time increases, the intensity of the absorption peak (characteristic absorption peak of MB at 660 nm) decreases.12a After 3 h of UV illumination, the absorption peak almost disappears. It is distinctly notable that the degradation rates are different for mesoporous TiO2 and P25.
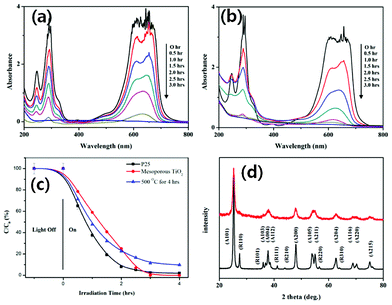 | ||
| Fig. 7 Absorption spectra of MB solution in the presence of mesoporous TiO2 (a) and P25 (b) under UV exposure. (c) Photodegradation rate of mesoporous TiO2 (●) in comparison with the Degussa P25 (■) and the sample calcinated at 500 °C for 4 h (▲). The comparative XRD spectra (d) of mesoporous TiO2 (red) and Degussa P25 (black). | ||
While the decrease of MB absorbance intensity is different at each interval of time for P25 (Fig. 7b), the decrease is almost constant for the case of mesoporous TiO2 (Fig. 7a). The rate of degradation of MB over UV irradiation time is shown in Fig. 7c. It can be noticed that the photodegradation rates for both the commercial Degussa P25 (TiO2 nanoparticles in mixed anatase and rutile phases) and particulate meso-titania (prepared by calcinating at 500 °C for 4 h) samples are higher than mesoporous TiO2 during the first 30 min of reaction. The difference probably comes from the difference in morphology of the samples. As both the Degussa P25 and meso-TiO2 prepared by calcinating at 500 °C are of a particulate nature, their initial photocatalytic activity is high, due to higher collision frequency with MB, according to collision theory in Arrhenius law.12b,13 The number of collisions decreases as the geometrical path increases. However, as the number of collisions over time in laminar shaped mesoporous TiO2 samples is constant, the rate of MB degradation also remains constant. After this initial period, the photodegradation rate of the ordered mesoporous TiO2 increased and 100% degradation could be achieved. As can be noticed, our ordered meso-TiO2 sample is laminar in nature with micrometric dimensions, and has a lower crystallinity than that of Degussa P25 (Fig. 7d). While Degussa P25 contains 80% anatase and 20% rutile, mesoporous TiO2 is purely of anatase phase. As the smaller particle size and higher crystallinity generate higher redox ability in metal oxides due to the larger band gap induced by both factors,13 an initial lower photocatalytic activity of meso-TiO2 is expected. However, due to a higher specific area of mesoporous TiO2, they should provide higher photo active catalytic sites and higher photocatalytic efficiency. Considering these two points, it can be noted that a photo catalyst with a high surface area and micrometric dimensions, as in the case of meso-TiO2, produced a photocatalytic activity very similar to the photocatalytic activity of TiO2 microspheres formed with nanocrystallites reported by Juan Matos’s group and Caue Ribeiro’s group.14
Fig. 8 shows the first-order kinetic plots comparing mesoporous TiO2 and Degussa P25 samples. The photodegradation of the dye and the photocatalytic reaction follow pseudo first-order kinetics, and both can be described by the relation ln(C0/C) = kt, where C and C0 are the actual and initial dye concentrations, and k is the photodegradation rate constant.13,15 Though it seems our mesoporous TiO2 sample has two distinct rate constants for the 0–2.0 h (k = 0.589 h−1) and 2.0–3.0 h (k = 3.436 h−1) time intervals, in comparison with Degussa P25 (k = 1.274 h−1), the average MB photodegradation rate constant for the mesoporous TiO2 (1.372 h−1) is much higher. On the other hand, the TiO2 sample calcinated at 500 °C for 4 h (of particulate nature, losing most of its mesoporous frameworks) revealed similar photocatalytic behavior to that of Degussa P25.
