Antimicrobial and antioxidant activity evaluation of tetrazolo[1,5-a]pyrimidines: A simple diisopropylammonium trifluoroacetate mediated synthesis
Chandran
Raju
a,
Kalaipriya
Madhaiyan
b,
Ramakrishnan
Uma
a,
Radhakrishnan
Sridhar
*b and
Seeram
Ramakrishna
b
aPachaiyappa’s College, University of Madras, Aminjikarai, Chennai 600 029, India. Tel: +91-9902100665
bNUSCNN Lab National University of Singapore, Singapore. Fax: 65-6773 0339; Tel: +65 97165929
First published on 1st October 2012
Abstract
A simple, efficient and economic synthesis of tetrazolo [1,5-a]pyrimidine-6-carboxylates was accomplished by three-component reaction of β-ketoesters with a mixture of aromatic aldehyde and 5-aminotetrazole using TFA:DIPEA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the catalyst. The synthesized tetrazole compounds are evaluated for their antimicrobial and antioxidant activity.
:1) as the catalyst. The synthesized tetrazole compounds are evaluated for their antimicrobial and antioxidant activity.
The pyrimidine core unit has attracted the attention of researchers owing to its biological and pharmaceutical activity.1,2 The chemistry behind the synthesis is well established, however a requirement for new molecules with the fundamental pyrimidine skeleton is still an area of active research. A ‘green’ method to synthesis the heterocyclic core unit is therefore of key importance. Dihydropyrimidines (DHPM) act as potential calcium channel blockers,3 inhibitors of mitotic kinesin, Eg5 for the treatment of cancer,4,5 TRPA1 modulators for treating pain,6etc. Marine alkaloids containing the DHPM unit exhibit antiviral, antibacterial and anti-inflammatory activities.7,8 Compounds containing a DHPM scaffold such as the purine derivatives are well-known for their pharmacological and therapeutic activity. DHTPM has been studied for its antimicrobial activity,9 farnesyl transferase inhibitory effect,10 as an antihypertensive,11 in KATP channel opening,12 and as a central nervous system stimulant,13etc.
Many methods have been employed for the synthesis of DHPMs,14 including (i) catalysed with Lewis acids such as InCl3,15 Cu(OTf)2,16etc, (ii) phenyl boronic acid17 catalysis of the Biginelli reaction in acetonitrile solvent under reflux/18h conditions, (iii) ammonium chloride18 solid supported solvent-free synthesis of DHPMs at 100 °C, (iv) green synthesis mediated by polystyrene sulfonic acid19 under microwave heating at 80 °C.
DHTPMs have been synthesised using iodine20, mineral acids,21 sulfamic acid22 and strontium chloride hexahydrate.23 We found that there are only limited reports dedicated to the synthesis of DHTPMs24–26 using 5-aminotetrazole, which has both endocyclic nitrogen and exocyclic amino groups, to form a fused heterocycle as a 1,3-binucleophile synthon to replace the urea of the Biginelli reaction. An iodine mediated synthesis has recently been reported as a multi-component reaction method for preparation of DHTPMs.27 Most synthetic protocols previously reported have the drawbacks of requiring high temperatures, prolonged reaction time, drastic reaction conditions, low yields or the use of expensive acid catalysts. Herein, we develop a simple and convenient protocol for the synthesis of the title compounds in higher yields by using trifluoroacetic acid and diisopropyl ethylamine salt (1:1) as a catalyst. Further, this economic catalyst is relatively less explored in organic synthesis reactions which encouraged us to investigate its use.
