Selective deoxygenation of stearic acid via an anhydride pathway†
Stefan A.W.
Hollak
a,
Johannes H.
Bitter
b,
Jacco van
Haveren
a,
Krijn P. de
Jong
b and
Daan S. van
Es
*a
aFood and Biobased Research, Wageningen University and Research Centre, P.O. Box 17, Wageningen, AA 6700, The Netherlands. E-mail: daan.vanes@wur.nl; Fax: (+31) 317 475347
bInorganic Chemistry and Catalysis, Utrecht University, Sorbonnelaan 16, Utrecht, CA 3584, The Netherlands
First published on 14th August 2012
Abstract
Stearic anhydride is proposed as reactive intermediate in the hydrogen free decarbonylation and ketonization of stearic acid over Pd/Al2O3 at 523 K. This information is crucial towards developing of a selective low temperature decarbonylation process of fatty acids towards olefins.
Introduction
The utilization of biomass for the production of transportation fuels is an important research area due to diminishing fossil fuel reserves, rising petroleum prices and increasing awareness of global warming.1 Several technologies are known to produce liquid fuels from biomass, summarized in recent reviews.2 Biodiesel is currently the most common biofuel in Europe and is produced by transesterification of triglycerides (main constituent of vegetable oils and animal fats) with methanol using a homogeneous base catalyst.3 Although the resulting fatty acid methyl esters (typical first generation biodiesel) have desirable fuel qualities such as good cetane number and lubricity, there are many technical issues associated with widespread use, like poor storage stability, lower heat content and engine compatibility issues compared to regular diesel fuels due to the high cloud and pour point.4Deoxygenation of vegetable oils or animal fats yields hydrocarbons similar to those found in regular diesel fuel. Such biofuels (the so-called second generation biodiesel) have higher energy densities and higher storage stabilities than first generation biodiesel because of the absence of oxygen containing functional groups and are fully compatible with existing vehicles and fuel infrastructures. In the literature, pyrolysis,5 hydrodeoxygenation6 and deoxygenation7 are reported for the production of second generation biodiesel.
The patent literature mainly describes the use of elevated hydrogen pressures and temperatures (3–10 MPa, 550–620 K) for the production of second generation biodiesel from vegetable oils.8 Especially the need for (non-renewable) hydrogen remains a challenge to overcome. Furthermore, the use of hydrogen results in the reduction of double bond functionalities present in unsaturated oils or fatty acids which reduces their potential application in high value chemical production. A further drawback of the current technology is the concomitant hydrodeoxygenation of glycerol to propane, which consumes valuable hydrogen and hinders potential valorization of the glycerol.
Stearic acid is a commonly used model compound in fatty acid deoxygenation reactions for more realistic feedstocks like rape seed and palm oil fatty acids. Many deoxygenation studies, concerning stearic acid or other closely related model compounds, have been published in recent years using various catalysts under inert,7d,9 hydrogen9d,10 or hydrothermal conditions.11 The deoxygenation of such free fatty acids can proceed via decarboxylation, decarbonylation–dehydration or hydrogenation yielding heptadecane, heptadecene or octadecane, depending on which reaction takes place. An overview of these possible deoxygenation reactions and thermodynamic data at various reaction temperatures are shown in the supporting information (Fig. S1†).
Palladium catalysts are often used in these deoxygenation reactions since Snåre et al. performed a catalyst screening study, which reported Pd/C as the most active and selective catalyst.7d Pd/γ-Al2O3 and Pt/γ-Al2O3 were the most effective of the metal oxide supported catalysts, giving only small amounts of ketonized or heavy byproducts.7d Mechanistic proposals for the catalytic deoxygenation of aliphatic esters under a hydrogen atmosphere over heterogeneous palladium catalysts were reported by Han et al. Here alkyl–oxygen and acyl–oxygen cleavage were proposed for decarboxylation and decarbonylation mechanisms.10a,12
In this work we report evidence for stearic anhydride as the reactive intermediate in the hydrogen free deoxygenation of stearic acid over Pd/Al2O3 at a relatively low reaction temperature of 523 K. Although stearic anhydride has been proposed previously for homogeneous catalysts using either Pd- or Rh-based catalysts,13 to the best of our knowledge no deoxygenation literature is reported, where stearic anhydride is suggested as intermediate product using heterogeneous catalysts. These fundamental understandings are valuable for optimizing deoxygenation activity and tuning the selectivity towards selective paraffin or olefin production.
Results and discussions
We have chosen Pd/γ-Al2O3 as the metal oxide based catalyst to allow for the possibility of thermal regeneration. A commercially available, well characterized catalyst was chosen in order to have a constant quality of catalyst over a broad range of experiments. Characterization data, before and after the catalytic reaction, are shown in the supporting information (Table S1, Fig. S2–S3†). A low reaction temperature is desired for designing a production process where biofuel is efficiently produced. Also, it was expected that ketonization, being an endothermic process, is more pronounced at higher temperatures.14 Since heavy products such as ketones were suggested to contribute to catalyst deactivation during deoxygenation reactions, it is desired to prevent ketonization as much as possible.9dCatalytic reactions over Pd/γ-Al2O3 under nitrogen pressure at different reaction temperatures showed that deoxygenation of stearic acid was successful upwards from 523 K (Fig. 1). The absence of unsaturated products at 523 K indicates that selective decarboxylation proceeds at this temperature. Experiments preformed in the absence of Pd/γ-Al2O3 did not give any conversion (entry 3, Table 1) and stearone was formed in the presence of bare γ-Al2O3 (entry 4, Table 1). These results confirm the catalytic activity of palladium in the decarboxylation reaction, as well as the possibility of stearic acid ketonization over alumina sites in the absence of palladium nanoparticles.
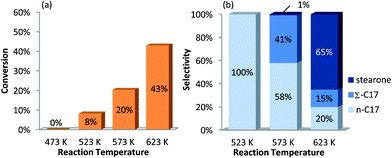 | ||
| Fig. 1 Stearic acid conversion (a) and product selectivity (b) as a function of reaction temperature, using Pd/Al2O3 as catalyst (6 h, 7 bar N2). Stearic acid concentration: 0.14 mol L−1. | ||
| Entry | Reactant | Reactant amount (g) | Catalyst | Reaction timea (h) | Conv.b (%) | Selectivity, (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n-C17 | Σ-C17 | n-C18 | SAc | C35d | Heavies | ||||||
| a 523 K, 7 bar N2. b Mass balance 95+% for all reactions. c Stearic acid selectivity when using stearic anhydride as feed. d Stearone. e Reaction at 473 K. f Neat reaction, no solvent used. | |||||||||||
| 1 | Stearic acid | 1.0 | Pd/γ-Al2O3 | 24 | 20 | 100 | 0 | 0 | 0 | 0 | 0 |
| 2 | Stearic acid | 10.0 | Pd/γ-Al2O3 | 24 | 10 | 79 | 11 | 0 | — | 10 | <1 |
| 3 | Stearic acid | 1.0 | None | 24 | 0 | 0 | 0 | 0 | — | 0 | 0 |
| 4 | Stearic acid | 1.0 | γ-Al2O3 | 24 | 6 | 0 | 0 | 0 | — | 100 | 0 |
| 5 | 1-Octadecanol | 1.0 | Pd/γ-Al2O3 | 24 | 100 | 86 | 0 | 14 | — | 0 | 0 |
| 6 | Octadecanal | 1.0 | Pd/γ-Al2O3 | 24 | 100 | 100 | 0 | 0 | — | 0 | 0 |
| 7 | Stearic anhydride | 1.0 | Pd/γ-Al2O3 | 24 | 100 | 27 | 25 | 0 | 46 | 2 | 0 |
| 8 | Stearic anhydride | 1.0 | Pd/γ-Al2O3 | 5 | 83 | 7 | 42 | 0 | 49 | 2 | 0 |
| 9e | Stearic anhydride | 1.0 | Pd/γ-Al2O3 | 24 | 48 | 4 | 46 | 0 | 49 | 0 | 0 |
| 10 | Stearic anhydride | 10.0f | Pd/γ-Al2O3 | 24 | 62 | 14 | 20 | 0 | 35 | 12 | 19 |
| 11 | Stearic anhydride | 1.0 | γ-Al2O3 | 24 | 100 | 0 | 1 | 0 | 71 | 28 | 0 |
| 12 | Stearic anhydride | 1.0 | None | 24 | 40 | 0 | 1 | 0 | 58 | 26 | 15 |
At 573 K C17 hydrocarbons (C17 HCs) were formed with 99% selectivity, of which 58% was heptadecane (n-C17) and 41% was unsaturated C17 (Σ-C17) hydrocarbons (Fig. 1). Only minor amounts of stearone, and no cracking products were formed (mass balance >98%). These findings are in contrast to those reported by Snåre et al. using Pd/γ-Al2O3 as the catalyst in the deoxygenation reaction of stearic acid, also at 573 K and with similar feed concentrations. These authors reported stearone formation with 48% selectivity and also cracking products and heavies were detected at similar conversion levels as reported in this study.
Investigating this remarkable difference in product selectivity fell outside the scope of our study, but could be due to differences in the catalyst preparation method (Al2O3 preparation, as well as metal deposition). Furthermore, the different reactor systems used (semi-continuous vs. batch in this work) could also have influenced the product selectivity.
The conversion and selectivity again significantly changed when the reaction temperature was further increased to 623 K (Fig. 1). Significant stearone formation lowered C17 HC selectivity dramatically. This shift in product selectivity towards ketonized products at increasing temperature is in line with the expectation mentioned before.
Next, the activity and selectivity as a function of initial feed concentration were investigated to determine reaction order in stearic acid concentration. Solvent free conditions are industrially preferred so were also included in the series. Subsequent reactions were performed at the lowest reaction temperature at which deoxygenation was observed (523 K). At low initial stearic acid concentration a 1st order dependence of TOF was found which plateaus at increasing feed concentrations, typical for a Langmuir–Hinshelwood dependence (Fig. 2). A gradual change in product selectivity as a function of initial stearic acid concentration was observed with significant formation of Σ-C17 and stearone at higher feed concentrations (Fig. 2, entry 1–2, Table 1). GC-MS analysis showed that various isomers of mono unsaturated C17 HCs, as well as multiple Σ-C17 HCs were present in the reaction mixture after reaction at increased feed concentrations at 523 K. A verification test with n-C17 (99%) under these reaction conditions showed that a negligible amount was converted to Σ-C17 HCs (<2% after 24 h). Hence, the observed amount of Σ-C17 species could not stem from n-C17 dehydrogenation. Decarbonylation of stearic acid towards 1-heptadecene (1-C17:1) could have occurred here. Various isomers of mono and multiple Σ-C17 HCs could subsequently be formed via double bond isomerization and dehydrogenation of 1-C17:1. This was confirmed by a stability test with 1-C17:1, which showed 91% conversion towards various isomers as well as n-C17 and various dehydrogenation products resulting from transfer hydrogenation. In the literature, palladium was also shown to be catalytically active in transfer hydrogenations.15
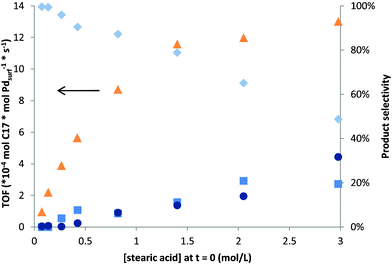 | ||
| Fig. 2 Turn over frequency (primary axes: ▲) and product selectivity (secondary axis: n-C17, ◆; Σ-C17, ■; stearone, ●) as function of initial stearic acid concentration (24 h, 7 bar N2). | ||
Increasing formation of stearone points to intramolecular reactions of stearic acid, which become more predominant at higher concentrations. The exact mechanism of the ketonization process is, however, still under debate, despite being extensively described in the literature. Various mechanisms were proposed over time which involved carbanions,16 free radicals,17 carbonium ions,18 and a four-center intermediate.19 A possible reaction pathway for the stearic acid ketonization could be the formation of stearic anhydride by condensation of adsorbed stearic acid and subsequent decarboxylation towards stearone.9g,20 Anhydride decarboxylation towards a ketone could be initiated on basic alumina sites, since the basic sites of the catalyst could act as an acceptor for the acidic CO2 by the highly negative framework charge of oxygen.9g Stearic anhydride formation would be an equilibrium, positively dependent on the stearic acid concentration. The existence of this anhydride intermediate for both the ketonization and the decarbonylation reaction (1a, Scheme 1), would explain the selectivity shift from n-C17 towards unsaturated C17 HCs and stearone at increasing initial stearic acid concentrations.
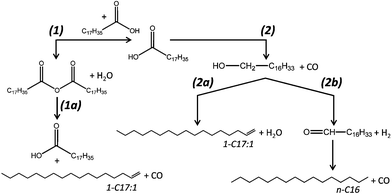 | ||
| Scheme 1 Potential decarbonylation pathways of stearic acid towards hydrocarbons in the absence of external hydrogen. | ||
Stearic anhydride is however, as mentioned in the introduction, until now only proposed as an intermediate in homogeneous decarbonylation catalysis.13 Indications for ketene formation on the surface of zirconia supported nickel catalysts during stearic acid hydrodecarboxylation studies were recently reported.21 Ketenes are generally very reactive and may subsequently react with acids to form anhydrides.22 Combining these reports with our findings resulted in the hypothesis that stearic anhydride is formed on the catalyst surface, as an intermediate for both the ketonization and the decarbonylation reaction, under our reaction conditions.
Catalytic reactions with stearic anhydride over the Pd/γ-Al2O3 catalyst were performed to investigate the reactivity and product selectivity of this proposed reaction intermediate. Full conversion of stearic anhydride to stearic acid, heptadecane, heptadecenes and stearone was achieved under standard reaction conditions (entry 7, Table 1). Heptadecene and stearic acid are the proposed anhydride decarbonylation products and heptadecane is formed by both decarboxylation of stearic acid and transfer hydrogenation of heptadecene. Also after a reaction time of 5 h, almost full conversion was achieved (entry 8, Table 1). Selectivity to the unsaturated hydrocarbons increased compared to the 24 h reaction, which confirms that heptadecane is a secondary product from stearic acid decarboxylation and transfer hydrogenation of heptadecene. These results also demonstrate that the formation of an anhydride intermediate from stearic acid would be the rate limiting step in the decarbonylation reaction under current reaction conditions.
Stearone formation from stearic anhydride was not observed in any significant amounts over the Pd/γ-Al2O3 catalyst at 523 K. However, considerable stearone formation was found when using γ-Al2O3 only (entry 11, Table 1). This confirms the catalytic activity of palladium in the decarbonylation reaction and furthermore shows the possibility of stearone formation from stearic anhydride on the alumina surface. Stearic anhydride was partly converted to stearone and other heavy products in solution at the reaction temperature of 523 K, thus in the absence of catalyst or support material (entry 12, Table 1).
Thus, the vicinity of palladium active sites during anhydride formation appears to determine whether decarbonylation to 1-heptadecene, stearone formation and/or the formation of other heavy products takes place. This is also clear from the wide range of products observed at increased stearic anhydride concentrations (entry 10, Table 1); besides decarbonylation products, significant amounts of stearone and other heavies are also formed.
Stearic anhydride decarbonylation was also effective at 473 K (entry 9, Table 1) with high selectivity towards unsaturated products, even after a reaction time of 24 h. This can be explained by lower transfer hydrogenation activity and reduced decarboxylation activity at this reaction temperature (Fig. 1). These results allow for selective stearic acid decarbonylation by shifting the anhydride equilibrium towards anhydride formation at this low reaction temperature in future investigations.
The nature of the catalytic sites of anhydride formation is, for that reason, an interesting point. This is possibly catalyzed by acid sites of alumina. Quenching of the reaction mixture with diethylamine, however, did not provide evidence for the existence of significant amounts of anhydride when performing a catalytic reaction on γ-alumina. Also during the reaction on Pd/γ-alumina, no significant amounts of anhydrides could be detected. A possible explanation for this could be the high reactivity of stearic anhydride under the reaction conditions.
In order to assess the thermodynamics of the anhydride pathway sequence, calculations were performed for the decarbonylation and ketonization reactions, using the HSC program.23 Butyric acid and butyric anhydride were used in the calculations since the thermodynamic data for stearic anhydride were not available. However, it is expected that these calculations on butyric acid give a good indication for the thermodynamics of stearic acid. A minor increase in Gibbs energy was shown for anhydride formation and both reactions have an overall negative Gibbs energy (Fig. S4†). The thermodynamic equilibrium between butyric acid and its anhydride is 5.3 × 10−4 at 523 K, based on these calculations. Although activation energies were not calculated, these data indicate that an anhydride intermediate is thermodynamically and kinetically feasible with low concentrations of the intermediate anhydride present during catalytic reactions. Until now we did not accomplish the quenching of formed anhydrides, or spectroscopically show these intermediates with ex situ IR. The high reactivity of stearic anhydride, in combination with the unfavourable thermodynamic equilibrium, could explain these complications. From current results it can furthermore be concluded that the Pd/Al2O3 catalyst significantly lowers the decarbonylation activation energy, resulting in a selectivity shift from ketonization (when conduction experiments with Al2O3) to selective decarbonylation with Pd nanoparticles present.
Alternatively, a decarbonylation pathway of stearic acid to 1-heptadecene could proceed via an intermediate alcohol (2a, Scheme 1). In order to verify this possibility, experiments were performed using 1-octadecanol (used here as a readily available model compound for 1-heptadecanol). As can be seen in entry 5 (Table 1), the main product from 1-octadecanol was heptadecane instead of octadecene. Apparently, dehydration of the alcohol does not occur to a large extent. The loss of one carbon atom can be rationalized by assuming that under the reaction conditions the primary alcohol is dehydrogenated yielding the corresponding aldehyde and hydrogen, followed by consecutive decarbonylation of the aldehyde (2b, Scheme 1). Since instead of octadecene, only octadecane was present in the reaction mixture, the hydrogen formed during the alcohol dehydrogenation is apparently consumed by octadecene hydrogenation.
In order to verify the aldehyde decarbonylation, octadecanal was subjected to the reaction conditions. The results were full conversion to heptadecane with 100% selectivity (entry 6, Table 1), which confirm that the heptadecane obtained from 1-octadecanol is formed via a dehydrogenation–decarbonylation mechanism. A similar dehydrogenation–decarbonylation mechanism is known to occur over the Pd/Al2O3 catalyst when using ethanol, with ethanal as the dehydrogenation intermediate and methane and carbon monoxide as the subsequent decarbonylation products.24
If stearic acid decarbonylation would occur via an intermediate alcohol, hexadecane is also expected as the dehydrogenation–decarbonylation product of the intermediate alcohol. Since no C16 components have been observed in our reactions using stearic acid as feedstock, we can conclude that a decarbonylation–dehydration mechanism via 1-octadecanol is also not occurring under current reaction conditions over the Pd/γ-Al2O3 catalyst.
In Scheme 2 the suggested deoxygenation reaction pathways are shown, which are proposed to occur in the hydrogen free deoxygenation of stearic acid over Pd/γ-Al2O3 at 523 K, including decarboxylation and the suggested decarbonylation and ketonization pathway via the anhydride intermediate.
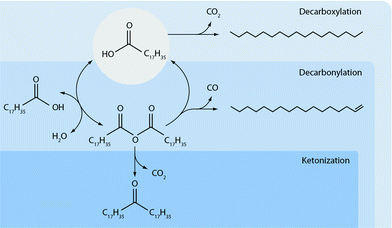 | ||
| Scheme 2 Suggested deoxygenation pathways of stearic acid at 523 K in the absence of external hydrogen. | ||
Conclusions
In conclusion, the product selectivity of stearic acid deoxygenation over Pd/γ-Al2O3 at 523 K is strongly dependent on the feed concentration with selective decarboxylation at the low end and increasing decarbonylation and ketonization activity at higher feed concentrations. We have provided evidence that this dependence is caused by the existence of stearic anhydride as the reactive intermediate in the decarbonylation and ketonization reaction. The observations of changing product selectivity as a function of feed concentration are in accordance with the anhydride intermediate since the high feed concentration is promotes anhydride formation. Model reactions also showed that decarbonylation–dehydration via 1-octadecanol does not occur under our conditions.Since stearic anhydride is readily converted, the rate limiting step in the formation of the unsaturated C17 hydrocarbons is the in situ formation of stearic anhydride. Furthermore, it was shown that stearic anhydride decarbonylation already proceeds at 473 K. We are currently exploring the possibilities to direct product selectivity by stimulating anhydride formation.
Acknowledgements
This research has been performed within the framework of the CatchBio program. The authors gratefully acknowledge the support of the Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science. We especially appreciate the contributions of Rob Gosselink and Frits van der Klis during this work. The authors also want to thank Bert Sandee (BASF) for supplying and characterizing the catalyst, Wouter Teunissen for his GC and GC-MS expertise, Cor van der Spek for performing TEM microscopy, Rajeesh Pazhavelikkakath Purushothaman for the ICP-MS data and Mariette Twilt for the graphical abstractReferences
- S. N. Naik, V. V. Goud, P. K. Rout and A. K. Dalai, Renewable Sustainable Energy Rev., 2010, 14, 578–597 CrossRef CAS.
- (a) J. R. Regalbuto, Biofuels, Bioprod. Biorefin., 2011, 5, 495–504 CrossRef CAS; (b) D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC; (c) J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 RSC.
- (a) US Pat., 20070033863, 2007; (b) P. Bondioli, Top. Catal., 2004, 27, 77–82 CrossRef CAS.
- (a) J. A. Kinast, Subcontractor report; NREL/SR-510-31460, Colorado, 2003; (b) S. M. Geyer, M. J. Jacobus and S. S. Lestz, Transactions of the ASAE, 1984, 27, 375–381 CAS; (c) A. Demirbas, Energy Convers. Manage., 2003, 44, 2093–2109 CrossRef CAS.
- (a) D. G. Lima, V. C. D. Soares, E. B. Ribeiro, D. A. Carvalho, E. C. V. Cardoso, F. C. Rassi, K. C. Mundim, J. C. Rubim and P. A. Z. Suarez, J. Anal. Appl. Pyrolysis, 2004, 71, 987–996 CrossRef CAS; (b) A. O. Adebanjo, A. K. Dalai and N. N. Bakhshi, Energy Fuels, 2005, 19, 1735–1741 CrossRef CAS; (c) K. D. Maher and D. C. Bressler, Bioresour. Technol., 2007, 98, 2351–2368 CrossRef CAS; (d) E. B. Higman, I. Schmeltz, H. C. Higman and O. T. Chortyk, J. Agric. Food Chem., 1973, 21, 202–204 CrossRef CAS.
- (a) T. V. Choudhary and C. B. Phillips, Appl. Catal., A, 2011, 397, 1–12 CrossRef CAS; (b) R. Sotelo-Boyaás, Y. Liu and T. Minowa, Ind. Eng. Chem. Res., 2011, 50, 2791–2799 CrossRef; (c) M. Stumborg, A. Wong and E. Hogan, Bioresour. Technol., 1996, 56, 13–18 CrossRef CAS; (d) I. Sebos, A. Matsoukas, V. Apostolopoulos and N. Papayannakos, Fuel, 2009, 88, 145–149 CrossRef CAS.
- (a) C. A. Fisk, T. Morgan, Y. Ji, M. Crocker, C. Crofcheck and S. A. Lewis, Appl. Catal., A, 2009, 358, 150–156 CrossRef CAS; (b) R. O. Idem, S. P. R. Katikaneni and N. N. Bakhshi, Fuel Process. Technol., 1997, 51, 101–125 CrossRef CAS; (c) T. Morgan, D. Grubb, E. Santillan-Jimenez and M. Crocker, Top. Catal., 2010, 53, 820–829 CrossRef CAS; (d) M. Snåre, I. Kubičková, P. Mäki-Arvela, K. Eränen and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708–5715 CrossRef.
- (a) T. Kalnes, T. Marker and D. R. Shonnard, Int. J. Chem. React. Eng., 2007, 5, 1–9 Search PubMed; (b) J. Myllyoja, P. Aalto, P. Savolainen, P. Purola, V. Alopaeus and J. Gronqvist, US Pat., US 2007/0010682 A1, 2007; (c) J. Myllyoja, P. Aalto and E. Harlin, PCT Pat., WO 2007/003708, 2007; (d) D. Murzin, I. Kubickova, M. Snåre, P. Mäki-Arvela, J. Myllyoja, PCT Pat., WO 2006/075057 A2, 2006; (e) J. R. Gomes, US Pat., US 2006/0186020 A1, 2006; (f) J. J. Scheibel and R. T. Reilman, US. Pat., US 2007/0281875 A1, 2007.
- (a) I. Simakova, O. Simakova, P. Mäki-Arvela and D. Y. Murzin, Catal. Today, 2010, 150, 28–31 CrossRef CAS; (b) H. Bernas, K. Eränen, I. Simakova, A.-R. Leino, K. Kordás, J. Myllyoja, P. Mäki-Arvela, T. Salmi and D. Y. Murzin, Fuel, 2010, 89, 2033–2039 CrossRef CAS; (c) J.-G. Na, B. E. Yi, J. N. Kim, K. B. Yi, S.-Y. Park, J.-H. Park, J.-N. Kim and C. H. Ko, Catal. Today, 2010, 156, 44–48 CrossRef CAS; (d) P. Do, M. Chiappero, L. Lobban and D. Resasco, Catal. Lett., 2009, 130, 9–18 CrossRef CAS; (e) S. Lestari, I. Simakova, A. Tokarev, P. Mäki-Arvela, K. Eränen and D. Y. Murzin, Catal. Lett., 2008, 122, 247–251 CrossRef CAS; (f) T. Danuthai, S. Jongpatiwut, T. Rirksomboon, S. Osuwan and D. E. Resasco, Appl. Catal., A, 2009, 361, 99–105 CrossRef CAS; (g) T. Sooknoi, T. Danuthai, L. L. Lobban, R. G. Mallinson and D. E. Resasco, J. Catal., 2008, 258, 199–209 CrossRef CAS.
- (a) J. Han, H. Sun, Y. Ding, H. Lou and X. Zheng, Green Chem., 2010, 12, 463–467 RSC; (b) J. G. Immer, M. J. Kelly and H. H. Lamb, Appl. Catal., A, 2010, 375, 134–139 CrossRef CAS; (c) S. Lestari, P. Mäki-Arvela, H. Bernas, O. Simakova, R. Sjöholm, J. Beltramini, G. Q. M. Lu, J. Myllyoja, I. Simakova and D. Y. Murzin, Energy Fuels, 2009, 23, 3842–3845 CrossRef CAS; (d) S. Lestari, P. Mäki-Arvela, I. Simakova, J. Beltramini, G. Lu and D. Murzin, Catal. Lett., 2009, 130, 48–51 CrossRef CAS; (e) S. Lestari, P. Mäki-Arvela, K. Eränen, J. Beltramini, G. Max Lu and D. Y. Murzin, Catal. Lett., 2010, 134, 250–257 CrossRef CAS; (f) P. Mäki-Arvela, M. Snåre, K. Eränen, J. Myllyoja and D. Y. Murzin, Fuel, 2008, 87, 3543–3549 CrossRef.
- (a) J. Fu, X. Lu and P. E. Savage, Energy Environ. Sci., 2010, 3, 311–317 RSC; (b) J. Fu, X. Lu and P. E. Savage, ChemSusChem, 2011, 4, 481–486 CrossRef CAS; (c) S. Matsubara, Y. Yokota and K. Oshima, Org. Lett., 2004, 6, 2071–2073 CrossRef CAS; (d) M. Watanabe, T. Iida and H. Inomata, Energy Convers. Manage., 2006, 47, 3344–3350 CrossRef CAS.
- J. Han, J. Duan, P. Chen, H. Lou and X. Zheng, Adv. Synth. Catal., 2011, 353, 2577–2583 CrossRef CAS.
- (a) T. A. Foglia and P. A. Barr, J. Am. Oil Chem. Soc., 1976, 53, 737–741 CrossRef CAS; (b) J. L. Nôtre, E. L. Scott, M. C. R. Franssen and J. P. M. Sanders, Tetrahedron Lett., 2010, 51, 3712–3715 CrossRef; (c) J. A. Miller, J. A. Nelson and M. P. Byrne, J. Org. Chem., 1993, 58, 18–20 CrossRef CAS; (d) J. A. Miller and J. A. Nelson, Organometallics, 1991, 10, 2958–2961 CrossRef CAS.
- (a) J. L. Ohlson, C. W. Hoerr, US Pat., 2697729, 1954; (b) M. Jayamani and C. N. Pillai, J. Catal., 1984, 87, 93–97 CrossRef CAS; (c) R. Pestman, R. M. Koster, A. van Duijne, J. A. Z. Pieterse and V. Ponec, J. Catal., 1997, 168, 265–272 CrossRef CAS.
- G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567–580 CrossRef CAS.
- (a) O. Neunhoeffer and P. Paschke, Ber. Dtsch. Chem. Ges. B, 1939, 72, 919–929 CrossRef; (b) A. L. Miller, N. C. Cook and F. C. Whitmore, J. Am. Chem. Soc., 1950, 72, 2732–2735 CrossRef CAS; (c) L. Ötvös and L. Noszkó, Tetrahedron Lett., 1960, 1, 19–22 CrossRef.
- J. Bell and R. I. Reed, J. Chem. Soc., 1952, 1383–1389 RSC.
- C. C. Lee and J. W. T. Spinks, J. Org. Chem., 1953, 18, 1079–1086 CrossRef CAS.
- R. I. Reed and M. B. Thornley, J. Chem. Soc., 1957, 3714–3717 RSC.
- M. Glinski, W. Szymanski and D. Lomot, Appl. Catal., A, 2005, 281, 107–113 CrossRef CAS.
- B. Peng, X. Yuan, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 9400 CrossRef CAS.
- C. D. Hurd and M. F. Dull, J. Am. Chem. Soc., 1932, 54, 3427–3431 CrossRef CAS.
- HSC Chemistry for Windows, version 4.0, ChemSW, Inc., 1999 Search PubMed.
- J. M. D. McGregor and L. K. Doraiswamy, Ind. Eng. Chem. Res., 2000, 40, 108–113 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: See DOI: 10.1039/c2ra21651e |
| This journal is © The Royal Society of Chemistry 2012 |