Electrochemical deposition and pseudocapacitive behavior in urea-based quasi-ionic liquid electrolytes studied with X-ray absorption spectra†
Ming-Jay
Deng
*a,
Jin-Ming
Chen
*a,
Kueih-Tzu
Lu
*a,
Cheng-Chia
Wang
b,
Jyh-Fu
Lee
a and
Jeng-Kuei
Chang
c
aNational Synchrotron Radiation Research Center, Hsinchu, Taiwan. E-mail: deng.mj@nsrrc.org.tw; jmchen@nsrrc.org.tw; ktlu@nsrrc.org.tw; Fax: +886-3-578-3813; Tel: +886-3-578-0281
bDepartment of Applied Science, National Hsinchu University of Education, Hsinchu, Taiwan
cInstitute of Materials Science and Engineering, National Central University, Taoyuan, Taiwan
First published on 10th September 2012
Abstract
Co(II), Sn(II), V(III), Ni(II), Mn(II), and Ru(III) species in urea-based quasi-ILs were investigated using cyclic voltammograms and X-ray absorption near-edge structure (XANES) spectra. The excellent pseudocapacitive characteristics of MnO2–graphene nanocomposite electrodes in ILs, associated with the large variation of Mn oxidation state during charge and discharge cycles, were also elucidated using in situ XANES spectra.
Room temperature ionic liquids (RTILs) are extensively employed in diverse devices for energy storage, such as lithium secondary batteries and electric double-layer capacitors because of their unique physical and chemical properties; these include being nonvolatile, nonflammable, low toxicity, stable thermally and electrochemically, highly conductive, having wide electrochemical windows and a varied combination of cations and anions.1–8 The wide electrochemical window of an IL enables the electrodeposition of active elements that are difficult to reduce in aqueous solutions.2 Some organic solvents, including propylene carbonate (PC) and diethyl carbonate, have already found practical application in contemporary electronic devices, such as in mobile telephones and portable computers, but all these organic solvents have potential drawbacks according to safety concerns about their flammable and volatile nature that can lead to explosions or incendiary accidents. Furthermore, lithium anodes with a high theoretical discharge capacity (3860 mA h g−1) are unusable in such solvents because of dendritic lithium deposition during the charging cycle. From a point of view of practical application, it is of great interest to seek ILs with satisfactory solubility, modest cost, small viscosity and wide potential windows. RTIL (or molten-salt) electrolytes based on urea-based mixtures have been extensively investigated since 2003.9–14 Especially, the complex systems based on urea–ChCl (choline chloride10,14) or urea–EMIC (1-ethyl-3-methylimidazolium chloride9) were considered to be promising electrolytes for electrochemical behavior because of their facile, environmentally friendly preparation without organic solvents, large ionic conductivity, satisfactory solubility and modest cost, which are practical advantages in diverse applications of electrodeposition9,10,14 and electrochemical double-layer capacitors.11,13 Such essential characteristics facilitate the preparation of transition-metal solutions for electrodeposition. One serious drawback of these urea–salt melts, particularly for electrochemical applications, is their large viscosity and unsatisfactory potential window. Finding a convenient preparation of a urea-based mixture with a wide potential window for practical electrochemical applications is accordingly highly desirable. To investigate the metal deposition, the coordination chemistry of metal ions and the charge storage mechanism of metal-oxide-based pseudocapacitors in various ILs, which can provide insight and control parameters for the preparation of metallic electrodeposition and high performance of pseudocapacitors, synchrotron-based X-ray spectroscopic techniques are highly effective. However, spectroscopic evidence of detailed electrodeposition chemistry and charge storage mechanism of metal-oxide-based pseudocapacitors in ILs is relatively less explored.
In this work, we describe facile and environmentally friendly preparations of urea-based quasi-ILs composed of an organic compound, urea, and a lithium salt, such as LiPF6 and LiTFSI, and devote attention to their electrochemical deposition and pseudocapacitive characteristics on MnO2–graphene, elucidated with synchrotron-based X-ray spectroscopic techniques (Fig. SI1†). The urea–LiPF6 quasi-IL exhibits wide electrochemical windows, ∼5.0 V, and ionic conductivity up to 2.8 mS cm−1 at 301 K. Their superior physicochemical properties enable them to serve as practical electrolytes in electrochemical pseudocapacitors. Using cyclic voltammograms (CV) and X-ray absorption near-edge-structure (XANES) spectra at the transition-metal K edge, we investigated the electrodeposition of representative metals, including Co(II), Sn(II), V(III), Ni(II), Mn(II), and Ru(III) species, in these quasi-IL. We elucidated the charge storage mechanism of MnO2–graphene nanocomposite (MGN) electrodes in the quasi-IL during charge and discharge cycles with in situ XANES spectra.
Binary mixtures of the corresponding urea salt and lithium salt LiPF6 and LiTFSI were readily developed as quasi-ILs to have high quality without organic solvents (see ESI for details). The compositions of urea–LiPF6 (molar ratio 3.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and urea–LiTFSI (molar ratio 3.2:1) quasi-ILs were prepared from LiPF6, LiTFSI and urea in this study. Urea–LiPF6 and urea–LiTFSI11,12 quasi-ILs were dried in vacuum at 383 K for 12 h before measurement with an air-tight dilatometer, a viscometer and a meter for electrical conductivity at 301 K. (Fig. SI2† shows the structures of urea–LiPF6 and urea–LiTFSI.11,12) The density of urea–LiPF6 quasi-IL was 1.26 g mL−1 and of urea–LiTFSI quasi-IL 1.38 g mL−1. The viscosities of urea–LiPF6 (103 cP) and urea–LiTFSI (239 cP) were smaller than that of urea–ChCl (450 cP).10 The conductivity of quasi-ILs is regarded as a crucial factor for their applications as electrolytes in diverse energy-storage devices, because their performance is governed mainly by the viscosity, density and formula mass of the IL.2 An IL with a small viscosity typically displays a large conductivity; for instance, the conductivities of urea–LiPF6 (2.8 mS cm−1) and urea–LiTFSI (2.2 mS cm−1) are greater than that of urea–ChCl (0.9 mS cm−1).10
:1) and urea–LiTFSI (molar ratio 3.2:1) quasi-ILs were prepared from LiPF6, LiTFSI and urea in this study. Urea–LiPF6 and urea–LiTFSI11,12 quasi-ILs were dried in vacuum at 383 K for 12 h before measurement with an air-tight dilatometer, a viscometer and a meter for electrical conductivity at 301 K. (Fig. SI2† shows the structures of urea–LiPF6 and urea–LiTFSI.11,12) The density of urea–LiPF6 quasi-IL was 1.26 g mL−1 and of urea–LiTFSI quasi-IL 1.38 g mL−1. The viscosities of urea–LiPF6 (103 cP) and urea–LiTFSI (239 cP) were smaller than that of urea–ChCl (450 cP).10 The conductivity of quasi-ILs is regarded as a crucial factor for their applications as electrolytes in diverse energy-storage devices, because their performance is governed mainly by the viscosity, density and formula mass of the IL.2 An IL with a small viscosity typically displays a large conductivity; for instance, the conductivities of urea–LiPF6 (2.8 mS cm−1) and urea–LiTFSI (2.2 mS cm−1) are greater than that of urea–ChCl (0.9 mS cm−1).10
Fig. 1(a,b) display CV curves of neat urea–LiPF6 and urea–LiTFSI quasi-IL, respectively, recorded with a Pt electrode. The urea–LiTFSI quasi-IL exhibits a cathodic potential limit near −3.1 V and an anodic potential limit near +2.2 V (vs. Fc/Fc+), whereas the urea–LiPF6 quasi-IL exhibits a cathodic potential limit near −3.2 V and an anodic potential limit near +2.0 V (vs Fc/Fc+). The cathodic and anodic limiting potentials are estimated by the potential at a current density of ±0.1 mA cm−2. (Fig. SI3† shows the enlarged CV.) The cathodic potential limit in the urea–LiPF6 quasi-IL, which is determined by the reduction of the cations, is comparable to that in the BMP-TFSI IL.15 The cathodic potential limit is recognized to be crucial for the electrodeposition of metals.
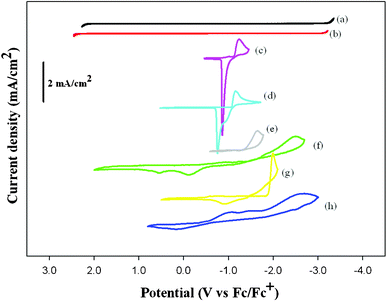 | ||
| Fig. 1 Cyclic voltammograms of (a) blank urea–LiPF6, (b) blank urea–LiTFSI, (c) 100 mM Co(II)/urea–LiTFSI solution, (d) 100 mM Sn(II)/urea–LiTFSI solution and (e) 30 mM Ni(II)/urea–LiTFSI solution at Pt electrode, respectively. (f) 100 mM Mn(II)/urea–LiPF6 solution, (g) 40 mM Ru(III)/urea–LiPF6 solution, (h) 100 mM V(III)/urea–LiPF6 solution at Pt electrode, respectively. Scan rate is 50 mV s−1 at 323 K. | ||
The electrochemical deposition of metals such as Co, Sn and Ni was investigated with the urea–LiTFSI quasi-IL. CoCl2 and SnCl2 dissolved more readily in the urea–LiTFSI quasi-IL than in the BMP-TFSI IL. Solutions containing Co(II) and Sn(II) (each 100 mM), were accordingly prepared on direct addition of these metal chlorides into the IL, but for Ni(II) (concentration greater than 30 mM) in urea–LiTFSI quasi-IL, NiCl2 became insoluble. The CV curves of Co(II), Sn(II), Ni(II) recorded on a platinum electrode in the urea–LiTFSI quasi-IL at 323 K are characteristic for metal electrodeposition in that redox waves were observed in each CV, as shown in Fig. 1(c)–1(e). Before and after the electrodeposition, the electroplating solution and the electrodeposited metal for Co, Sn and Ni were characterized with K-edge XANES spectra, shown in Fig. 2(a)–2(c), respectively. (Fig. SI4† shows the schematic illustration of the electrochemical cell.) These XANES results in Fig. 2 clearly indicate that CoCl2, SnCl2 and NiCl2 dissolved in the urea–LiTFSI quasi-IL to produce transition-metal ions with a larger absorption edge of the K-edge spectra; the corresponding metal layers were deposited on a Pt substrate surface with a smaller absorption edge of the K-edge spectra. The electrodeposition of Ni metal is notably difficult because of either insolubility or great difficulty of dissolution in common BF4−, PF6− and TFSI-based counterparts IL.14 The electrodeposition of Ni metal from an IL was achieved only in a Lewis-acidic IL such as chloroaluminate and chlorozincate, other than the urea–ChCl eutectic10,14 and the DCA-based IL.16 This capability to electrodeposit Ni metal in the urea–LiTFSI quasi-IL is thus of great significance.
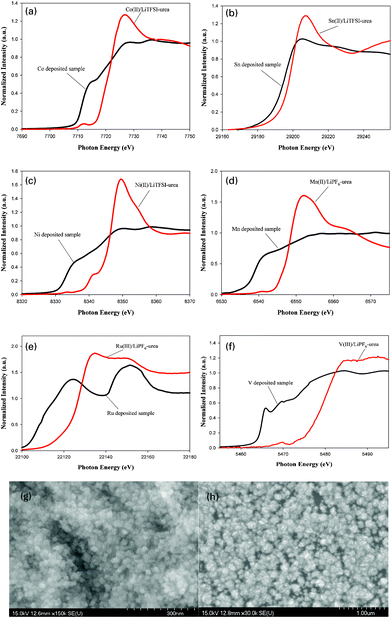 | ||
| Fig. 2 (a)–(f) K-edge XANES spectra of electroplating solutions and the corresponding electrodeposited samples for Co, Sn, Ni, Mn, Ru and V, respectively. SEM micrographs of the surface as deposited of (g) Mn and (h) Ru, obtained from the urea–LiPF6 quasi-IL. | ||
Because the urea–LiPF6 quasi-IL shows greater solubility than the urea–LiTFSI quasi-IL, it was chosen for electrodeposition of the metals Mn, Ru and V. The CV curves of these metal ions recorded with a Pt electrode at 323 K exhibit characteristics typical for metal electrodeposition.
The CV shown in Fig. 1(f) indicates that MnCl2 (100 mM) dissolves readily in the urea–LiPF6 quasi-IL. The electrode was scanned initially from the open circuit potential to the negative direction at a sweep rate 50 mV s−1. A broad reductive wave of Mn(II) is coupled with two small stripping peaks, and a current loop indicative of a nucleation process is observed. The area of the oxidation wave is much smaller than that of the reduction wave, indicating that the metallic Mn electrodeposits produced during the cathodic scan were not reoxidized completely. A similar behavior was observed for the electrodeposition of metallic Mn in other ILs.2,15,16
The CV curve of Ru(III) (40 mM) in urea–LiPF6 quasi-IL exhibits a prominent reduction wave and a smaller oxidation wave, as shown in Fig. 1(g). Electrolysis of Ru(III) at −2.0 V resulted in the deposition of metallic Ru. The CV curve of Ru(III) in the urea–LiPF6 quasi-IL is more straightforward than that in the BMI-TFSI IL.17
The CV curve of V(III) (100 mM) in urea–LiPF6 quasi-IL exhibits two shoulders visible in the cathodic sweep around −1.0 V, followed by a clear reduction peak at −2.5 V, as shown in Fig. 1(h). The reduction of V(III) to V(0) might occur in one step or through an intermediate oxidation state, as indicated by the shoulder at −1.0 V in the reduction. A stripping peak is observed at 0 V in the anodic scan.
Fig. 2(d)–2(f) show XANES spectra at the transition-metal K edge recorded for the electroplating solution and electrodeposited samples for Mn, Ru and V, respectively. The significant downward energy shift of XANES spectra in Fig. 2 indicates clearly that MnCl2, RuCl3 and VCl3 dissolved in the urea–LiPF6 quasi-IL to produce transition-metal ions with an increased rising edge for absorption; the corresponding metal layers were deposited on the substrate surface with a decreased rising edge for absorption. SEM micrographs of representative deposition nanoparticles of Mn and Ru are shown in Fig. 2(g) and 2(h), respectively. (Fig. SI5† shows the other SEM images.) Further spectra such as XANES and EXAFS measurements in situ are required to produce detailed information on the coordination chemistry of metal ions with these quasi-ILs.
To evaluate the pseudocapacitive performance of the low-cost RTIL (or molten salt) as an electrolyte is of great interest. As an electrochemical device for charge storage near 300 K, the preparation of RTIL electrolytes in pseudocapacitors is restricted by other factors. For example, LiTFSI was one of the most reactive salts, causing corrosion of the aluminum in battery electrolytes.18 Thus, this salt must be used with caution and thoroughly considered before implementation, and the investigation of novel electrolytes matching the requirement in developing electrochemical charge-storage devices is still a hot topic of extensive research. Considering their popular use in lithium-ion batteries, the high oxidation capability of the ClO4− anion and the higher melting-point temperature of electrolyte with the B(C2O4)2− anion will restrict the use of ClO4− and B(C2O4)2− anions in the electrolyte of electrochemical charge-storage devices. Considering the popular use in lithium-ion batteries and the intrinsic properties of LiPF6, especially for its large conductivity and stable voltage, and its low side effects on the environment, we developed a simple, cheap and ecologically compatible preparation of the urea–LiPF6 quasi-IL more readily than the well known BMP-TFSI IL. This urea–LiPF6 quasi-IL exhibits an electrochemical window ∼5.0 V. The urea–LiPF6 quasi-IL contains Li(urea)n+ cations13 as working ions in the IL electrolyte, but neither protons nor alkali cations (Li+, Na+, K+) exist in the BMP-TFSI IL. The crucial point for the effective pseudocapacitance performances of ILs is whether the constituent ions can be incorporated into the electrode, leading oxide materials to proceed via a redox reaction. For an investigation of the pseudocapacitive behavior of MnO2–graphene nanocomposite (MGN) electrodes, the urea–LiPF6 quasi-IL is accordingly a candidate likely superior to the BMP-TFSI IL. A TEM image in Fig. 3(a) reveals that Mn oxide particles were uniformly distributed and adhered well on graphene. Fig. 3(b) shows the CV of MGN electrodes, prepared from MnO2 deposited from KMnO4 solution (details are provided in the ESI),19,20 in the urea–LiPF6 and BMP-TFSI at a potential sweep rate of 5 mV s−1. The CV curve with rectangular shapes and mirror-image symmetric characteristics in urea–LiPF6 exhibits a capacitive behavior with a potential range approximately 2.5 V from −1.5 to +1.0 V, which is nearly triple that observed in traditional aqueous electrolytes. As noted, the CV response current remained almost constant during the forward and backward scans but directly changed its flow direction when the potential was reversed. These results indicate clearly that high capacitance and reversibility of the oxide electrode are more readily achievable in urea–LiPF6 than in BMP-TFSI.
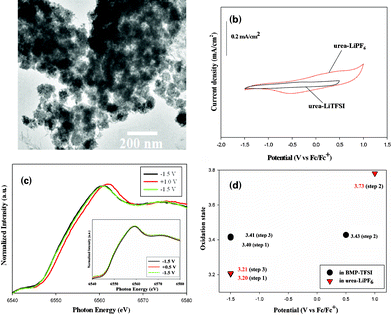 | ||
| Fig. 3 (a) TEM image of the MGN electrode. (b) CV of the MGN electrode recorded in the urea–LiPF6 and BMP-TFSI, respectively. (c) In situ Mn K-edge XANES spectra of the MnO2 electrode measured at the applied potentials in sequence −1.5 V, +1.0 V and then back to −1.5 V in the urea–LiPF6 quasi-IL. The inset shows the MGN electrode measured at the applied potentials in sequence −1.5 V, +0.5 V, and then back to −1.5 V in BMP-TFSI IL. (d) Variation of the oxidation state of Mn ions in the MGN electrode with respect to the applied potential in ILs. | ||
The specific capacitance (C) of Mn oxide is calculated with this equation,
| C = Qm/ΔV | (1) |
Fig. 3(c) shows the in situ Mn K-edge XANES spectra of the MGN electrode measured under three applied potentials in a sequence −1.5 V, then +1.0 V and finally −1.5 V in urea–LiPF6 and −1.5 V, then +0.5 V and finally −1.5 V in BMP-TFSI, respectively. The adsorption rising edge of the Mn K-edge spectra of the MGN electrode shifted toward higher energy with increasing applied potential and returned nearly to the initial position when the potential was switched back, as clearly recognized in urea–LiPF6 but not in BMP-TFSI (as shown in the inset in Fig. 3(c)). The energy (E0) at the absorption threshold, which is obtained from the first inflection point on the absorption edge, is known to be linearly correlated to the valence state of the transition metal in materials.26,27 Based on the E0 derived from X-ray absorption spectra in Fig. 3(b), the average Mn valance of the MGN electrode in the IL was determined in a sequence, as shown in Fig. 3(d). It is noteworthy that the variation of Mn valence between −1.5 V and +1.0 V electrodes in the urea–LiPF6 quasi-IL is much greater than that in the BMP-TFSI IL. The results evidently confirm that a continuous and reversible faradic redox transition of MnO2 indeed occurred in the urea–LiPF6 quasi-IL, contributing the significant capacitance observed in Fig. 3(b). It indicates that Li(urea)n+ cations as the primary working species reversibly insert into and exit from the tunnels between the [MnO6] octahedral subunits28 (not only absorb on the surface of MnO2 films) and cause the large variations of the oxidation state of Mn. It accordingly confirms that the cations were the working species that compensate the Mn valency changes upon charging and discharging in the quasi-IL electrolyte. The search for more suitable working ions in IL electrolytes that can further improve the charge storage properties of metal-oxide-based pseudocapacitors is in progress.
In summary, our results reveal that urea-based quasi-ILs, prepared by a facile and environmentally friendly procedure, are practical as potential electrolytes for electrodeposition and supercapacitors, as characterized by synchrotron-based X-ray spectroscopic techniques. The urea–LiPF6 quasi-IL exhibits an electrochemical window of ∼5.0 V and greater ionic conductivity than those in the well-known ILs. The potential electrolytes of these quasi-ILs for electrochemical application are indicated with the prominent electrodeposition of representative metals including Co(II), Sn(II), V(III), Ni(II), Mn(II), Ru(III) species. In addition, the charge storage mechanism of MGN electrode in these quasi-ILs during charge and discharge cycles was elucidated using in situ XANES spectra. A continuous and reversible Faradic redox transition with significant variations of Mn valence of MnO2 was observed in the urea–LiPF6 quasi-IL electrolyte that exhibited an outstanding pseudocapacitive performance. A comprehensive understanding of electrodeposition and pseudocapacitive behavior in urea-based quasi-IL electrolytes is of great benefit for electrochemical research and future practical applications. More detailed investigations of metal deposition, the coordination chemistry of metal ions and the charge storage mechanism of metal-oxide-based pseudocapacitors is in progress and will be shortly reported.
Acknowledgements
We thank the NSRRC staff for their technical support. This research is supported by the NSRRC and the National Science Council of the Republic of China under grant numbers NSC 99-2113-M-213-007, NSC 99-2218-E-008- 007-MY3 and NSC 99-2113-M-213-006.References
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS.
- F. Endres, D. R. MacFarlane and A. P. Abbott, Electrodeposition from Ionic Liquids, Wiley–VCH, Weinheim, Germany, 2008 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS.
- J. F. Wishart, Energy Environ. Sci., 2009, 2, 956 CAS.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863 CrossRef CAS.
- J. F. Huang and H. Y. Chen, Angew. Chem., Int. Ed., 2012, 51, 1684 CrossRef CAS.
- T. Tsuda, K. Kondo, T. Tomioka, Y. Takahashi, H. Matsumoto, S. Kuwabata and C. L. Hussey, Angew. Chem., Int. Ed., 2011, 50, 1310 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- T. Tsuda, T. Tomioka and C. L. Hussey, Chem. Commun., 2008, 2908 RSC.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70 RSC.
- R. Chen, F. Wu, H. Liang, L. Li and B. Xu, J. Electrochem. Soc., 2005, 152, A1979 CrossRef CAS.
- H. Y. Liang, H. Li, Z. X. Wang, F. Wu, L. Q. Chen and X. J. Huang, J. Phys. Chem. B, 2001, 105, 9966 CrossRef CAS.
- R. Chen, F. Wu, L. Li, B. Xu, X. P. Qiu and S. Chen, J. Phys. Chem. C, 2007, 111, 5193 Search PubMed.
- A. P. Abbott and K. J. McKenzie, Phys. Chem. Chem. Phys., 2006, 8, 4265 RSC.
- M. J. Deng, P. Y. Chen and I. W. Sun, Electrochim. Acta, 2007, 53, 1931 CrossRef CAS.
- M. J. Deng, P. Y. Chen, T. I. Leong, I. W. Sun, J. K. Chang and W. T. Tsai, Electrochem. Commun., 2008, 10, 213 CrossRef CAS.
- M. Jayakumar, K. A. Venkatesan, T. G. Srinivasan and P. R. V. Rao, Electrochim. Acta, 2009, 54, 6747 CrossRef CAS.
- S. W. Song, T. J. Richardson, G. R. V. Zhuang, T. M. Devine and J. W. Evans, Electrochim. Acta, 2004, 49, 1483 CAS.
- Y. Ma, J. Luo and S. L. Suib, Chem. Mater., 1999, 11, 1972 CrossRef CAS.
- C.-Y. Chen, C. Y. Fan, M. T. Lee and J. K. Chang, J. Mater. Chem., 2012, 22, 6274 RSC.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L. C. Qin, Carbon, 2011, 49, 2917 CrossRef CAS.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366 CrossRef CAS.
- G. H. Yu, L. B. Hu, M. Vosgueritchian, H. L. Wang, X. Xie, J. R. McDonough, X. Cui, Y. Cui and Z. N. Bao, Nano Lett., 2011, 11, 2905 CrossRef CAS.
- H. L. Wang, H. S. Casalongue, Y. Y. Liang and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 7472 CrossRef CAS.
- Z. S. Wu, D. W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H. M. Cheng, Adv. Funct. Mater., 2010, 20, 3595 CrossRef CAS.
- M. Belli, A. Scafati, A. Bianconi, S. Mobilio, L. Palladino, A. Reale and E. Burattini, Solid State Commun., 1980, 35, 355 CrossRef CAS.
- P. Ghigna, G. Flor and G. Spinolo, J. Solid State Chem., 2000, 149, 252 CrossRef CAS.
- R. Baddour-Had Jean and J. P. Pereira-Ramos, Chem. Rev., 2010, 110, 1278 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21379f |
| This journal is © The Royal Society of Chemistry 2012 |