Results and discussion
The catalyst acts to impart the required acidity to catalyse the three component coupling reaction shown in (Scheme 1). Further, microwave irradiation at 90 °C for 30 min to 45 min effectively completes the reaction achieving good to excellent yields (Table 1). A plausible mechanism for the reaction is via the crotonate intermediate depicted in the schematic (Scheme 2). The crotonate intermediate reacts with the aldehyde functionality to form DHTPM. The method is worked and optimized not only for aromatic aldehydes but also for functional heteroaromatic aldehydes. Aldehydes with both electron withdrawing and electron donating substituents were investigated under the same reaction conditions to ensure a broad range use of the catalyst.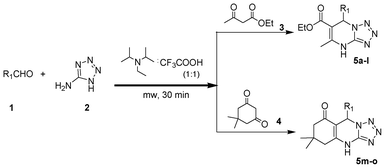 | ||
| Scheme 1 Diisopropylammonium trifluoroacetate mediated synthesis of tetrazolo pyrimidines. | ||
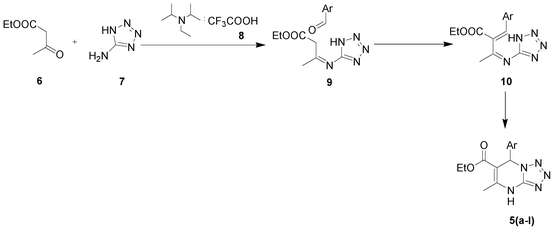 | ||
| Scheme 2 Plausible mechanism of DHTPM synthesis. | ||
| Product | IR (KBr) (cm−1) | NMR | Ar | Time (min) | Mass m/z | Yield (%)a | M.P. (°C) | |
|---|---|---|---|---|---|---|---|---|
| 5a | 3226 | 3160 |
1H NMR (300MHz, DMSO-d6): δ 0.97 (3H, t, J = 7.08 Hz), 2.48 (3H, s, CH3–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 3.93 (2H, q, J = 7.08 Hz, OCH2), 6.81 (1H, s, CH), 7.49–7.47 (1H, d), 7.53–7.50 (2H, m), 7.89–7.87(4H, m), 11.3 (1H, brs, NH). C), 3.93 (2H, q, J = 7.08 Hz, OCH2), 6.81 (1H, s, CH), 7.49–7.47 (1H, d), 7.53–7.50 (2H, m), 7.89–7.87(4H, m), 11.3 (1H, brs, NH). |
2-Naphthyl | 20 | 335.36 M-1 | 90 | 255-256 |
| 3051 | 2944 | 13C NMR: (75 MHz, DMSO-d6): δ 165.07, 148.9, 147.3, 138.7, 133.2, 133.0, 129.0, 128.5, 128.0, 127.0, 126.9, 125.2, 98, 60.1, 59.5, 18.9, 14.3. | ||||||
| 1696 | 1572 | |||||||
| 1230 | 1124 | |||||||
| 1018 | 785 | |||||||
| 5b | 3382 | 3188 |
1H NMR (300MHz, DMSO-d6): δ 0.82 (3H, t, J = 7.08 Hz), 2.33 (3H, s, CH3–CC), 3.89 (2H, q, J = 7.08 Hz, OCH2), 6.66 (1H, s, CH), 7.21–7.18 (1H, t, J = 8.5 Hz, 3.54 Hz), 7.49–7.33 (8H, m), 11.3 (1H, brs, NH). |
2-Biphenyl | 25 | 361.4 M-1 | 80 | 210–212 |
| 3059 | 2955 | 13C NMR: (75 MHz, DMSO-d6): δ 14.4, 18.9, 56.6, 60.2, 98.6, 127.7, 128.5, 128.8, 128.9, 138.7, 140.2, 141.5, 146.8, 148.7, 164.8. | ||||||
| 1699 | 1562 | |||||||
| 1222 | 1097 | |||||||
| 1068 | 756 | |||||||
| 5c | 3226 | 3162 |
1H NMR(300MHz, DMSO-d6): δ 1.01 (3H,t, J = 7.08 Hz), 2.05 (3H, s, CH3–CC), 2.48 (3H, s, CH3-Ph), 4.03–3.86 (2H, m, J = 7.08 Hz, OCH2), 6.58 (1H, s), 7.10–7.04 (3H, m, H-Ph), 7.24–7.18 (1H, m, H-Ph), 11.2 (1H, brs, NH). |
3-MeC6H4 | 15 | 299.3 M+1 | 95 | 218–220 |
| 3049 | 2947 | 13C NMR: (75 MHz, DMSO-d6): δ 14.2, 18.8, 21.3, 59.2, 60, 98.2, 24.7, 128.1, 129, 129.6, 138.3, 141.3, 146.9, 148.9, 165. | ||||||
| 1698 | 1447 | |||||||
| 1301 | 1225 | |||||||
| 1097 | 1018 | |||||||
| 703 | ||||||||
| 5d | 3183 | 3055 |
1H NMR (300MHz, DMSO-d6): δ 1.04–0.99 (3H, t, J = 7.08 Hz), 2.49–2.40 (3H, s, CH3–CC), 3.89 (3H, s, OCH3), 3.94–3.90 (2H, q, J = 7.08 Hz, OCH2), 6.80 (1H, s, CH), 6.93–6.90 (1H, m), 6.99–6.96 (1H, d), 7.29–7.25 (2H, m), 11.2 (1H, brs, NH). |
2-MeOC6H4 | 25 | 315.33 M-1 | 75 | 199–201 |
| 2941 | 2842 | 13C NMR: (75 MHz, DMSO-d6): δ 14.2, 18.8, 55.6, 56.1, 59.9, 97.2, 112.3, 120.7, 128.8, 129.9, 130.5, 147.2, 149.5, 157.4,165.2. | ||||||
| 1704 | 1655 | |||||||
| 1569 | 1273 | |||||||
| 1099 | 990 | |||||||
| 762 | ||||||||
| 5e | 3230 | 3162 |
1H NMR (300MHz, DMSO-d6): δ 1.03 (3H, t, J = 7.05 Hz), 2.48 (3H, t, CH3–CC), 3.71(1H, s, OCH3), 3.95 (2H, q, J = 7.08 Hz, OCH2), 6.61(1H, s, CH), 6.88–6.81 (3H, m), 7.27–7.22 (1H, m), 11.2 (1H, brs, NH). |
3-MeOC6H4 | 17 | 315.33 M-1 | 95 | 158–159 |
| 3051 | 2945 | 13C NMR: (75 MHz, DMSO-d6): δ 14.3, 18.8, 55.6, 59, 60.1, 98.1, 113.9, 114, 119.6, 130.4, 142.8, 147, 149, 159, 165. | ||||||
| 2845 | 1697 | |||||||
| 1569 | 1263 | |||||||
| 1121 | 777 | |||||||
| 706 | ||||||||
| 5f | 3229 | 3167 |
1H NMR (300MHz, DMSO-d6): δ 1.03 (3H, t, J = 7.08 Hz, CH3 ethyl). 2.06 (3H, s, COCH3), 2.47 (3H, s, CH3–CC), 3.95 (2H, q, J = 7.08 Hz, OCH2), 6.67 (1H, s, CH), 7.10–7.08 (2H, d, J = 8.61 Hz), 7.36–7.33 (2H, d, 8.61 Hz), 11.3 (1H, brs, NH). |
4-OCOCH3C6H4 | 42 | 343.34 M-1 | 90 | 227–28 |
| 3056 | 2953 | 13C NMR: (75 MHz, DMSO-d6): δ 14.3,18.9, 21.2, 21.2, 58.6, 60.1, 98, 115.7, 122.5, 128.9, 138.9, 147.2, 148.8, 150.8, 165, 169.5. | ||||||
| 1762 | 1703 | |||||||
| 1572 | 1226 | |||||||
| 1095 | 910 | |||||||
| 696 | ||||||||
| 5g | 3227 | 3057 |
1H NMR (300MHz, DMSO-d6): δ 1.00 (3H, t, J = 7.08 Hz), 2.50 (3H, s, CH3–CC), 3.94 (2H, q, J = 7.08 Hz, OCH2), 6.88 (1H, s, CH), 7.21–7.16 (2H, m), 7.40–7.34 (2H, m), 11.36 (1H, brs, NH). |
2-FC6H4 | 20 | 303.29 M+1 | 92 | 182–184 |
| 2986 | 2945 | 13C NMR: (75 MHz, DMSO-d6): δ 14.2, 18.9, 53.8, 60.1, 96.7, 116, 116.2, 125.2, 125.3, 128.2, 128.3, 130.1, 130.2, 131.2, 131.3, 147.7, 148.9, 158.9, 161.4, 164.8. | ||||||
| 1657 | 1569 | |||||||
| 1491 | 1333 | |||||||
| 1270 | 1126 | |||||||
| 1021 | 855 | |||||||
| 754 | ||||||||
| 5h | 3672 | 3227 |
1H NMR (300MHz, DMSO-d6): δ 1.09 (t, 3H, J = 7.08 Hz), 2.49 (3H, s, CH3–CC), 3.98 (2H, q, J = 7.08 Hz, OCH2), 6.68 (1H, s, CH), 7.21 (1H, m), 7.48–7.39 (2H, m), 11.3 (1H, brs, NH). |
3,4-F2 C6H4 | 15 | 321.28 M-1 | 95 | 208–209 |
| 3160 | 3050 | 13C NMR: (75 MHz, DMSO-d6): δ 13.8, 18.6, 57.8, 59.7, 96.6, 116.9, 117.7, 117.9, 124.4, 138.7, 147.4, 148.2, 150.6, 164.5. | ||||||
| 2944 | 1697 | |||||||
| 1570 | 1282 | |||||||
| 1100 | 777 | |||||||
| 5i | 3475 | 3182 |
1H NMR(300MHz, DMSO-d6): δ 0.98 (3H, t, J = 7.08 Hz), 2.49 (3H, s, CH3–CC), 3.97–3.90 (2H, s, J = 7.08 Hz, OCH2), 6.74 (1H, s, CH), 7.59–7.54 (1H, m), 7.70–7.67 (1H, d, J = 6.84 Hz), 7.80–7.77 (1H, q), 7.90 (1H, m), 11.38 (1H, brs, NH). |
3-CNC6H4 | 20 | 310.31 M-1 | 98 | 191–192 |
| 3045 | 2941 | 13C NMR: (75 MHz, DMSO-d6): δ 14.2, 19.1, 58.6, 60.2, 97, 112.1, 118.8, 130.6, 131.6, 132.8, 142.9, 148.1, 148.7, 164.8. | ||||||
| 2231 | 1716 | |||||||
| 1567 | 1219 | |||||||
| 1097 | 774 | |||||||
| 5j | 3728 | 2988 |
1H NMR (300MHz, DMSO-d6): δ 0.98 (3H, t, J = 7.08 Hz). 2.49–2.45 (3H, s, CH3–CC), 3.97–3.92 (2H, q, J = 7.08 Hz, OCH2), 6.74 (1H, s, CH), 7.39–7.35 (1H, m), 7.71–7.69 (1H, d, J = 7.98 Hz), 8.51–8.49 (1H, m), 8.62–8.61 (1H, d, J = 2.16 Hz), 11.2 (1H, s). |
3-Pyridyl | 25 | 286.29 M-1 | 85 | 238–240 |
| 2905 | 2782 | 13C NMR: (75 MHz, DMSO-d6): δ 14.3, 19, 57.2, 59.8, 60.2, 97.2, 124.5, 135.4, 137.1, 147.9, 148.8, 149.1, 150.1, 164.8. | ||||||
| 1713 | 1648 | |||||||
| 1568 | 1220 | |||||||
| 1024 | 840 | |||||||
| 711 | ||||||||
| 5k | 3787 | 3157 |
1H NMR (300MHz, DMSO-d6): δ 1.12 (3H, t, J = 7.08 Hz). 2.40 (3H, s, CH3–CC), 4.08 (2H, q, J = 7.08 Hz, OCH2), 6.30 (1H, s, CH), 6.67 (1H, s), 7.56 (1H, s), 7.69 (1H, s), 11.3 (1H, brs, NH). |
3-Furyl | 33 | 275.26 M-1 | 74 | 189–192 |
| 3086 | 2935 | 13C NMR: (75 MHz, DMSO-d6): δ 14.4, 18.8, 51.1, 60.2, 97.4, 109.4, 126.2, 141.1, 144.5, 147.1, 149.1, 165.05. | ||||||
| 1707 | 1567 | |||||||