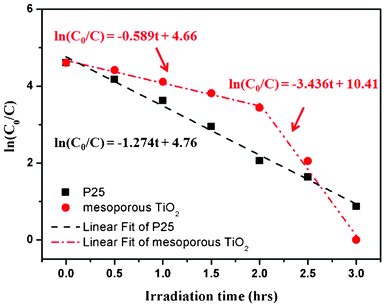 | ||
| Fig. 8 First order kinetics plots of MB degradation by the mesoporous TiO2 (red line) and Degussa P25 (black line) under UV irradiation. | ||
The surface area, pore size and MB photodegradation rate constant values for the ordered mesoporous TiO2 sample (calcinated at 350 °C for 4 h) are presented in Table 1 and compared with the corresponding values of commercial Degussa P25. Even with high photocatalytic activity, the long term stability is a critical issue for the practical applications of these catalysts. While nanocrystalline TiO2 particles like Degussa P25 present high photocatalytic activity due to their large surface to volume ratios, it is very difficult to separate them from the reaction solution. Industrial application of catalysts needs their separation from the reaction solution and recovering for further utilization.13,14 Although centrifugation or filtration could be carried out to separate them; the recovered amount of recyclable catalyst remains limited. On the other hand, mesoporous TiO2 can be easily precipitated within 10 min because of its micrometric particle size, and hence can be directly applied as a recyclable photocatalyst. In Fig. 9, the results of 5 times reutilization of the mesoporous TiO2 catalyst are presented. After 5 cycles of photodegradation, their photocatalytic activity did not decrease. As can be seen, the MB solution became completely colorless after 5 cycles. As the photocatalytic activity of the meso-TiO2 did not deteriorate from its initial photocatalytic activity even after 5 cycles, it is expected to be reused for even larger cycles.
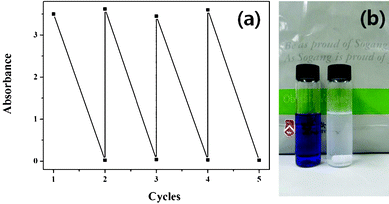 | ||
| Fig. 9 Absorbance values of the MB solution before (upper points) and after (lower points) photodegradation using the same mesoporous TiO2 sample 5 times (a); the photograph in (b) shows how the color of the dye solution changes (from blue to colorless) after each photodegradation cycle of 3 h. | ||
The results obtained from the TEM, SAXS, and N2 adsorption/desorption analyses of the samples indicate that the calcination conditions are extremely important for obtaining crystalline mesoporous metal oxides with ordered pores. Nucleation and growth of the crystallites occur in the amorphous TiO2 framework in the process of removing the polymer during calcination. Due to the fragile nature of the pore walls in mesoporous transition-metal oxides and Td > Tc, calcination at a temperature close to Td of the used SDA causes a collapse of the porous framework, converting the laminar ordered porous structures to crystalline particles. Therefore, while calcinating in ambient air, the calcination temperature must be fixed below the Td of SDA and the process should be performed for a longer time. The effects of calcination conditions on the formation of crystalline ordered meso-structures and aggregated particles of transition-metal oxides are illustrated schematically in Fig. 10.
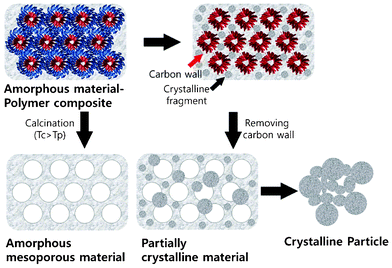 | ||
| Fig. 10 Schematic illustration of the formation of mesoporous metal oxide structures during the crystallization process. Tc and Td correspond to the crystallization temperature of metal oxide and the dissociation temperature of the polymer template used as SDA. | ||
Conclusions
The porosity and stability of mesoporous TiO2 frameworks synthesized using Pluronic F-127 as SDA in the process of their crystallization depend on calcination conditions. While a calcination treatment can be useful both for removing the used SDA and crystallization of the mesoporous structures, it is a trivial process for the metal oxide meso-structures for which Tc (SiO2, 1200 °C) > Td (F-127, 400 °C). For fragile metal oxide meso-structures with Td > Tc (TiO2, 350 °C), a complete removal of the residual carbon arising from SDA is quite difficult as a thermal treatment close to Td would collapse the ordered porous frameworks, forming fragments or aggregated porous particles. Mesoporous TiO2 calcinated at 350 for 4 h exhibited highly ordered pore arrays with a stable crystalline framework. The sample manifests high photocatalytic activity for MB degradation. The high surface area of mesoporous TiO2 structures provide higher photo active catalytic sites, thereby enhancing the photodegradation rate constant in comparison with TiO2 in nanocrystalline form. Moreover, mesoporous TiO2 can be directly applied industrially as a recyclable photocatalyst.Acknowledgements
This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) grant funded from the Ministry of Education, Science and Technology (MEST) of Korea for the Center for Next Generation Dye-sensitized Solar Cells (No. 2012-0000591).References
- (a) T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 63, 988–992 CrossRef CAS; (b) S. Inagaki, Y. Fukushima and K. Kuroda, J. Chem. Soc., Chem. Commun., 1993, 680–682 RSC.
- (a) M. Xu, D. Feng, R. Dai, H. Wu, D. Zhao and G. Zheng, Nanoscale, 2011, 3, 3329–3333 RSC; (b) E. R-Hernndez, A. L-Noriega, D. Arcos, I. I-Barba, O. Terasaki and M. V-Reg, Chem. Mater., 2007, 19, 3455–3463 CrossRef; (c) S. H. Joo, J. Y. Park, C-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS; (d) M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS; (e) A. Popat, S. B. Hartono, F. Stahr, J. Liu, S. Z. Qiao and G. Q. Lu, Nanoscale, 2011, 3, 2801–2818 RSC; (f) D. Bruhwiler, Nanoscale, 2010, 2, 887–892 RSC.
- (a) C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartili and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS; (b) A. Vinu, T. Mori and K. Ariga, Sci. Technol. Adv. Mater., 2006, 7, 753–846 CrossRef CAS; (c) S. Angloher, J. Kecht and T. Bein, Chem. Mater., 2007, 19, 3568–3574 CrossRef CAS; (d) D. Baute, H. Zimmermann, S. Kababya, S. Vega and D. Goldfarb, Chem. Mater., 2005, 17, 3723–3727 CrossRef CAS; (e) M. Bandyopadhyay, A. Birkner, M. W. E. van den Berg, K. V. Klementiev, W. Schmidt, W. Grnert and H. Gies, Chem. Mater., 2005, 17, 3820–3829 CrossRef CAS.
- (a) O. C. Gobin, Y. Wan, D. Zhao, F. Kleitz and S. Kaliaguine, J. Phys. Chem. C, 2007, 111, 3053–3058 CrossRef CAS; (b) S. A. Bagshaw, E. Prouset and T. J. Pinnavaia, Science, 1995, 269, 1242–1244 Search PubMed; (c) D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS; (d) D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS; (e) U. Ciesla and F. Schuth, Microporous Mesoporous Mater., 1999, 27, 131–149 CrossRef CAS; (f) B. Smarsly and M. Antonietti, Eur. J. Inorg. Chem., 2006, 9, 1111–1113 CrossRef; (g) Y. Deng, T. Yu, Y. Wan, Y. Shi, Y. Meng, D. Gu, L. Zhang, Y. Huang, C. Liu, X. Wu and D. Zhao, J. Am. Chem. Soc., 2007, 129, 1690–1697 CrossRef CAS; (h) Y. Deng, J. Liu, C. Liu, D. Gu, Z. Sun, J. Wei, J. Zhang, L. Zhang, B. Tu and D. Zhao, Chem. Mater., 2008, 20, 7281–7286 CrossRef CAS; (i) H-C. Kim, S-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS.
- (a) A. Vinu, J. Dedeeek, V. Murugesan and M. Hartmann, Chem. Mater., 2002, 14, 2433–2435 CrossRef CAS; (b) A. Vinu and M. Hartmann, Chem. Lett., 2004, 33, 588–589 CrossRef CAS; (c) A. Vinu, T. Krithiga, V. Murugesan and M. Hartmann, Adv. Mater., 2004, 16, 1817–1821 CrossRef CAS; (d) H. Sun, J. Han, Y. Ding, W. Li, J. Duan, P. Chen, H. Lou and X. Zheng, Appl. Catal., A, 2010, 390, 26–34 CrossRef CAS; (e) M. Selvaraj and D. W. Park, Appl. Catal., A, 2010, 388, 22–30 CrossRef CAS; (f) Y. Horiuchi, H. Ura, T. Kamegawa, K. Mori and H. Yamashita, Appl. Catal., A, 2010, 387, 95–99 CrossRef CAS.
- (a) R. C. Hayward, B. F. Chmelka and E. J. Kramer, Adv. Mater., 2005, 17, 2591–1595 CrossRef CAS; (b) H. Shibata, T. Ogura, T. Mukai, T. Ohkubo, H. Sakai and M. Abe, J. Am. Chem. Soc., 2005, 127, 16396–16397 CrossRef CAS; (c) F. Schuth, Chem. Mater., 2004, 13, 3184–3195 CrossRef.
- N. Arconada, Y. Castro and A. Duran, Appl. Catal., A, 2010, 385, 101–107 CrossRef CAS.
- (a) M. S. Wong and J. Y. Ying, Chem. Mater., 1998, 10, 2067–2077 CrossRef CAS; (b) P. Liu, I. L. Moudrakovski, J. Liu and A. Sayari, Chem. Mater., 1997, 9, 2513–2520 CrossRef CAS; (c) G. J. A. Soler-Illia, P. C. Angelome, M. C. Fuertes, D. Grosso and C. Boissiere, Nanoscale, 2012, 4, 2549–2566 RSC; (d) J. Y. Cho, W. H. Nam, Y. S. Lim, W-S. Seo, H-H. Park and J. Y. Lee, RSC Adv., 2012, 2, 2449–2453 RSC; (e) T-Y. Ma, H. Li, T-Z. Ren and Z-Y. Yuan, RSC Adv., 2012, 2, 2790–2796 RSC.
- (a) R. Zhang, W. Ding, B. Tu and D. Zhao, Chem. Mater., 2007, 19, 4379–4381 CrossRef CAS; (b) Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–3361 CrossRef CAS; (c) H. Yin, M. Zhen, H. Zhu, M. Chi and S. Dai, Appl. Catal., A, 2010, 386, 147–156 CrossRef CAS.
- J. N. Kondo and K. Domen, Chem. Mater., 2008, 20, 835–847 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotty, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- (a) J. H. Boer, The Structure and Properties of Porous Materials, Butterworths, London, 1958, p. 68 Search PubMed; (b) V. Luca, M. Osborne, D. Sizgek, C. Griffith and P. Z. Arauzo, Chem. Mater., 2006, 18, 6132–6138 CrossRef CAS.
- (a) M. Ye, Q. Zhang, Y. Hu, J. Ge, Z. Lu, L. He, Z. Chen and Y. Yin, Chem.–Eur. J., 2010, 16, 6243–6250 CrossRef CAS; (b) Y. Yu and D. Xu, Appl. Catal., B, 2007, 73, 166–171 CrossRef CAS.
- (a) J. Matos, A. Garcia, L. Zhao and M. M. Titirici, Appl. Catal., A, 2010, 390, 175–182 CrossRef CAS; (b) H. Mao, A. R. Malagutti and C. Ribeiro, Appl. Catal., A, 2010, 382, 284–292 CrossRef.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1–14 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |