DOI:
10.1039/C2RA21333H
(Paper)
RSC Adv., 2012,
2, 11976-11987
Biocompatible amphiphilic hyperbranched nanocapsules with a functional core: Synergistic encapsulation and asynchronous release properties towards multi-guest molecules†
Received
24th May 2012
, Accepted 6th October 2012
First published on 29th October 2012
Abstract
To achieve an encapsulation–release property towards multi-guest molecules, amphiphilic hyperbranched nanocapsules (AHN) consisting of a functional hyperbranched poly(β-cyclodextrin) [HBP(β-CD)] core and a methoxy polyethylene glycol shell was first constructed by click chemistry. The encapsulation–release capacity and corresponding mechanism of AHN towards multi-guest molecules were investigated by fluorescence and UV-vis spectroscopy. The results indicated that the encapsulation–release properties of AHN were pH dependent and can be applied to multi-guest systems. The multi-guest encapsulation capacity of AHN was derived from the synergistic encapsulation phenomenon of different guest molecules and the molecular recognition property of the HBP(β-CD) core. Compared with single-guest systems, AHN displays a sustained release characteristic accompanied by an “asynchronous release phenomenon” in multi-guest release systems. The release rate can also be effectively controlled because of the molecular recognition property of the HBP(β-CD) core. Both in vitro cell apoptosis and in vivo systematic toxicity assays confirmed that AHN possessed good biocompatibility.
Introduction
The encapsulation–release property of amphiphilic polymer nanocapsules is one of the most attractive areas in contemporary supramolecular host–guest chemistry because of its biotechnological and pharmaceutical applications.1–4 In particular, amphiphilic linear block polymers are known for their ability to self-assemble into polymeric micelle-based nanocapsules in selective solvents above the critical micelle concentration (CMC). Thus, these polymers are considered good candidate host molecules for encapsulation–release investigations and applications, because they lead to improved water solubility and stability in physiological environments.2 However, the formed micelles are unstable and easily dissociate into free chains with altered environmental parameters, such as temperature and concentration.5 To overcome this problem, amphiphilic hyperbranched nanocapsules (AHN) have been applied in the encapsulation–release field of hydrophobic compounds, water-soluble dyes, and metal colloids.1,3,4,6,7 AHN consist of a hyperbranched polymer as the core and external substituents or linear polymers as the shell, which has different solubilities from the core. Compared with their linear counterparts, AHN can stably exist as unimolecular micelles8,9 or multimolecular micelles10,11 in an aqueous solution under different environmental conditions because of their unique chemical structure and amphiphilic nature.
Almost all encapsulation–release studies on AHN have focused on the single-guest molecule.1,12 Frey et al.1,13–15 reported on the synthesis and encapsulation properties of an amphiphilic “molecular nanocapsule” consisting of a hyperbranched polyglycerol core, whereas the same activity is not shown by linear polymers. This finding suggests that the encapsulation is related to the architecture of the hyperbranched polymer. Novel unimolecular reversed micelles consisting of AHN exhibit encapsulation and slow-release abilities for hydrophilic molecules. The effects of the shell density and size selectivity on the molecular encapsulation properties were also elucidated.16–19 Wan et al.6,20,21 also reported that the core of AHN can highly selectively encapsulate a guest from a mixture derived from the molecular recognition properties and independent from the sequence of guest addition. Recently, Gong et al.22–25 also studied the synthesis and encapsulation–release properties of amphiphilic hyperbranched block copolymers. They indicated that these polymers have significant potential use in tumor-targeted drug delivery AHN. However, with the development of host–guest chemistry, the demand for the encapsulation–release ability of the host molecule with multi-guest molecules is increasing considerably.3,6,19,26,27 The establishment of double- or multi-drug delivery systems is highly necessary with the development of drug delivery systems.28,29 Different encapsulation–release phenomena for double-guest molecules from AHN have been observed by other researchers3,6,30 and our group.31–33 However, the following questions remain unanswered. First, can the encapsulation–release property of AHN be applied to multi-guest molecules, or is it limited to only one or two guest molecules? Second, if the former is possible, what is the corresponding encapsulation–release mechanism? Third and last, does AHN has sufficient biocompatibility for application in real drug delivery systems?
To address these issues, our idea is to construct core–shell structured AHN by conjugating hydrophilic polyethylene glycol monomethyl ether (mPEG) segments onto the periphery of hydrophobic hyperbranched poly(β-cyclodextrin) [HBP(β-CD)] to examine the encapsulation–release properties in an aqueous solution. According to our previous work,34,35 synthesised HBP(β-CD) possesses two types of molecular cavity from β-CD and the topographical structure of the hyperbranched polymer. This special structure enables the encapsulation of different guest molecules within one polymer system. On the other hand, mPEG chains were used as the shell component to achieve water solubility and biocompatibility, to minimize the immunogenic potential, and to increase the blood circulation half-life of the resulting nanocarriers. This strategy has been proven successful in the development of stealth-liposomes.36,37 Therefore, our study can help extend the application of AHN in supramolecular host–guest chemistry and complex drug delivery systems.
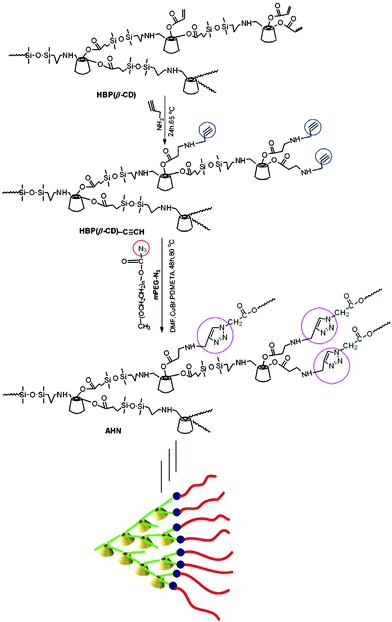
Herein, a novel AHN consisting of an HBP(β-CD) core and an mPEG shell was first synthesised by the copper-catalysed azide–alkyne click reaction (CuAAC) (Scheme 1). The aqueous solution morphology of AHN with a core–shell structure was carefully observed by dynamic light scattering (DLS) and transmission electron microscopy (TEM). Using lonidamine (LND), levofloxacin lactate (LL), and rhodamine B (RhB) as multi-guest molecules, fluorescence spectrophotometry (FL) was used to investigate the encapsulation capacity of AHN based on the synergistic encapsulation phenomenon from different guest molecules and the molecular recognition property of the HBP(β-CD) core. The special “asynchronous release phenomenon” from AHN was confirmed by UV-vis spectroscopy for the first time. The corresponding release kinetics depending on the diffusion-controlled mechanism were also studied in detail. Finally, in vitro cell apoptosis and in vivo systematic toxicity of AHN assays were conducted to determine the biocompatibility.
 |
| | Scheme 1 Synthetic routes for AHN. | |
Results and discussion
Synthesis and morphology of AHN
To obtain AHN, HBP(β-CD) was first synthesised using an ABx-type β-CD monomer according to our previous work34 (the NMR data are listed in the Experimental section). Afterwards, click chemistry was used as a versatile strategy to prepare AHN because of its high specificity, high yield, and near-perfect fidelity.38–40 Consequently, alkynyl groups were incorporated onto the periphery of HBP(β-CD) by the Michael addition reaction of propargylamine with double bonds. Propargylamine was used as the reactant and solvent in this reaction for raising reaction efficiency. mPEG was then sequentially modified using chloracetyl chloride and sodium azide (NaN3) to obtain azido groups. Polymeric structures of HBP(β-CD)–C≡CH and mPEG–N3 were characterised by FTIR and NMR (Fig. S1–S3, ESI†). Lastly, AHN was synthesised by CuAAC between HBP(β-CD)–C≡CH and mPEG–N3. The synthetic routes of AHN are shown in Scheme 1.
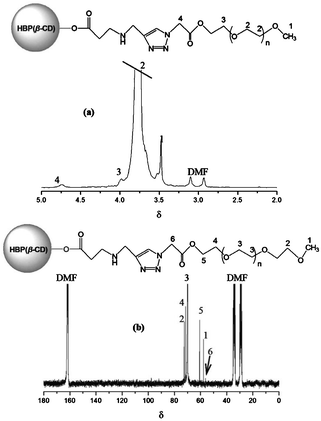
The NMR spectra of AHN are shown in Fig. 1. The chemical shifts at δ 3.76, 3.98 (–O–CH2–), and 3.48 (–O–CH3) are attributed to protons originating from mPEG segments (Fig. 1a). A new additional chemical shift at δ 4.74 (–CH2–COO–) assigned to characteristic protons on clickable azido groups is also observed. The same conclusion can be drawn from the 13C NMR spectrum with carbons at δ 60.72, 70.03, 71.50, 72.55 (–O–CH2–), 57.73 (–O–CH3), and 56.53 (–CH2–COO–) (Fig. 1b). The NMR spectra indicate that AHN have been successfully synthesised by the clickable reaction. Notably, both protons and carbons from the core layer HBP(β-CD) overlap because of the existence of mPEG segments in Fig. 1a. The molecular weights and distribution of AHN determined by SEC/MALLS measurements are 8.24 × 104 (Mn), 2.03 × 105 (Mw), and 2.46, respectively. The arm number of mPEG linked on each HBP(β-CD) is about 29, as determined by the Mn values of AHN, HBP(β-CD)–C≡CH, and mPEG-N3.
The potential applications of a polymer in biomaterials-related fields are known to be generally related to the aqueous media. Therefore, to examine the encapsulation–release property of the synthesised AHN in an aqueous solution, their morphology was first observed by DLS and TEM. The typical intensity diameter distribution of AHN at 25 °C is shown in Fig. 2a. These particles with a diameter of 353 nm are considered as multimolecular micelles formed from the self-assembly of unimolecular micelles, which is driven by the strong hydrophobic–hydrophilic interactions in the inner core and outer shell (Fig. 2).10,11 To examine visually the size and morphology of AHN, the typical TEM image is presented in Fig. 2b. Spherical micelles with a concentration of 10 mg mL−1 are found to be uniformly dispersed in the aqueous solution. These micelles, constructed from AHN, show a black dense core surrounded by a light, thin corona, presenting a typical micellar characteristic. This result indicates that these micelles consist of hydrophobic HBP(β-CD) as the inner core and hydrophilic short mPEG chains as the external shell. Notably, the preparation and formation of these multimolecular micelles are much easier than those of their counterparts. Generally, the micelles can be prepared by adding water to an organic solution of the polymers until the required amount of water for micelle formation is obtained.41 In this work, the stable multimolecular micelles can be formed by direct dissolution of polymer into water. The self-assembly process is fast and does not require any other special conditions. Du and Armes42 also reported that patchy multi-compartment micelles form by the direct dissolution of a primary amine-based poly(ethylene oxide)-block-poly(ε-caprolactone)-block-poly(2-aminoethyl methacrylate) triblock copolymer in water. Therefore, from the preparation method viewpoint, this multimolecular micelle system is more advanced than other micelle systems.
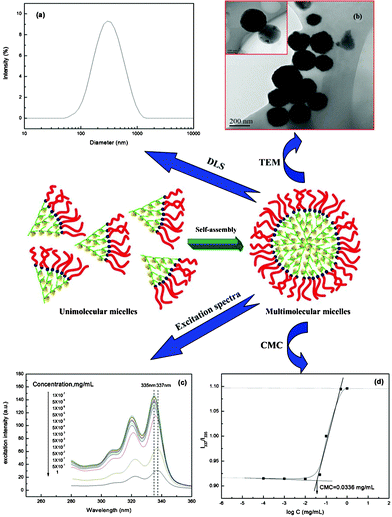 |
| | Fig. 2 Typical intensity diameter distribution of AHN at 25 °C in aqueous solution (a); TEM images of multimolecular micelles of AHN in aqueous solution (b); excitation spectra of pyrene as a function of the AHN concentration in aqueous solution (c); change of intensity ratio (I337/I335) versus logarithm of the AHN concentration (d). | |
To confirm quantitatively the formation of multimolecular micelles, the CMC was estimated by FL using pyrene as a hydrophobic probe. The excitation spectra of pyrene in the AHN solutions with various concentrations are shown in Fig. 2c. A red shift from 335 nm to 337 nm is observed with increased AHN concentration, indicating the formation of micelles.24–26 The intensity ratio (I337/I335) of the pyrene excitation spectra versus the logarithm of the copolymer concentration is shown in Fig. 2d. The CMC is obtained from the intersection of the baseline and tangent of the rapidly rising I337/I335 curve in Fig. 2d. The value is determined as 0.0336 mg mL−1. These results confirm the existence of AHN as multimolecular micelles in an aqueous solution.
Multi-guest encapsulation capacity and mechanism
AHN consisting of a hydrophobic HBP(β-CD) core and a hydrophilic mPEG shell are constructed based on the above discussion. Subsequently, using the anti-cancer drug LND, antibacterial drug LL, and fluorochrome RhB as guest molecules (Scheme 2), we attempt to answer the three questions mentioned in the Introduction. These molecules are selected because their main absorption peaks do not overlap with one another in a mixed solution. Thus, their absorption intensities can be detected by fluorescence emission spectroscopy in an aqueous solution (Fig. S4, ESI†).
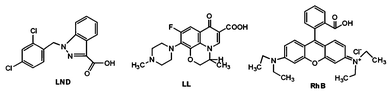 |
| | Scheme 2 Chemical structures of LND, LL and RhB. | |
After selecting the guest molecules, single-guest encapsulation experiments are initially conducted to confirm the ability of AHN to encapsulate steadily with three kinds of guest molecules. Accordingly, Fig. S5 (ESI†) presents evidently decreased peak intensities of LND, LL, and RhB with gradually increased polymer concentration. Then, one-step multi-guest encapsulation experiments are conducted, and the results are shown in Fig. 3 and 4. Fig. 3 shows that with increased polymer concentration, the peak intensities of LND, LL, and RhB solutions obviously decrease. This finding indicates that all molecules are simultaneously encapsulated into AHN at various pH values, i.e. 7.4, 10, and 1.4. Given the strong peak intensities of LL, the peak intensities of LND and RhB are not very obvious at pH 1.4, whereas the corresponding value at the top of the peaks is still observed. After a comparison of peak intensities under different pH values, the AHN are found to be likely to encapsulate LND, LL, and RhB at pH 10, 7.4, and 7.4, respectively. Consequently, the decreased peak intensities at λmax of the guest molecules are most obvious at these pH values with increased polymer concentration in Fig. 4. Thus, the encapsulation property of AHN can be applied to multi-guest molecule systems.
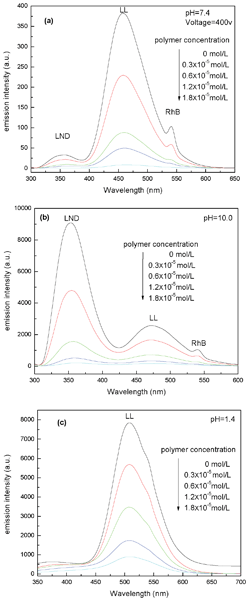 |
| | Fig. 3 Fluorescence emission spectra of LND + LL + RhB solution at pH = 7.4 (a), 10 (b) and 1.4 (c) in the presence of different AHN concentrations (the test voltage of Fig. 3a was set to 400 V because the peak of LL is too strong to be visible at default voltage 700 V, the same below). | |
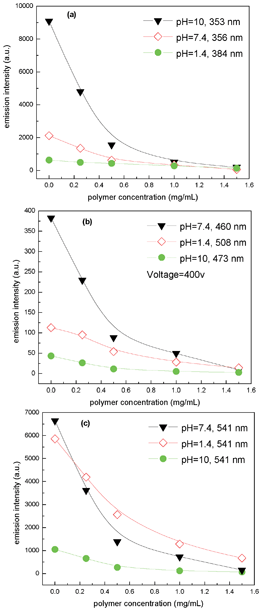 |
| | Fig. 4 Emission intensities at λmax of LND (a), LL (b) and RhB (c) solution as a function of polymer concentrations at different pH values. | |
To further investigate the encapsulation capacity and mechanism of AHN towards multi-guest molecules, gradual multi-guest encapsulation experiments are conducted. One guest molecule is first encapsulated into the AHN solution, and the other one is then gradually added (Fig. 5). Fig. 5a and 5b show that the peak intensities of RhB in the AHN solution gradually decrease with the addition of LND or LL solution. This phenomenon reveals that more RhB molecules are encapsulated into the cavities from the core layer of AHN with the cooperation effect of LND or LL, which has been called a synergistic encapsulation phenomenon.3 In contrast to the RhB+AHN solution system, Fig. 5c and 5d show that the peak intensities of LL+AHN increase after the addition of LND or RhB solution. Thus, the LL molecules encapsulated in AHN are displaced by LND or RhB, and then return to the bulk buffer solution again. Generally, the functional core of the amphiphilic hyperbranched system can highly selectively encapsulate a guest from a mixture derived from the molecular recognition properties, independent from the sequence of guest addition.6,20,21 More recently, a fuzzy recognition mechanism based on the statistical accumulation of elementary host–guest interactions has been proposed by Wan et al.43–45 Thus, LL loses the ability to consume the polymer cavity because of the molecular recognition property of the hyperbranched poly(β-CD) core, as shown in Fig. 5c and 5d. Interestingly, the synergistic encapsulation phenomenon of the guest molecules and the molecular recognition property of the hyperbranched poly(β-CD) core are found to co-exist in the LND+AHN solution system, as shown in Fig. 5e and 5f. The peak intensities of LND decrease after the addition of LL solution, but increase after the addition of RhB solution. An appropriate explanation is that different guest molecules show different synergistic encapsulation abilities based on their molecular structure and polarity.43 In this paper, LL may enhance the encapsulation effect of LND, whereas RhB may weaken it in the process of encapsulation. On the other hand, the molecular recognition property of the core layer is determined by the size/shape match effect between the host and guest, as well as the cooperative effects of hydrophobic, electrostatic, and hydrogen bonding interactions.46,47 During the guest encapsulation process, both guest and host molecules considerably rearrange their conformations to maximize the complementary host/guest interactions based on a fuzzy recognition mechanism.44,45 Therefore, the conformations of the LND+AHN system seems to be more stable with the addition of LL, whereas adding RhB breaks the dynamic balance of the LND+AHN system, leading to the release of LND from the core layer of the AHN. In summary, the gradual double-guest encapsulation phenomenon depends on the balance between the two kinds of interactions in the polymer solution system.
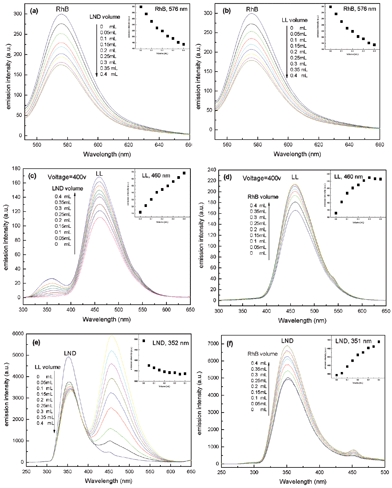 |
| | Fig. 5 Fluorescence emission spectra of RhB+AHN (a,b), LL+AHN (c,d) and LND+AHN (e,f) solutions with gradual adding of LND (a,c), LL (b,e), or RhB (d,f) solutions at pH = 7.4. Inset pictures (IP): emission intensities of RhB+AHN (a,b), LL+AHN (c,d) and LND+AHN (e,f) as a function of added volume of LND (a,c-IP), LL (b,e-IP), or RhB (d,f-IP) solutions. | |
Two guest molecules are simultaneously encapsulated into the AHN solution; the third one is gradually added to this double-guest system (Fig. 6). Obviously, both peak intensities from the double-guest molecules increase after the addition of the third one in Fig. 6a and 6b. For example, Fig. 6a shows that the emission intensities at the λmax values of LL and RhB increase from 111 to 140 and from 23 to 31 with the gradual addition of LND, respectively. By contrast, after the addition of LL solution, the peak intensities of LND decrease but those of RhB increase (Fig. 6c). These results further indicate that the synergistic encapsulation phenomenon and molecular recognition property as the main driving force and interaction mechanism of multi-guest encapsulation still simultaneously exist in the three-guest system. Thus, the encapsulation property of AHN can be applied to multi-guest molecules and presents a certain pH dependence. The encapsulation capacity and interaction mechanism of AHN towards multi-guest molecules are derived from the synergistic encapsulation phenomena of different guest molecules and the molecular recognition property of the hyperbranched poly(β-CD) core.
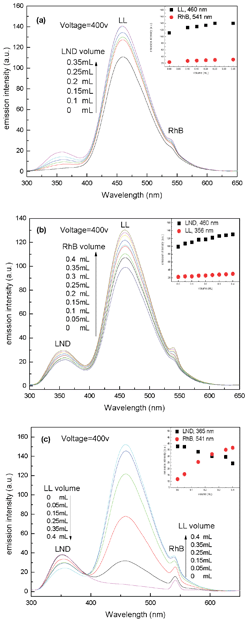 |
| | Fig. 6 Fluorescence emission spectra of LL+RhB+AHN (a), LL+LND+AHN (b), and LND+RhB+AHN (c) solutions with gradual adding of LND (a), RhB (b), or LL (c) solutions at pH = 7.4. Inset picture (IP): emission intensities of LL+RhB+AHN (a), LL+LND+AHN (b), and LND+RhB+AHN (c) as a function of added volume of LND (a-IP), RhB (b-IP), or LL (c-IP) solutions. | |
Multi-guest release behaviour and kinetics
To discuss and further confirm the first and second questions in the Introduction from the viewpoint of release behaviour and mechanism, the release system of AHN towards multi-guest molecules is established under simulated physiological conditions (pH 1.4 and 7.4, T = 37 °C). LND, LL, and RhB are still used to make a comparison with the encapsulation behaviour of AHN. The release profiles at pH 7.4 and 37 °C of the guest molecules are shown in Fig. 7a–7c. Compared with the single-guest system, no significant burst release occurs either from LND, LL, or RhB in the multi-guest system. The release rate of LND, LL, or RhB in the single-guest system is also higher and almost constant, whereas sustained release is observed for the multi-guest system. The difference in the release behaviour can be mainly attributed to the hydrophobic–hydrophobic interactions between the different guest molecules and the hydrophobic core layer.24–26 In other words, these interactions restrain the release of guest molecules from samples, which results in decreased release rates. Thus, the release property of AHN towards multi-guest molecules is completely achieved.
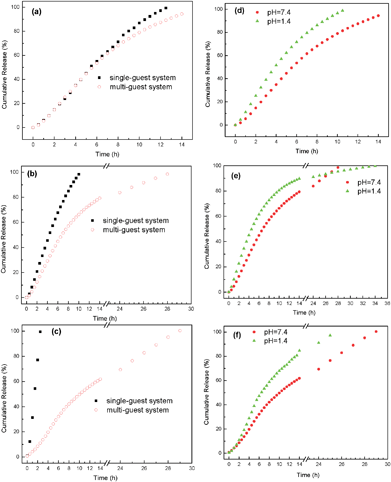 |
| | Fig. 7 Release profiles of LND (a), LL (b) and RhB (c) as the single-guest and multi-guest system from AHN under pH = 7.4 conditions at 37 °C. Release profiles of LND (d), LL (e) and RhB (f) as a multi-guest system from AHN under pH = 1.4 and pH = 7.4 conditions at 37 °C, respectively. | |
On the other hand, the release rates of the three guest molecules in the multi-guest system totally differ at pH 1.4, pH 7.4, and 37 °C (Fig. 7d–7f). Generally, LND shows the fastest release rate, followed by LL and then RhB. For example, the cumulative amounts of LND, LL, and RhB are 94.3%, 79.3%, and 61.8% at 14 h and pH 7.4. This phenomenon indicates that the release rate of the three guest molecules can be adjusted or controlled because of the molecular recognition property of the hyperbranched poly(β-CD) core based on the above discussion. Compared with the synergistic encapsulation phenomenon, the new phenomenon can be termed as an “asynchronous release phenomenon.” To the best of our knowledge, the controlled release methods of AHN mainly focus on breaking the shell layer,16–18 degradation,48 changing the NaCl concentration in solution,30 and cellular membrane transport.49 This paper is the first to report the use of the molecular recognition property of the hyperbranched poly(β-CD) core to control the release rate of different guest molecules. The release profiles of multi-guest molecules from the samples observed in Fig. 7d–7f also clearly reveal that the pH of the solution strongly affects the release behaviour. The cumulative release amount and rate of multi-guest molecule release at pH 1.4 are much higher than those at pH 7.4. For example, the cumulative release amount of LND quickly reaches 96.5% at pH 1.4, whereas the amount is only 78.9% at pH 7.4 within the same 10 h period. This finding can be explained by the protonation of tertiary amino groups in LND, LL, and RhB molecular structures as well as the much faster degradation of micelle cores at extremely low pH.50
To elucidate the release behaviours of the multi-guest system, the release mechanism of AHN towards multi-guest molecules is hereby proposed. Based on the above discussion, Fig. 8 shows the initial burst followed by a controlled release at pH 1.4, pH 7.4, and 37 °C for multi-guest molecules from the samples. This finding indicates that the release may obey a diffusion-controlled mechanism.24,25 This mechanism is further confirmed by a study on the release kinetics using a simple semi-empirical equation (eqn (1)) and a modified equation (eqn (2)) to describe the release behaviour of polymeric micelles.26,51,52
| | 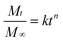 | (1) |
| | 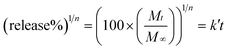 | (2) |
where
Mt and
M∞ are the cumulative amounts of guest molecule released at time
t and infinity, respectively,
k is the release constant,
k′ is the constant proportional to
k, and
n describes the kinetic and release mechanism. For a diffusion–degradation controlled release system,
n for spherical particles is usually between 0.43 and 0.85. When
n is close to 0.43, diffusion called the “Fickian diffusion” is the major driving force. When
n is close to 0.85, the release is mainly controlled by degradation.
51,52 Based on the above deduction about the diffusion-controlled mechanism,
n should be 0.43.
Eqn (2) rearranged from
eqn (1) is more suitable for this release system.
53 After establishing the release kinetic equation, the relationship between the cumulative release amount and release time is presented.
Fig. 8 shows that the cumulative release amount and release time of the multi-guest release system have a good linear correlation within the given range of time (such as 0–14 h) because the
R2 value is equal or close to 0.99.
26 This result indicates that the release mechanism of AHN towards multi-guest molecules is in accordance with “Fickian diffusion”, which is mainly dominated by the diffusion-controlled mechanism. The same release mechanism for the multi-guest release system under different pH conditions can be concluded from
Fig. 8. Therefore, the release property of AHN can be also applied to the multi-guest system with the sustained release characteristic and “asynchronous release phenomenon”, compared with the single-guest system. The release rate can be controlled by the molecular recognition property of the hyperbranched poly(β-CD) core and depends on the pH of the solution. Lastly, the release behaviour is in accordance with the diffusion-controlled mechanism.
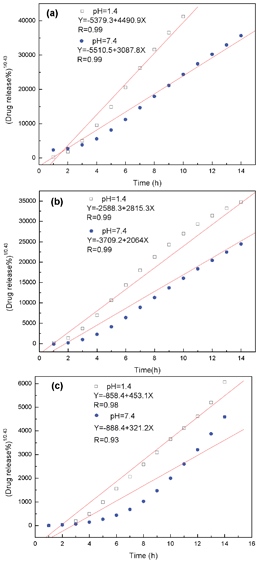 |
| | Fig. 8 Release kinetics of LND (a), LL (b) and RhB (c) as the multi-guest system from AHN under pH = 1.4 and pH = 7.4 conditions at 37 °C. | |
In vitro cell apoptosis
To evaluate the viability of L929 fibroblast cells after contact with the extract liquid of AHN for 24 h, cell apoptosis was first measured by AnnexinV-FITC/PI staining, which can distinguish early apoptotic from late apoptotic/necrotic cells. Fig. 9 shows the typical CLSM images of the Annexin V-FITC/PI staining of the negative control, 100% sample, and positive groups, respectively. Almost no visible early apoptosis and dead cells are found in the negative group, and few of these cells are observed in the 100% sample group. These results indicate that AHN have good biocompatibility with L929 fibroblast cells. To further confirm the cell viability of the different groups, flow cytometry measurements were conducted. The flow cytometry quadrantal diagrams in Fig. 10 show the lower left, lower right, and upper right quadrants denoting the viable, early apoptotic, and late apoptotic/necrotic regions, respectively. No significant visible apoptosis and necrosis are observed in the blank group (100% viability) and negative group (99.7% viability). By contrast, the apoptotic and necrotic rates slightly increase with increased extract liquid concentration of AHN in the test groups (99.4%, 99.5%, 98.4%, and 96.6% viability for 12.5%, 25%, 50%, and 100%, respectively). Obviously, late apoptosis and necrosis are found in the positive group (64.4% viability). In summary, the flow cytometry results are in accordance with the CLSM results, further confirming the biocompatibility of AHN.
![CLSM images of Annexin V-FITC/PI staining of negative control group, 100% sample group and positive group [a: Annexin V-FITC; b: PI; c: Annexin V-FITC/PI (green represents early apoptosis cells, red represents dead cells, both green and red represent late apoptosis cells); d: images of differential interference contrast microscope of the same field].](/image/article/2012/RA/c2ra21333h/c2ra21333h-f9.gif) |
| | Fig. 9 CLSM images of Annexin V-FITC/PI staining of negative control group, 100% sample group and positive group [a: Annexin V-FITC; b: PI; c: Annexin V-FITC/PI (green represents early apoptosis cells, red represents dead cells, both green and red represent late apoptosis cells); d: images of differential interference contrast microscope of the same field]. | |
![Flow cytometry quadrantal diagrams of L929 fibroblast cells after contact with the extract liquid of AHN for 24 h. [Tube 1: blank; tubes 2–5: sample groups 12.5%, 25%, 50%, 100%; tube 6: negative; tube 7: positive. Survival cells are located in Q3, early apoptotic cells in Q4 (Annexin V-FITC positive), late apoptotic/necrotic cells in Q2 (Annexin V-FITC/PI double positive) and necrotic cells/nuclear fragments in Q1 (PI single positive).]](/image/article/2012/RA/c2ra21333h/c2ra21333h-f10.gif) |
| | Fig. 10 Flow cytometry quadrantal diagrams of L929 fibroblast cells after contact with the extract liquid of AHN for 24 h. [Tube 1: blank; tubes 2–5: sample groups 12.5%, 25%, 50%, 100%; tube 6: negative; tube 7: positive. Survival cells are located in Q3, early apoptotic cells in Q4 (Annexin V-FITC positive), late apoptotic/necrotic cells in Q2 (Annexin V-FITC/PI double positive) and necrotic cells/nuclear fragments in Q1 (PI single positive).] | |
In vivo systematic toxicity
The systematic toxicity of AHN in vivo is also evaluated. The AHN samples suspended in saline are injected through the tail vein into ICR mice with a single dose of 40 mg kg−1. Gross anatomy and pathomorphology examinations show that all the organs of the test group, including lung (Fig. 11f), liver (Fig. 11g), kidney (Fig. 11h), heart (Fig. 11i), and spleen (Fig. 11j), undergo no obvious change in morphology compared with those of the control group (Fig. 11a–11e), including the lymphoid follicles or the area of the white pulp. During the entire experimental period, no death, obvious weight change, or unusual behaviour is observed, including vocalisations, laboured breathing, difficulty moving, hunching, or unusual interactions with cage mates. These results further prove that AHN is biocompatible.
![Systematic toxicity of AHN (40 mg kg−1, single dose, i.v.) on healthy ICR mice. [Histological section of (a,f) lung (bronchiole), (b,g) liver (hepatic lobules), (c,h) kidney (glomerule), (d,i) heart (cardiac muscle), and (e,j) spleen (splenocyte) stained with H&E; (a–e) control group, (f–j) test group.]](/image/article/2012/RA/c2ra21333h/c2ra21333h-f11.gif) |
| | Fig. 11 Systematic toxicity of AHN (40 mg kg−1, single dose, i.v.) on healthy ICR mice. [Histological section of (a,f) lung (bronchiole), (b,g) liver (hepatic lobules), (c,h) kidney (glomerule), (d,i) heart (cardiac muscle), and (e,j) spleen (splenocyte) stained with H&E; (a–e) control group, (f–j) test group.] | |
Conclusions
AHN consisting of a functional HBP(β-CD) core and mPEG shells are constructed by click chemistry. The aqueous solution morphology of AHN presents spherical micelles. The encapsulation–release property of AHN can be applied to multi-guest molecules and present a certain pH dependence. The multi-guest encapsulation capacities of AHN towards multi-guest molecules are derived from the synergistic encapsulation phenomenon of different guest molecules and the molecular recognition property of the HBP(β-CD) core. The multi-guest release behaviour of AHN based on the diffusion-controlled mechanism presents the sustained release characteristic and “asynchronous release phenomenon” compared with the single-guest system. The release rate can be controlled by the molecular recognition property of the HBP(β-CD) core. AHN also possesses good biocompatibility both in vitro and in vivo.
Materials and methods
Materials
mPEG (Mn = 1300, degree of polymerization is about 29) was purchased by Liaoyang Kelong Chemical Ltd. (Liaoyang City, China). Propargylamine (more than 98.0% purity) was provided by Shanghai Ruibo Chemical Ltd. (Shanghai City, China). NaN3 was purchased from Xinxing Chemical Reagents Plant (Beijing City, China). (Dimethylamino)-pyridine (DMAP) was provided from Zhejiang Jintan Chemical Plant (Jintan City, China). N,N,N′′,N′′,N′′-pentamethyl diethylenetriamine (PMDETA) was supplied by Yutian Chemical Ltd. (Liyang City, China) and used as received without further purification. LL was purchased from Xi'an University of Architecture & Technology (Xi'an City, Shaanxi Province, China). LND was purchased from Changzhou Ruiming Pharmaceutical Ltd. (more than 98% purity) (Changzhou City, China). RhB was purchased from Wako Pure Chemical Industries Ltd. (Osaka City, Japan). DMF used as the eluent for SEC/MALLS (HPLC grade) was received from Dima Tech (USA). Chloracetyl chloride and other reagents were all purchased from Tianjin Kermel Chemical Reagents Development Center (Tianjin City, China) and dried with 4 Å grade molecular sieves before use.
Characterization
FTIR spectra were obtained on a Nicolet iS10 spectrometer (Nicolet, USA), casting samples into thin films on KBr. Transition mode was used and the wavenumber range was set from 4000 to 400 cm−1 (resolution 4 cm−1, measuring time 16 s). The 1H NMR and 13C NMR spectra were conducted on a Bruker Avance 300 spectrometer (Bruker BioSpin, Switzerland) operating at 300 MHz (1H) in CDCl3 or DMF-d6. The molecular structure parameters of hyperbranched polymers were determined on a DAWN EOS size exclusion chromatography/multiangle laser light scattering (SEC/MALLS) instrument equipped with a viscometer (Wyatt Technology, USA), HPLC grade DMF containing LiCl (0.01 mol L−1) (at 40 °C) was used as the eluent at a flow rate of 0.5 mL min−1. The chromatographic system consisted of a Waters 515 pump, differential refractometer (Optilab rEX) and one column, MZ 103 Å 300 × 8.0 mm. The MALLS detector (DAWN EOS), quasi-elastic light scattering (QELS), and differential viscosity meter (ViscoStar) were placed between the SEC and the refractive index detector. The molecular weight (Mw) and molecular weight distribution (MWD) were determined by SEC/DAWN EOS/Optilab rEX/QELS model. ASTRA software (Version 5.1.3.0) was utilized for the acquisition and analysis of data. The particle size and particle size distribution index (PDI) were measured by Zetasizer Nano-ZS DLS (Malvern Instruments, UK). AHN with a concentration of 10 mg mL−1 were kept at equilibrium at a predetermined temperature for 5 min before data collection and were determined at 25 °C. TEM experiments were carried out on a JEOL JEM-3010 instrument (JEOL, Japan). AHN was firstly dissolved in water (10 mg mL−1), and then the solution was dropped onto copper grids and dried at room temperature before measurement. FL measurements were performed on a Hitachi F-4600 model spectrophotometer at room temperature (Hitachi, Japan). All measurements were performed on air-equilibrated solutions at 25 °C. The emission spectra were recorded from 243 to 600 nm for LND, 288 to 600 nm for LL, 552 to 700 nm for RhB with excitation wavelengths of 223 nm, 268 nm and 532 nm, respectively. The spectra were recorded with a scan rate at 1200 nm min−1. UV measurements were performed on a Shimadzu UV-2550 model spectrometer (Shimadzu, Japan). All solutions withdrawn were kept at 37 °C for 48 h prior to measurements. The spectra were recorded from 200 to 650 nm.
(a) Synthesis of HBP(β-CD).
HBP(β-CD) was synthesized using a ABx-type β-CD monomer according to our previous work.341H NMR (DMF-d6, δ, ppm): 3.34–3.80 (β-CD protons); 4.86 [C(1) –H]; 2.84 (β-CD–CH2–NH–); 2.84 (–NH–CH2–); 1.05 (protons from α addition); 1.75, 2.03 (protons from β addition); −0.06–0.00 [–Si(CH3)2–O–Si(CH3)2–]; 5.90–6.27 (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2). 13C NMR (DMF-d6, δ, ppm): 102.1 (C-1); 82.4 (C-4); 73.1, 72.2, 71.0 (C-3, 5, 2); 70.4 (C-3′); 69.1 (C-5′); 60.5 (C-6); 2.84 (–NH–CH2–); 20.9, 34.2, 45.4, 170.0 (carbons from α addition); 28.8, 30.9, 52.5, 165.5 (carbons from β addition); 0.2–1.2 [–Si(CH3)2–O–Si(CH3)2–]; 162.5 (–COO–); 125.6 (–CHCH2); 128.3 (–CHCH2). SEC/MALLS: Mn = 41
CH2). 13C NMR (DMF-d6, δ, ppm): 102.1 (C-1); 82.4 (C-4); 73.1, 72.2, 71.0 (C-3, 5, 2); 70.4 (C-3′); 69.1 (C-5′); 60.5 (C-6); 2.84 (–NH–CH2–); 20.9, 34.2, 45.4, 170.0 (carbons from α addition); 28.8, 30.9, 52.5, 165.5 (carbons from β addition); 0.2–1.2 [–Si(CH3)2–O–Si(CH3)2–]; 162.5 (–COO–); 125.6 (–CHCH2); 128.3 (–CHCH2). SEC/MALLS: Mn = 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 850, Mw = 83290, MWD = 1.99, ηn = 5.0. Degree of branching (DB) is about 0.55.
850, Mw = 83290, MWD = 1.99, ηn = 5.0. Degree of branching (DB) is about 0.55.
(b) Synthesis of HBP(β-CD)–C≡CH.
HBP(β-CD) (0.5 g) was dissolved in an excess amount of propargylamine (5.5 g, 0.1 mol) under vigorous stirring, and the reaction was kept at 65 °C for 24 h. After the reaction was completed, the excess amount of propargylamine was evaporated under reduced pressure at 60 °C/10−4 bar. Then the precipitate was repeatedly dissolved in DMF, and poured into a large amount of acetone several times. The HBP(β-CD)–C≡CH obtained was dried at 40 °C in a vacuum oven for 3 d. 1H NMR (DMF-d6, δ, ppm): 2.21 (–C≡CH); 2.01 (–NH–, protons from propargylamine); 2.36 (–COO–CH2–). 13C NMR (DMF-d6, δ, ppm): 69.8 (–C≡CH); 76.5 (–C≡CH); 43.3 (–NH–CH2–, carbons from propargylamine); 45.1 (–COO–CH2–NH–); 172.7 (–COO–). SEC/MALLS: Mn = 42270, Mw = 84960, MWD = 2.01.
(c) Synthesis of mPEG–N3.
mPEG (Mn = 1300, 15 g), pyridine (15.8 g, 0.2 mol) and DMAP (10 mg) were completely dissolved in THF (100 mL) under a N2 atmosphere. Chloroacetyl chloride (22.6 g, 0.2 mol) dissolved in THF (30 mL) was added dropwise to the above solution in an ice-water bath. The pyridinium hydrochloride could be found as a white precipitate in the early stages and then it changed to a yellowish solid. After the reaction was kept at ambient temperature for 24 h, the precipitate was filtered out and the crude product was left in the filtrate. The solvent was evaporated under reduced pressure at 50 °C/10−4 bar. The product was obtained, and was redissolved in CH2Cl2 and then precipitated into cold diethyl ether three times. mPEG–Cl obtained was dried at 30 °C in a vacuum oven for 5 d. 1H NMR (CDCl3, δ, ppm): 3.38 (CH3–O–); 3.65 (–O–(CH2)2–); 3.88 (–O–CH2–CH2–COO–); 4.12 (–O–CH2–CH2–COO–); 4.35 (–CH2–Cl). FTIR (KBr): 1748 cm−1 (v CO). Thus, NaN3 (3.9 g, 0.06 mol) was added to a solution of the obtained mPEG–Cl (5 g) in dry DMF (30 mL) under a N2 atmosphere, and the reaction mixture was stirred vigorously at 120 °C for 6 h. After the mixture was cooled to room temperature, 30 mL H2O was added, and then extracted by CH2Cl2 three times. The combined CH2Cl2 solution was washed with cool water three times, condensed and dried with anhydrous Na2SO4 overnight, and then precipitated in cold diethyl ether. The mPEG–N3 obtained was dried at 30 °C in a vacuum oven for 5 d. 1H NMR (DMF-d6, δ, ppm): 3.38 (CH3–O–); 3.65 (–O– (CH2)2–); 3.92 (–O–CH2–CH2–COO–); 2.96 (–CH2–N3). FTIR (KBr): 2106 cm−1 (v C–N3), 1741 cm−1 (v CO).
(d) Synthesis of AHN.
In a flask equipped with magnetic stirring, HBP(β-CD)–C≡CH (0.1 g), mPEG–N3 (1.57 g), CuBr (146 mg, 1 mmol) and PMDETA (163 mg, 1 mmol) were first dissolved in DMF (4 mL). The mixed solution was bubbled with nitrogen gas for 45 min, and sealed under vacuum. The reaction was conducted at 80 °C for 48 h. After the mixture was cooled to room temperature, it was dialyzed in a dialysis bag (molecular weight cut off: 3500) against distilled water for 7 d. It was refreshed at an interval of 12 h. The dialyzed product AHN was lyophilized and kept in glassware under vacuum for further characterization. 1H NMR (DMF-d6, δ, ppm): 3.76, 3.98 (–O–CH2–); 3.48 (–O–CH3); 4.74 (–CH2–COO–). 13C NMR (DMF-d6, δ, ppm): 60.72, 70.03, 71.50, 72.55 (–O–CH2–), 57.73 (–O–CH3), 56.53 (–CH2–COO–). SEC/MALLS: Mn = 8.24 × 104, Mw = 2.03 × 105, MWD = 2.46.
Preparation of multimolecular micelles
Preparations of multimolecular micelles was carried out at room temperature using water as a solvent. AHN (20 mg) was first dissolved in distilled water (20 mL), and then the system was continuously stirred for 12 h. Later, the mixture was subjected to centrifugation for the removal of bigger aggregations.
Determination of CMC
To obtain the CMC of AHN solution, the solid polymer was initially dissolved in water with a certain content of pyrene. Then the polymer solution was diluted step by step to various desired concentrations (from 1 × 10−6 mg mL−1 to 10 mg mL−1), while keeping the pyrene concentration at around 6 × 10−6 mol L−1. The excitation spectra were recorded from 280 to 360 nm with an emission wavelength of 390 nm, and then the I337/I335 ratio values of all spectra were calculated.
Guest molecule encapsulation
(a) Single-guest encapsulation.
Single-guest encapsulation of AHN was measured by FL, using LL (5 × 10−5 mol L−1), LND (5 × 10−5 mol L−1) or RhB (5 × 10−5 mol L−1) as guest molecules in a buffer solution with ionic strength equal to 0.1 mol L−1 and pH = 7.4. Typically, the polymer solution was diluted step by step to various desired concentrations (from 0 mol L−1 to 1.8 × 10−5 mol L−1) using the different guest molecules solution. All solutions were maintained for more than 12 h to ensure the binding equilibrium and then stirred prior to measurement.
(b) One-step multi-guest encapsulation.
One-step multi-guest encapsulation of AHN was measured by FL, using LL (5 × 10−5 mol L−1), LND (5 × 10−5 mol L−1) and RhB (5 × 10−5 mol L−1) as guest molecules in a buffer solution with ionic strength equal to 0.1 mol L−1 and pH = 1.4, 7.4 or 10. Typically, a solution containing LL, LND and RhB was mixed with an equal volume, and then the polymer solution was diluted step by step to various desired concentrations (from 0 mol L−1 to 1.8 × 10−5 mol L−1) using the mixed solution. All solutions were maintained for more than 12 h to ensure the binding equilibrium and then stirred prior to measurement.
(c) Gradual multi-guest encapsulation.
Gradual double-guest encapsulation of AHN was measured by FL, using LL (5 × 10−5 mol L−1), LND (5 × 10−5 mol L−1) and RhB (5 × 10−5 mol L−1) as guest molecules in a buffer solution with ionic strength equal to 0.1 mol L−1 and pH = 7.4. When LL was encapsulated in AHN with the manner of single-guest encapsulation (polymer concentration is 0.6 × 10−5 mol L−1), the solution of LND or RhB (from 0 mL to 0.4 mL) was gradually added into the bottle several times with given volumes each time. Other gradual double-guest encapsulation procedures were similar to the above steps. Then, gradual three-guest encapsulation of AHN was measured as follows. When LL and LND were encapsulated in AHN in the manner of double-guest encapsulation (polymer concentration is 0.5 mg mL−1), the solution of RhB (from 0 mL to 0.4 mL) was gradually added into the bottle several times with given volumes each time. Other gradual three-guest encapsulation procedures were similar to the above steps. All solutions were maintained for about 10 min prior to measurement.
Guest molecules loading
(a) Single-guest loading.
LL, LND, RhB and AHN were dissolved in appropriate THF under vigorous stirring for 12 h, respectively. The weight ratio of LL, LND or RhB to polymer is 6:100. The mixture was subjected to centrifugation for the removal of free guests, and then, the solution containing guest-loaded polymer and the solid consisting of free guests were separated. THF in the solution was volatilized at 40 °C in a vacuum oven for 2 d, and then sample slices of 0.2 mm in thickness consisting of two model guests and polymer were prepared.
(b) Multi-guest loading.
LL, LND, RhB and AHN were dissolved simultaneously in appropriate THF under vigorous stirring for 12 h. The weight ratio of LL, LND, RhB and the polymer is 2:2:2:100. The following procedures were similar to the single-guest loading.
Guest molecules release behaviors
The sample slices were sealed separately using a dialysis bag of 4 cm in length. The dialysis bag was used because these polymers have a significantly higher molecular weight, so that they would stay inside the dialysis bag (molecular weight cut off: 3500), whereas those guests of low molecular weight would readily diffuse out. The bags were immersed in 40.0 ml buffer solution with ionic strength equal to 0.1 mol L−1 and pH = 1.4 or 7.4 at 37 °C. In a certain time interval, 5.0 ml buffer solution was withdrawn and replaced with 5.0 ml of fresh buffer. All release measurements were carried out in triplicate for each sample, and an average value was adopted. The cumulative release was calculated by using eqn (3) as follows.| | 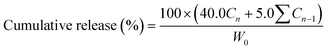 | (3) |
Where W0 (mg) is the weight of the guest in the polymer; Cn (mg ml−1) is the concentration of LL, LND and RhB in buffer solution, which was withdrawn n times, Cn−1 (mg ml−1) is the concentration of LL, LND and RhB in buffer solution, which was withdrawn n−1 times.
In vitro cell apoptosis assays of AHN
Cell apoptosis experiments were performed on flow cytometry and confocal laser scanning microscopy (CLSM). Mouse fibroblast L929 cells (ATCC CCLl, NCTC Clone 929, Clone of Strain L) were first proliferated at 37 °C, 5% CO2 in RPMI 1640 (GIBCO BRL) supplemented with 10% fetal bovine serum. The extract liquid of AHN was diluted by RPMI 1640, 100%, 50%, 25% and 12.5%. Apoptosis analysis of cells was conducted after contact with extract liquid for 24 h. Apoptosis was measured using the annexin V-FITC apoptosis detection kit from Keygentec (Nanjing, China). This method detects externalization of phospholipids normally located on the cytoplasmic surface of the cell membrane via binding with the modified anticoagulant Annexin V. Cells were also stained with propidium iodide (PI) to detect dead cells. Unstained cells were classified as survival, and cells stained for Annexin V only (early apoptosis) and cells stained for both Annexin V and PI (late apoptosis) were combined and called apoptosis, and cells stained for PI only were dead.
CLSM studies: stirred cell suspension was pipetted into a 24-well plate with sterilized microscopic glass with l × 105 cells/well, incubated at 37 °C, 5% CO2 for 4 h. Then 200 μL of the extract liquid (containing 10% FBS) with different dosage was added into each well, and another well adding RPMI 1640 (containing 10% FBS) were used as a blank control. One well adding the extract liquid of polystyrene was used as a negative control, and the other well with 0.5% phenol (ultimate concentration) was used as a positive control. After being cultured at 37 °C, 5% CO2 for 24 h, it was then washed two times with PBS, and 100 μL 1× binding buffer and 2 μL Annexin V-FITC on each microscopic glass were added. The solution was incubated in the dark for 20 min at room temperature, and then 1 μL PI for staining was added for 5 min. It was then washed three times with PBS, and the glasses were tied onto the glass slide, mounted by 90% glycerine diluted with PBS. After that, each glass was observed with CLSM.
For flow cytometry: the stirred cell suspension was pipetted into a 24-well plate with l × 105 cells/well, and incubated at 37 °C, 5% CO2 for 4 h. Then the supernatant was discarded and 200 μL of the extract liquid (containing 10% FBS) with different dosage was added into 4 × 3 parallel wells, and another 3 wells adding RPMI 1640 (containing 10% FBS) were used as blank controls. 3 wells with the extract liquid of polystyrene were used as a negative control, and another 3 wells with 0.5% phenol (ultimate concentration) were used as a positive control. After being cultured at 37 °C, 5% CO2 for 24 h, the L929 cells in each well were digested with 2.5 g L−1 trypsin, washed 2 times with PBS, 300 μL 1× binding buffer was added to suspension cells, and the cell suspension was adjusted for 1 × l09 L−1 concentration. 5 μL Annexin V-FITC was added and the mixture was incubated in the dark for 20 min at room temperature, and then 5 μl PI was added for staining for 5 min before testing. Apoptosis analysis of cells in each group was tested by flow cytometry. The cell viability percentage was calculated by comparing with the blanks (the cell viability percentage of the blanks was represented as 100%).
In vivo systematic toxicity of AHN
ICR mice (supplied by Experimental Animal Center, School of Medicine, Xi'an Jiao Tong University) weighing 20 ± 2 g were kept in an environment complying with NIH guidelines for the care and use of laboratory animals. All animal procedures were performed following protocol approved by the Institutional Animal Care. A total of 20 healthy ICR mice (half male and half female) were equally allocated into two groups and intravenously administrated with (a) test group (40 mg kg−1, AHN was dispersed in saline) and (b) control group (40 mg kg−1, saline) twice daily (8 h interval every time) for a total of 2 d. Body weights and clinic manifestation were observed and recorded carefully throughout the experimental period of 14 d. Tissues recovered from the necropsy were fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) for histological examination using standard techniques. After hematoxylin/eosin staining, the slides were observed and photos were taken using an optical microscope.
Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (No. 21004049), China Postdoctoral Science Foundation for Specialized Funded Project (No. 201003684), Specialized Research Fund for the Doctoral Program of Higher Education of the Ministry of Education (No. 20106102120053), NPU Fundamental Research Foundation (No. JC201270) and Opening Project of Shaanxi Provincial Key Laboratory of Polymer Science and technology for financial support.
References
- S. E. Stiriba, H. Kautz and H. Frey, J. Am. Chem. Soc., 2002, 124, 9698 CrossRef CAS.
- N. Nasongkla, E. Bey, J. Ren, H. Ai, C. Khemtong, J. S. Guthi, S. Chin, A. D. Sherry, D. Boothman and J. Gao, Nano Lett., 2006, 6, 2427 CrossRef CAS.
- C. H. Liu, C. Gao and D. Y. Yan, Macromolecules, 2006, 39, 8102 CrossRef CAS.
- M. Irfan and M. Seiler, Ind. Eng. Chem. Res., 2010, 49, 1169 CrossRef CAS.
- H. J. Liu, Z. Shen, S. E. Stiriba, Y. Chen, W. Q. Zhang and L. H. Wei, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4165 CrossRef CAS.
- D. C. Wan, G. C. Wang, H. T. Pu and M. Jin, Macromolecules, 2009, 42, 6448 CrossRef CAS.
- T. Satoh, Soft Matter, 2009, 5, 1972 RSC.
- C. N. Moorefield and G. R. Newkome, C. R. Chim., 2003, 6, 715 CrossRef CAS.
- G. Chen and Z. Guan, J. Am. Chem. Soc., 2004, 126, 2662 CrossRef CAS.
- H. Y. Hong, Y. Y. Mai, Y. F. Zhou, D. Y. Yan and J. Cui, Macromol. Rapid Commun., 2007, 28, 591 CrossRef CAS.
- J. Mao, P. H. Ni, Y. Y. Mai and D. Y. Yan, Langmuir, 2007, 23, 5127 CrossRef CAS.
- C. M. Paleos, D. Tsiourvas, Z. Sideratou and L. Tziveleka, Curr. Top. Med. Chem., 2008, 8, 1204 CrossRef CAS.
- A. Sunder, M. Kramer, R. Hanselmann, R. Mulhaupt and H. Frey, Angew. Chem., Int. Ed., 1999, 38, 3552 CrossRef CAS.
- M. Q. Slagt, S. E. Stiriba, R. J. M. K. Gebbink, H. Kautz, H. Frey and G. van Koten, Macromolecules, 2002, 35, 5734 CrossRef CAS.
- V. M. Garamus, T. V. Maksimova, H. Kautz, E. Barriau, H. Frey, U. Schlotterbeck, S. Mecking and W. Richtering, Macromolecules, 2004, 37, 8394 CrossRef CAS.
- T. Satoh, M. Tamaki, Y. Kitajyo, T. Maeda, H. Ishihara, T. Imai, H. Kaga and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 406 CrossRef CAS.
- Y. Kitajyo, T. Imai, Y. Sakai, M. Tamaki, H. Tani, K. Takahashi, A. Narumi, H. Kaga, N. Kaneko, T. Satoh and T. Kakuchi, Polymer, 2007, 48, 1237 CrossRef CAS.
- Y. Kitajyo, Y. Nawa, M. Tamaki, H. Tani, K. Takahashi, H. Kaga, T. Satoh and T. Kakuchi, Polymer, 2007, 48, 4683 CrossRef CAS.
- T. Satoh, Y. Kitajyo, R. Sakai, A. Narumi, K. Takahashi, H. Kaga, N. Kaneko and T. Kakuchi, Macromol. Symp., 2009, 279, 145 CrossRef CAS.
- D. C. Wan, H. T. Pu and X. Y. Cai, Macromolecules, 2008, 41, 7787 CrossRef CAS.
- D. C. Wan, J. J. Yuan and H. T. Pu, Macromolecules, 2009, 42, 1533 CrossRef CAS.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Q. Gong, Biomaterials, 2009, 30, 3009 CrossRef CAS.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Q. Gong, Macromol. Biosci., 2009, 9, 515 CrossRef CAS.
- S. Aryal, M. Prabaharana, S. Pilla and S. Q. Gong, Int. J. Biol. Macromol., 2009, 44, 346 CrossRef CAS.
- X. Q. Yang, J. J. Grailer, S. Pilla, D. A. Steeber and S. Q. Gong, Bioconjugate Chem., 2010, 21, 496 CrossRef CAS.
- S. Sengupta, D. A. Eavarone, I. Capila, G. Zhao, N. Watson, T. Kiziltepe and R. Sasisekharan, Nature, 2005, 436, 568 CrossRef CAS.
- Y. Liang, D. C. Wan, X. Y. Cai, M. Jing and H. T. Pu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 681 CrossRef CAS.
- S. Salmaso, A. Semenzato, S. Bersani, P. Matricardi, F. Rossi and P. Caliceti, Int. J. Pharm., 2007, 345, 42 CrossRef CAS.
- H. Y. Chen, Y. Zhao, Y. L. Song and L. Jiang, J. Am. Chem. Soc., 2008, 130, 7800 CrossRef CAS.
- L. A. Tziveleka, C. Kontoyianni, Z. Sideratou, D. Tsiourvas and C. M. Paleos, Macromol. Biosci., 2006, 6, 161 CrossRef CAS.
- W. Tian, X. D. Fan, J. Kong, Y. Y. Liu, T. Liu and Y. Huang, Polymer, 2010, 52, 2556 CrossRef.
- W. Tian, X. D. Fan, J. Kong, Y. Y. Liu, W. H. Zhang, G. W. Cheng and M. Jiang, Macromol. Chem. Phys., 2009, 210, 2107 CrossRef CAS.
- W. Tian, X. D. Fan, T. Liu, Y. Y. Liu, L. Sun, M. Jiang and Y. Huang, Chem J Chin Univ, 2009, 30, 632 CAS.
- W. Tian, X. D. Fan, J. Kong, T. Liu, Y. Y. Liu, Y. Huang, S. J. Wang and G. B. Zhang, Macromolecules, 2009, 42, 640 CrossRef CAS.
- W. Tian, X. Y. Lv, C. G. Mu, W. H. Zhang, J. Kong, Y. Y. Liu and X. D. Fan, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 759 CrossRef CAS.
- Y. Akiyama, Y. Nagasaki and K. Kataoka, Bioconjugate Chem., 2004, 15, 424 CrossRef CAS.
- C. Kontoyianni, Z. L. Sideratou, T. Theodossiou, L. A. Tziveleka, D. Tsiourvas and C. M. Paleos, Macromol. Biosci., 2008, 8, 871 CrossRef CAS.
- M. A. White, J. A. Johnson, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 11356 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS.
- G. W. Cheng, X. D. Fan, W. Tian, Y. Y. Liu and J. Kong, Polym. Int., 2010, 59, 543 CrossRef CAS.
- J. Rodriguez-Hernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691 CrossRef CAS.
- J. Z. Du and S. P. Armes, Soft Matter, 2010, 6, 4851 RSC.
- D. C. Wan, H. T. Pu and M. Jin, Macromolecules, 2010, 43, 3809 CrossRef CAS.
- D. C. Wan, H. T. Pu, M. Jin, G. W. Wang and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2373 CrossRef CAS.
- D. C. Wan, L. Lai, M. Jin and H. T. Pu, Macromol Chem Phys, 2011, 212, 1910 CAS.
- Y. Inoue, T. Hakuhsi, Y. Liu, L. H. Tong, B. J. Shen and D. S. Jin, J. Am. Chem. Soc., 1993, 115, 475 CrossRef CAS.
- Y. Inoue, Y. Liu, L. H. Tong, B. J. Shen and D. S. Jin, J. Am. Chem. Soc., 1993, 115, 10637 CrossRef CAS.
- J. H. Zou, W. F. Shi, F. Wang and B. Jun, Macromol. Biosci., 2005, 5, 662 CrossRef CAS.
- S. Chen, X. Z. Zhang, S. X. Cheng, R. X. Zhuo and Z. W. Gu, Biomacromolecules, 2008, 9, 2578 CrossRef CAS.
- K. Kataoka, T. Matsumoto, M. Yokoyama, T. Okano, Y. Sakurai, S. Fukushima, K. Okamoto and G. S .Kwon, J. Controlled Release, 2000, 64, 143 CrossRef CAS.
- J. Siepmann and N. A. Peppas, Adv. Drug Delivery Rev., 2001, 48, 139 CrossRef CAS.
- S. Zuleger and B. C. Lippold, Int. J. Pharm., 2001, 217, 139 CrossRef CAS.
- D. Lemoine, C. Francois, F. Kedzierewicz, V. Preat, M. Hoffman and P. Maincent, Biomaterials, 1996, 17, 2191 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21333h |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.