| 1383 | 1273 | |||||||
| 1069 | 778 | |||||||
| 5l | 3728 | 3091 | 1H NMR (300MHz, DMSO-d6): δ 1.08 (3H, t, J = 7.08 Hz), 2.48 (3H, s), 4.03 (2H, q, J = 7.08 Hz, OCH2), 7.12 (1H, s, CH), 7.71 (2H, d, J = 3.21Hz), 7.75 (2H, q, J = 3.21Hz), 11.5 (1H, brs, NH). | 2-Thiazolyl | 35 | 291.33 M-1 | 62 | 238–239 |
| 3043 | 2939 | 13C NMR: (75 MHz, DMSO-d6): δ 14.4, 18.9, 55.5, 60.4, 96.8, 122.4, 143.1, 148.4, 149.2, 164.7, 167.9. | ||||||
| 1706 | 1650 | |||||||
| 1567 | 1307 | |||||||
| 1276 | 1098 | |||||||
| 872 | ||||||||
| 5m | 3171 | 3056 | 1H NMR (300MHz, DMSO-d6) : δ 1.05 (3H, s, CH3), 1.07 (3H, s, CH3), 2.24 (2H, q, CH2–CO), 2.68 (2H, q, CH2–C), 7.67–7.34 (5H, m), 7.88 (1H, d, J = 8.13 Hz), 7.97 (1H, d, J = 8.13 Hz), 8.5 (1H, brs), 11.5 (1H, brs, NH). | 2-Naphthyl | 24 | 354.4 M+1 | 65 | 260–262 |
| 2958 | 2958 | 13C NMR: (75 MHz, DMSO-d6): δ 27.3, 28.1, 32.3, 49.8, 106, 123.5, 125.4, 125.9, 126.6, 128, 4, 128.9, 130.5, 133.3, 136.9, 148.2, 150.8, 193.1. | ||||||
| 2183 | 1644 | |||||||
| 1578 | 1365 | |||||||
| 1226 | 778 | |||||||
| 5n | 3477 | 3385 | 1H NMR (400MHz, DMSO-d6) : δ 1.05 (3H, s, CH3), 1.07 (3H, s, CH3), 2.24 (2H, q, CH2–CO), 2.68 (2H, q, CH2–C), 6.66 (1H, s, CH), 7.63–7.61 (4H, m), 7.47–7.34 (5H, m), 11.6 (1H, brs, NH). | 4-Biphenyl | 18 | 371.44 M+2 | 70 | 287–288 |
| 3191 | 2921 | 13C NMR: (100 MHz, DMSO-d6): δ 27.1, 27.8, 32.3, 49.8, 57.2, 105.8, 126.7, 126.9, 127.8, 128.9, 139.5, 140.2, 148.4, 150.6, 193.1. | ||||||
| 2336 | 1644 | |||||||
| 1578 | 1370 | |||||||
| 1051 | 750 | |||||||
| 5o | 2963 | 2361 | 1H NMR (400MHz, DMSO-d6) : δ 1.01 (3H, s, CH3), 1.05 (3H, s, CH3), 2.2 (2H, q, CH2–C), 2.24 (3H, s, COCH3), 2.60 (2H, q, CH2–CO), 6.62 (1H, s, CH), 7.09–7.07 (2H, m), 7.34–7.32 (2H, d, J = 8.4 Hz), 11.6 (1H, brs, NH). | 4-OCOCH3C6H4 | 20 | 353.38 M-1 | 60 | 250–252 |
| 1766 | 1644 | 13C NMR: (100 MHz, DMSO-d6): 21.3, 27.6, 28.6, 32.8, 50.3, 57.4, 105.9, 122.4, 128.8, 138.4, 148.9, 150.7, 151.2, 169.5, 193.5. | ||||||
| 1577 | 1369 | |||||||
| 1196 | 563 | |||||||
It was noted that the nature of the substituents in the aromatic aldehydes affected the yield and reaction time. Increased reaction time and decreased yield was observed in the case of o-substituted aromatic aldehydes and was attributed to sterical hindrance associated with ortho substitution. Increase in yields was observed with decreased reaction time for aromatic aldehydes with any substituent in the p-position. It was evident that under the reaction conditions or in the presence of catalyst, that the viable ester moieties were stable and did not hydrolyse. Heteroaromatic aldehydes with the triflate salt (TFA:DIPEA salt) were reacted using the catalysed protocol to generalize the condition for every system. Compared to the aromatic systems, heteroaromatic aldehydes had lower yields and required prolonged reaction times. The versatility of TFA:DIPEA over other triflate salts is clear from the Table 2. It resulted in good to excellent yield of DHTPMs in both ethanol and acetonitrile with microwave heating. Decreased reaction times were realized due to increased reactivity of the reactants under microwave conditions.
| Entry | Triflate saltab | Temp (°C) | Time (min) | Yield (%) |
|---|---|---|---|---|
|
a All reactions were carried out in a 1:1 ratio of base and acid.
b All reactions were carried out in both acetonitrile and ethanol.
c Trifluoacetic acid.
d Diisopropylamine.
e Ammonium trifluoroacetate.14
f Diisopropyl ethyl amine.
|
||||
| 1 | TFAc:Et3N | 90 | 50 | 58 |
| 2 | TFA:DIPAd | 90 | 35 | 72 |
| 3 | CF3COONH4e | 90 | 40 | 92 |
| 4 | TFA:DIPEAf | 90 | 25 | 95 |
Antimicrobial activity
All the screened DHTPMs showed moderate to good antibacterial and antifungal activity for the tested stains. It was observed that DHTPMs 5a, 5b, 5d, 5e, 5h and 5k inhibited gram positive bacteria Staphylococus aureus on par with the standard. Among the compounds tested the 3-furyl derivative, 5k and 3-methoxy phenyl, 5e were the most potent showing better activity than the standard at 25 μg/ml concentration Table 3. In the case of Bacillus subtilis inhibition, except for the biphenyl derivative, 5b and the 3-methoxy phenyl, 5e all other DHTPMs were less potent compared to the standard. Similarly for inhibition of Salmonellae Typhi all DHTPMs showed moderate activity and were less potent than the standard. When considering antifungal activity the 3-methyl derivative, 5c exerted better activity compared to the standard. The 3-furyl derivative, 5k and 4-biphenyl derivative, 5n exhibited moderate activity. Cyano, pyridyl compounds showed no activity against the tested stains and neither did the carboxylate derivatives of DHTPMs. Biphenyl substituted derivatives showed better activity than naphthyl substituents. Methyl and methoxy substitutions at the aromatic group enhanced activity of the DHTPMs and fluoro mono substitution at the 2-position of the phenyl ring did not enhance markedly activity when compared with the 3,4-difluoro substitution. Of the heteroaromatic substitutions at the pyrimidine moiety 3-furyl was the most potent and of all the DHTPMs tested. Since all tetrazolo compounds showed moderate to good activity against all screened stains, it can be concluded that DHTPMs is a good potential core moiety to exert antimicrobial activity. Hence, there is a need to further explore the core with a larger number of varying functionalities and libraries of compounds.| Compound | Zone of inhibition (mm) at 25 μg/mL | |||
|---|---|---|---|---|
| Staphylococus aureus | Bacillus subtilis | Salmonellae typhi | Candida albicans | |
| Strep | 10.8 ± 0.31 | 25.8 ± 0.01 | 26.6 ± 0.15 | — |
| Amph-B | — | — | — | 13.5±0.5 |
| 5a | 10 ± 1.9 | 6.1 ± 1.08 | 11.4 ± 0.21 | 3 ± 0.58 |
| 5b | 11 ± 0.5 | 23.6 ± 1.9 | 14.1 ± 1.52 | 5 ± 0.8 |
| 5c | 8.7 ± 0.3 | 10.6 ± 0. 6 | 8.1 ± 0.21 | 11.8 ± 0.58 |
| 5d | 11.5 ± 0.85 | 16.7 ± 0.96 | 11.4 ± 0.68 | — |
| 5e | 13.5 ± .97 | 19.8 ± 0.92 | 14.5 ± 0.25 | — |
| 5f | — | — | — | — |
| 5g | 8 ±0.2 | 1 ± 0.56 | 8 ± 0.69 | —— |
| 5h | 10 ± 0.12 | 7 ± 0.2 | 12 ± 1.03 | 5 ± 0.3 |
| 5i | — | — | — | — |
| 5j | — | — | — | — |
| 5k | 12 ± 1.52 | 9.2 ± 1.86 | 13.2 ± 2.1 | 7.1 ± 3.85 |
| 5l | 6 ± 0.25 | 12 ± 2.4 | 4 ± 1.34 | 4.1 ± 4.0 |
| 5m | 9 ± 0.84 | 6.7 ± 0.29 | 3 ± 1.5 | 5.6 ± 2.4 |
| 5n | 9.7 ± 1.8 | 10.1 ± 1.25 | 16.3 ± 0.87 | 8.7 ± 0.25 |
| 5o | — | — | — | — |
Antioxidant activity
Antioxidants exhibit free radical scavenging activity and hence are useful in the management of diseases. DPPH method,28 reducing power assay and superoxide scavenging method29 were carried out to ascertain the antioxidant potency of synthesised DHTPMs.34Table 4 shows the amount of each compound required for 50% inhibition of DPPH free radical (IC50) or ferric reducing power or scavenging of H2O2. Only potent active DHTPMs of the eight compounds and the standard were tested for in vitro antioxidant activity, all eight DHTPMs showed good activity on par with the standard except 5c and 5n. The 2-naphthyl derivative, 5a and thiazolyl derivative, 5l were identified as the most potent of the DHTPMs with a low IC50 value. DHTPMs 5b, 5m, 5h, 5k exhibited similar potency in comparison with the standard ascorbic acid in terms of the antioxidant activity. Since DHTPMs exerted good antioxidant activity with almost all the functional groups screened, it may be valuable to further investigate the potency of the basic skeleton or the DHTPM core moiety by further deriving the libraries of compounds. The general plausible mechanism (Scheme 3) for the antioxidant activity of the DHTPMs is explained via the oxidation of the dihydro pyrimidines ring leading to aromatisation of the DHTPMs.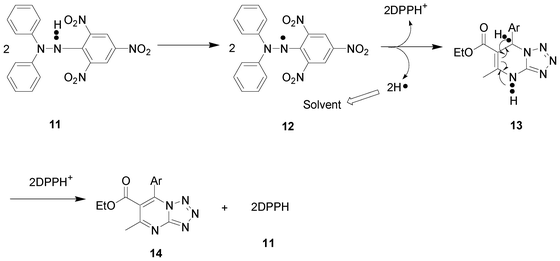 | ||
| Scheme 3 Plausible mechanism for DPPH-radical scavenging activity. | ||
| Entry | DPPH activity | Super oxide scavenging activity | Reducing power assay |
|---|---|---|---|
| 5a | 18 ± 0.2 | 14 ±1.4 | 12 ± 0.36 |
| 5b | 32 ± 0.65 | 28 ± 0.3 | 31 ± .82 |
| 5c | 81 ±.0.15 | 79 ± 0.54 | 80 ± 0.41 |
| 5h | 45 ± 0.1 | 40 ± 0.8 | 38 ± 1.65 |
| 5k | 50 ± .0.4 | 52 ± 0.52 | 52 ±0.21 |
| 5l | 21 ± 0.1 | 22 ± 2.0 | 24 ± 0.74 |
| 5m | 40 ± 0.3 | 52 ±0.1 | 41 ± 2.0 |
| 5n | 65 ± 1.2 | 70 ±1.5 | 61 ± 1.62 |
| Std | 46 ± 0.15 | 34 ± 0.29 | 39 ± 0.22 |
CONCLUSIONS
A new protocol for the synthesis of tetrazolopyrimidines was established using triflate salts as catalysts. In general the reaction times for the synthesis were reduced using microwave-assisted synthesis providing an easier to work up protocol. The antibacterial activity of the DHTPMs against gram-positive bacteria, typhoid causative gram-negative bacteria and antifungal activity to Candida albicans were studied. It was observed that the basic skeleton was active against the bacterial and fungal stains investigated in this study. In addition, the potent antioxidant activity of this active core moiety, i.e. the basic DHTPM skeleton, will be further investigated in our future work.Experimental
Synthesis and characterisation
Common reagents and solvents were purchased from commercial sources. 1H NMR (300 or 400 MHz) and 13C (75 or 100 MHz) NMR spectra were recorded in DMSO-d6 on a Bruker DPX 300 and 400 MHz spectrometer with chemical shifts being reported in parts per million (δ) relative to internal standard tetramethylsilane. IR spectra were obtained as mineral- oil mulls on a Spekord N80 spectrophotometer in the frequency of absorption (cm−1). Low resolution mass spectra were recorded at ionizing voltage (eV) by electron impact. Melting points were determined with Buchi melting B-545. Representative procedure for one-pot synthesis of tetrazolo[1,5-a]pyrimidine-6-carboxylates: A mixture of aldehyde (1 mmol), β-diketo ester/ dimedone (1 mmol), 5-amino tetrazole (1 mmol) and TFA:DIPEA ( 0.5 mmol) was taken in a microwave vial and irradiated at 90 °C for 30 to 45 min. After cooling, solid formed was filtered and washed with cold water (2 X 10ml) followed by diethyl ether, if necessary recrystallised from ethanol or ethyl acetate to afford pure product. Ethyl 5-methyl-7-(naphthalen-2-yl)-4, 7-dihydrotetrazolo [1, 5-a] pyrimidine-6-carboxylate, 5a. The title compound was obtained as a crystalline colourless solid (90%), mp 255-256 °C. IR (KBr) νmax/cm−1 3226 (broad NH), 3160, 3051 (CC), 2944 (C–H), 1696 (CO), 1572 (CN), 1230, 1018 (C–O and/or C–N ). 1H NMR (300MHz, DMSO-d6): δH 0.97 (3H, t, J = 7.08 Hz), 2.48 (3H, s, CH3–CC), 3.93 (2H, q, J = 7.08 Hz, OCH2), 6.81 (1H, s, CH), 7.49–7.47 (1H, d), 7.53–7.50 (2H, m), 7.89–7.87(4H, m), 11.3 (1H, brs, NH).13C NMR: (75 MHz, DMSO-d6): δc 14.3, 18.9, 59.5, 60.1, 98, 125.2, 126.9, 127.0, 128, 128.5, 129, 133, 133.2, 138.7, 147.3, 148.9, 165.07 (CO). MS (ESI) m/z 335.36 (MH−). Microbiology: All the compounds were screened for their antibacterial activity against Gram positive stains of Staphylococcus aureus and Bacillus subtilis; Gram negative bacteria, Salmonella typhi and antifungal activity against Candida albicans. Compounds 5a-o with various substituent’s in the aromatic ring were synthesised and screened to understand the influence of steric and electronic effects on the biological activity. Culture of microorganisms: For evaluating antibacterial activity Streptomycin was used as the standard drug. In general all the synthesized compounds exert a wide range of modest antibacterial activity in vitro against the tested organisms. Antimicrobial activity was carried out using disc-diffusion method.30 Petri plates were prepared with 20 mL of sterile Mueller-Hinton agar (MHA) (Hi-media, Mumbai) and Potato Dextrose Agar for C. albicans. The test cultures, 100 {*BLOB:P*}mL of suspension containing 108 CFU/mL bacteria (CFU= colony forming unit) were swabbed on the top of the solidified media and allowed to dry for 10 min. The tests were conducted with the DHTPMs (dissolved in 5% dimethyl sulfoxide (DMSO), respectively, 10 μg, 25 μg, 50 μg and 100 μg per well). The medium was sterilized by autoclaving at 120 °C. About 30 ml of the medium (nutrient agar medium) with the respective strains of bacteria and fungi was transferred aseptically into each sterilized Petri plate. The Plates were left at room temperature for solidification and 6 mm diameter was made using a sterile borer. The compounds were freshly reconstituted with Dimethyl Sulphoxide and tested at various concentrations. The samples and the control (0.1ml) were placed in 6-mm diameter well. Antibacterial assay plates were incubated at 37 ± 2 °C for 24 h, antifungal assay plates were incubated at 28 ± 2 °C for 48 h. Zone of inhibition was recorded in millimeters and the experiment was repeated twice for concordant results. Antioxidant activity: Reducing power The reducing power of the targets was assessed by the method of developed by Oyaizu.31 Various concentrations of the DHTPMs (2.5 ml) were mixed with 2.5 ml of 200 mM sodium phosphate buffer (pH 6.6) and 2.5 ml of 1% potassium ferricyanide. The mixture was incubated at 50 °C for 30 min. After incubation, 2.5 ml of 10% trichloroacetic acid (w/v) was added; the mixture was centrifuged at 1000 rpm for 10 min. The upper layer (5 ml) was mixed with 5 ml of deionised water and 1 ml of 0.1% of ferric chloride, and the absorbance was measured spectrophotometrically at 700 nm.32 Blank sample was prepared using distilled water instead of DHTPM. The values are presented as the means of triplicate analyses. The DHTPM concentration providing half of the original absorbance (EC50) was calculated from the graph of absorbance at 700 nm against DHTPM concentration. Ascorbic acid was used as standards. DPPH radical scavenging activity: Antioxidant assay is based on the measurements of the scavenging ability of DHTPMs towards the stable 2, 2-diphenyl-1-picrylhydrazyl radical (DPPH).33 The disappearance of this commercially available radical is measured spectrophotometrically at 517 nm in ethanolic solution. The antioxidant activity was expressed as the 50% inhibitory concentration (IC50) based on the amount of compound required for a 50% decrease of the initial DPPH radical concentration. As presented in Table 4, lower IC50 value indicates greater antioxidant activity. The potent antibacterial DHTPMs scavenged DPPH radical significantly in a concentration-dependent manner. Their comparable scavenging activities were expressed in IC50 values. Various concentrations of DHTPMs (0.3 ml) were mixed with 2.7 ml of 70% ethanol solution containing DPPH radicals (40 μg/ml). The mixture was shaken vigorously and left to stand for 60 min in the dark (until stable absorbance value was obtained). The reduction of the DPPH radical was determined by reading the absorbance at 517 nm. The radical-scavenging activity (RSA) was calculated as a percentage of DPPH discoloration, using the equation: RSA = [(ADPPH-AS)/ADPPH] × 100%.
Where AS is the absorbance of the solution when DHTPM was added at a particular level, and ADPPH is the absorbance of the DPPH solution. The DHTPM concentration providing 50% of radical-scavenging activity (IC50) was calculated from the graph of RSA percentage against extract concentration. Ascorbic acid was used as standards. Superoxide Anion Radical Scavenging Activity in PMS-NADH Systems: Measurement of superoxide anion scavenging activity of DHTPMs was based on the method described by;35 with slight modification.36 Superoxide radicals are generated in PMS-NADH systems by oxidation of NADH and assayed by the reduction of nitroblue tetrazolium (NBT). Tris-HCl buffer (3 ml, 16 mM, pH 8.0) containing 1 ml NBT (50 μM) solution, 1 ml NADH (78 μM) solution and a DHTPM solution in water were mixed. The superoxide radical-generating reaction was started by the addition of 1 ml of phenazine methosulfate (PMS) solution (10 μM) to the mixture. The reaction mixture was incubated at 25 °C for 5 min, and the absorbance was read at 560 nm using a UV-Vis spectrophotometer (Jasco V-530, Japan Servo Co. Ltd., Japan) and measured against blank samples. L-ascorbic acid was used as a control. Decreased absorbance of the reaction mixture indicated increased superoxide anion scavenging activity. The percent inhibition of superoxide anion generation was calculated using the following formula37
%Inhibition = [(Ao − A1)/Ao] × 100
where Ao was the absorbance of the control and A1 was the resultant absorbance of DHTPM addition.
Acknowledgements
Author C.R is thankful to the Principal, Pachaiyappa’s College, (an affiliate of the University of Madras) for providing facilities to undertake this work.References
- (a) C. O. Kappe, Tetrahedron, 1993, 49, 6937 CrossRef CAS; (b) A. K. Sharma, S. Jayakumar, M. S. Hundal and P. M. Mahajan, J. Chem. Soc., Perkin Trans. 2002, 1, 774 Search PubMed; (c) S. Sasaki, N. Cho, Y. Nara, M. Harada, S. Endo, N. Suzuki, S. Furuya and M. Fujino, J. Med. Chem., 2003, 46, 113 CrossRef CAS.
- C. O. Kappe, Acc. Chem. Res., 2000, 33, 879 CrossRef CAS.
- I. S. Zorkun, S. Sarac, S. Celebi and K. Erol, Bioorg. Med. Chem., 2006, 14, 8582 CrossRef CAS.
- J. C. Cochran, J. E. Gatial, T. M. Kapoor and S. P. Gilbert, J. Biol. Chem., 2005, 280, 12658 CrossRef CAS.
- S. Brier, D. Lemaire, S. DeBonis, E. Forest and F. Kozielski, Biochemistry, 2003, 43, 13072 CrossRef.
- M. M. Moran, C. Fanger, J. A. Chong, C. McNamara, X. G. Zhen and J. Mandel-Brehm, WO Patent 2,007,073,505, 2007; Chem.Abstr., 2007, 147, 118074 Search PubMed.
- B. B. Snider and Z. Shi, J. Org. Chem., 1993, 58, 3828 CrossRef CAS.
- L. E. Overman, M. H. Rabinowitz and P. A. Renhowe, J. Am. Chem. Soc., 1995, 117, 2657 CrossRef CAS.
- A. Aly, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 1285 CrossRef CAS.
- P. T. Lansbury and Z. H. Liu, Austria Patent 2,006,230,674,2006; Chem. Abstr., 2007, 146, 309–356 Search PubMed.
- M. A. H. Ismail, M. N. Y. Aboul-Einein, K. A. M. Abouzid and S. B. A. Kandil, Alex. J. Pharm. Sci., 2002, 16, 143 CAS.
- I. Drizin, M. W. Holladay, L. Yi, H. Q. Zhang, S. Gopalakrishnan, M. Gopalakrishnan, K. L. Whiteaker, S. A. Buckner, J. P. Sullivan and W. Carroll, Bioorg. Med. Chem. Lett., 2002, 12, 1481 CrossRef CAS.
- S. I. Nagai, T. Ueda, S. Sugiura, A. Nagatsu, N. Murakami, J. Sakakibara, M. Fujita and Y. Hotta, J. Heterocycl. Chem., 1998, 35, 325–327 CrossRef CAS.
- C. Raju, R. Uma, K. Madhaiyan, R. Sridhar and S. Ramakrishna, ISRN Organic chemistry, 2011 DOI:10.5402/2011/273136.
- B. C. Ranu, A. Hajra and A. U. Jana, J. Org. Chem., 2000, 65, 6270–6276 CrossRef CAS.
- A. S. Paraskar, G. K. Dewker and A. Sudalai, Tetrahedron Lett., 2003, 44, 3305–3308 CrossRef CAS.
- A. Debache, B. Boumoud, M. Amimour, A. Belfaitah, S. Rhouati and B. Carboni, Tetrahedron Lett., 2006, 47, 5697–5699 CrossRef CAS.
- A. Shaabani, A. Bazgir and F. Teimouri, Tetrahedron Lett., 2003, 44, 857–859 CrossRef CAS.
- V. Polshettiwar and R. S. Varma, Tetrahedron Lett., 2007, 48, 7343–7346 CrossRef CAS.
- L. Y. Zeng and C. Cai, J. Comb. Chem., 2010, 12, 35–40 CrossRef CAS.
- O. V. Fedorova, M. S. Zhidovinova, G. L. Rusinov and I. G. Ovchinnikova, Russian Chem. Bull., Int. Edn, 2003, 52, 1768–1769.
- C. Yao, S. Lei, C. Wang, C. Yu and S. Tu, J. Heterocyclic Chem., 2008, 45, 1609 CrossRef CAS.
- S. Chitra, D. Devanathan and K. Pandiarajan, Euro. J. med chem., 2010, 45, 367–371 CrossRef CAS.
- V.L. Gein, L. F. Gein, E. P. Tsyplyakova and O. S. Panova, Russ. J. Org. Chem., 2007, 43, 1382 CrossRef CAS.
- V. A. Chebanov, S. M. Sesenko and T. W. Gurley, Six- Membered Azaheterocycles Based on 1,3-Binucleophiles; In Azaheterocycles Based on R, -Unsaturated Carbonyls; Springer: Berlin Heidelberg, 2008, 83–107 Search PubMed.
- (a) V. A. Chebanov, Y. I. Sakhno, S. M. Desenko, S. V. Shishkina, V. I. Musatov, O. V. Shishkin and I. V. Knyazeva, Synthesis, 2005, 2597 CrossRef CAS; (b) C. S. Yao, S. Lei, C. H. Wang, C. X. Yu and S. J. Tu, J. Heterocycl. Chem., 2008, 45, 1609 CrossRef CAS.
- L. Y. Zeng, F. Ji and C. Cai, J. Heterocycl. chem., 2012, 49, 237–241 CrossRef CAS.
- (a) I. I. Koleva, T. A. Van Beek, J. P. H. Linseen, A. De Groot and L. N. Evstatieva, Phytochem. Anal., 2002, 13, 8–17 CrossRef CAS; (b) P. K. Suresh, S. Sucheta, V. D. Sudarshana, P. Selvamani and S. Latha, Afr. J. Biotechnol., 2008, 7, 1826–1828 Search PubMed.
- B. Halliwell and J. M. C. Gutteridge, Lipid peroxidation: a radical chain reaction. In: Free radicals in biology and medicine. Oxford University Press, Oxford ( 1985) 139–189 Search PubMed.
- M. Wettasinghe and F. Shahidi, Food Chem., 2000, 70, 17–26 CrossRef CAS.
- R. S. L. Taylor, N. P. Manandhar, J. B. Hudson and G. H. N. Towers, J. Ethnopharmacol., 1995, 546, 153–159 CrossRef.
- (a) M. Oyaizu, Japanese Journal of Nutrition, 1986, 40, 307–315 CrossRef; (b) W. Brand-Williams, M. Cuvelier and C. Berset, Lebensmittel-Wissenschaft und -Technologie, 1995, 28, 25–30 CrossRef CAS; (c) L. Barros, P. Baptista and I. C. F. R. Ferreira, Food and Chemical Toxicology, 2007, 45, 1731–1737 CrossRef CAS.
- W. M. Zhang, B. Li, L. Han and H. D. Zhang, Antioxidant activities of extracts from areca (ARECA CATECTU L.) flower, husk and seed. EJEAFChe, 2009, 8, 740–748 Search PubMed.
- C. Sánchez-Moreno, Food Sci., and Tech. Intl., 2002, 8, 121–137 Search PubMed.
- F. Liu, V. E. C. Ooi and S. T. Chang, Life Sci., 1997, 60, 763–771 CrossRef CAS.
- I. Gulcin, M. Oktay, E. Kirecci and O. I. Kufrevioglu, Food Chem., 2002, 83, 371–382 CrossRef.
- X. Y. Ye, H. X. Wang, F. Liu and T. B. Ng, Int. J. Biochem. Cell B, 2000, 32, 235–241 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |