En route towards α-benzotriazoyl nitroso derivatives†
Jean-Christophe M.
Monbaliu
ab,
Lucas K.
Beagle
a,
Judit
Kovacs
a,
Matthias
Zeller
c,
Christian V.
Stevens
b and
Alan R.
Katritzky
*ad
aCenter for Heterocyclic Compounds, Department of Chemistry, University of Florida, Gainesville, FL 32611-7200, USA. E-mail: katritzky@chem.ufl.edu
bDepartment of Sustainable Organic Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, B-9000 Ghent, Belgium
cDepartment of Chemistry, Youngstown State University, Youngstown, OH-44555, USA
dChemistry Department, King Abdulaziz University, Jeddah, 21589 Saudi Arabia
First published on 6th August 2012
Abstract
A new class of geminally-substituted nitroso compounds, i.e. α-benzotriazoyl nitroso derivatives, is presented. These compounds display a rather different behavior than other related nitroso compounds bearing a geminal electron-withdrawing group. An unexpected and spontaneous oxidation to the nitro analog is observed in solution.
1 Introduction
Nitroso compounds have emerged during the last decades as prominent reagents for the hydroxylation and amination of a wide variety of substrates, leading to useful 1,2-oxazines, nitrones and other aminohydroxylated building blocks.1a–e Numerous examples of nitroso Diels–Alder cycloaddition (nDA),2a–c nitroso-ene3a–c and nitroso-aldol4a–b reactions are already known. Recently, Srivastava and coworkers reported a promising gold-catalyzed annulation of nitroso arenes and alkynes.5 The nDA has been one of the most successfully incorporated nitroso reactions as the key step in the total synthesis of a broad range of biologically active molecules.1a,6a–m A wide variety of nitroso reagents have emerged, ranging from the very reactive (transient) acyl nitroso compounds to the rather stable nitroso arenes.7a,b Among these, geminally functionalized nitroso compounds (α-EWG-NO) are of particular interest: they have offered synthetic advantages in affording free 1,2-oxazines upon solvolysis of the initial Diels–Alder cycloadduct,7b and also found biological applications for the release of nitrogen oxide (NO) and nitroxyl (HNO).8a–e α-Chloro-9 and α-acetoxy nitroso10a–c derivatives are amongst the most commonly used α-EWG-NO dienophiles, although α-cyanonitroso dienophiles have been also reported.11Their in vitro and in vivo NO/HNO-related vasodilating activities were spotlighted in 2000 by Gasco and coworkers.8e A few years later, a seminal study by King8d led to the development of a series of promising α-acetoxy nitroso compounds which slowly release HNO upon hydrolysis under physiological conditions and have been proven to be vasorelaxant.8a,b In 2009, Toone et al. demonstrated that α-cyano nitroso compounds behave as NO donors.8c
This context has prompted us to assess α-benzotriazoyl nitroso derivatives as a new class of reagents. 1H-Benzotriazole has been shown to be an extremely versatile synthetic auxiliary in organic chemistry.12a–d We now document the synthesis, properties and reactivity of a new class of α-EWG nitroso compounds. X-ray, kinetic and computational studies rationalise the phenomena observed.
2 Results and discussion
2.1 Preparation of α-benzotriazoyl nitroso derivatives
Our first attempt to synthesize α-benzotriazoyl nitroso derivatives from their corresponding oximes 1a–c (Scheme 1) utilized 1-chloro-1H-benzo[d][1,2,3]triazole (1-CBT, 2) as an oxidizer.13a–d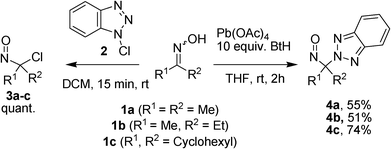 | ||
| Scheme 1 Reactivity of 1-CBT and preparation of α-benzotriazoyl nitroso derivatives. | ||
1-CBT (2) was prepared on a large scale according to a reported procedure14 and obtained in high purity after recrystallization.15 1-CBT (2) displays global16a–h and local17 electronic properties of interest for an oxidizer (Fig. 1): a moderate electrophilicity (ω = 1.7 eV), in contrast with other commonly used oxidizers which often lead to the overoxidation of oximes to nitro,18a,b and a strongly electropositive chlorine atom (f+Cl = 0.64) combined with the potential release of the nucleophilic benzotriazolate anion in situ.
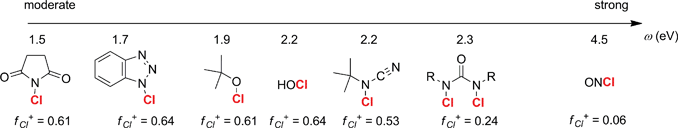 | ||
| Fig. 1 Global and local electronic properties for a selected series of nitrosation reagents (see refs 18a,b and 19a–d for experimental details). The compounds are scaled according to their global electrophilicity (ω) and local Fukui functions f+Cl are indicated for the chlorine atom. The global electrophilicity was computed according to the procedure introduced by Domingo16a–h and the Fukui functions were obtained using the procedure of Contreras.17 | ||
Despite these very promising features, the reaction of several oximes (1 in Scheme 1) in the presence of 1-CBT led exclusively to the formation of the corresponding α-chloro nitroso derivatives 3a–c in quantitative yields (Scheme 1). The incorporation of various additives (1H-benzo[d][1,2,3]triazole, sodium benzotriazolate) in the protocol did not affect the reaction. Computed heterolytic and homolytic bond dissociation enthalpies of 1-CBT revealed that the likelihood of a radical mechanism is much higher (ΔHhomo = 48.4, 44.8 and 44.7 kcal mol−1 in gas phase, THF and DCM, respectively) than the expected heterolytic mechanism (ΔHhetero = 318.9, 206.8, 203.7 kcal mol−1 in gas phase, THF and DCM, respectively), in agreement with our experimental observations (see Supporting Information† for details).
Recent work by King8d and by Kouklovsky and Vincent10b suggested PbIV(OAc)4 and iodobenzene diacetate-based reagents, and we obtained the best results from a preformed mixture of lead tetraacetate and benzotriazole (10 equiv.). Alkyl oximes derived from acetone (1a), 2-butanone (1b), and cyclohexanone (1c) led to their corresponding α-benzotriazoyl nitroso derivatives 4a (55%), 4b (51%) and 4c (74%) in dry THF at room temperature (Scheme 1).20 In dry dichloromethane, the corresponding yields were considerably lower.
2.2 Properties of α-benzotriazoyl nitroso derivatives
The behavior of nitroso compounds 4a,b differed from that of 4c. Compounds 4a,b formed white powders, but bright blue solutions, suggesting that the azodioxy dimer is the major form in the solid state but dissociates in solution into the monomeric nitroso derivative. This was confirmed by the observation of a signal at ∼1290 cm−1 in the solid state IR spectra. Contrastingly, compound 4c was isolated as a monomeric nitroso derivative. The shelf-life of compound 4c was over 4 months at room temperature, in deep contrast with the short shelf-life of its chloro- and acetoxy-analogs. The chloro-analog 3c decomposed after 3 days at −10 °C.Compound 4c was submitted for single crystal X-ray diffraction, resulting in interesting information about the solid state and confirming the monomeric nitroso structure (see Supporting Information†). The benzotriazoyl moiety appeared to be exclusively bonded at the quaternary carbon via the N2 position. From the collected X-ray data sets, a single endo conformer of compound 4c was detected (with nitroso group in an equatorial position and endo versus the benzotriazoyl system). This specific conformation could result either from the emergence of favorable π-system interactions, electrostatic repulsion between the lone pair of the nitrogen and the aromatic benzotriazoyl system (exo lone pair effect),21 or a combination of both phenomena. Computations at the B3LYP/6-31+G* level of theory showed that the conformer bearing the 2Bt substituent in an axial position and the nitroso moiety in a relative equatorial endo position was indeed the most stable. This specific relative position allows for the antiperiplanar alignment of the NO nitrogen lone pair and the σ*C-Bt2 and therefore increased vicinal negative hyperconjugation.22 A NBO23 analysis confirmed this assumption and showed that the nitrogen N2 lone pair on the benzotriazole substituent was similarly involved in a strong stabilizing negative hyperconjugation with the σ*C-NO, consequently weakening the corresponding σ bond.
2.3 Reactivity of α-benzotriazoyl nitroso derivatives
The computed global electrophilicity (ω) of α-benzotriazoyl nitroso compound 4c allowed its ranking on the electrophilicity scale.16a–h Compound 4c showed up in the same electrophilicity range (ωBt = 2.6 eV) as its chloro- and acetoxy-analogs (ωCl = 2.6 eV and ωAcO = 2.9 eV), i.e. as a moderate electrophile. Interestingly, compound 4c was inactive as a dienophile even after prolonged reaction time in the presence of an excess of 2,3-dimethylbutadiene or cyclopentadiene in various solvents (see Supporting Information†), while its chloro- and acetoxy-congeners have been known as dienophiles for decades.7b,9,10a-c This experimental observation contrasted with the rather similar computed activation barriers for the cycloaddition of model 2-nitrosopropane (A), benzotriazoyl- (B), chloro (C)- and acetoxy-derivatives (D) (Fig. 2). The cycloaddition step was computed for 2-nitrosopropane (A) in order to scout the impact of the leaving group on the cycloaddition step.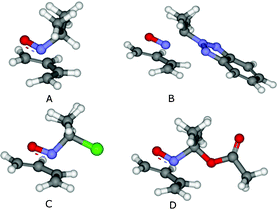 | ||
Fig. 2 Picture of the TSs associated with the cycloaddition of the selected model nitroso compounds onto butadiene. For each situation, 4 isomeric TSs have been isolated due to the relative endo/exo approach of the dienophile and the relative syn/anti orientation of the EWG group vs. N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. The results presented are relative to the most stable TS (endo/anti; see details in the Supporting Information†). Activation free energies (ΔG≠) are indicated in kcal mol−1: A: 26.6; B 27.3; C 27.2; D: 26.5. O. The results presented are relative to the most stable TS (endo/anti; see details in the Supporting Information†). Activation free energies (ΔG≠) are indicated in kcal mol−1: A: 26.6; B 27.3; C 27.2; D: 26.5. | ||
The activation barriers for the cycloaddition step (B3LYP/6-31+G*) were relatively independent of the nature of the leaving group (Fig. 2). To explain the observed lack of dienophile character for compound 4c, two main hypotheses have risen: (i) a more pronounced impact of the nature of the leaving group on the subsequent steps (i.e. elimination and solvolysis) (ii) the existence of a competitive reaction pathway for 4c in solution. The first hypothesis was dismissed on the basis of the excellent abilities of benzotriazole to act as leaving group in a plethora of reactions.12a–d The second hypothesis materialized upon the observation of various side-products depending on the reaction conditions. When solutions of compound 4c in various solvents were left in an open flask for 15 h, compound 4c was quantitatively converted into 2-(1-nitrocyclohexyl)-2H-benzo[d][1,2,3]triazole 6 (Scheme 2), whereas in degassed and dry THF a complex mixture was observed after 15 h, likely arising from the spin-trapping of radicals by 4c.8c,24 Addition of TEMPO to a degassed solution of 4c led to the progressive disappearance of the typical blue coloration of 4c and formed a complex mixture of spin-trapped compounds.8c,24
![Proposed mechanism for the formation of 2-(1-nitrocyclohexyl)-2H-benzo[d][1,2,3]triazole 6 from 4c.](/image/article/2012/RA/c2ra21311g/c2ra21311g-s2.gif) | ||
| Scheme 2 Proposed mechanism for the formation of 2-(1-nitrocyclohexyl)-2H-benzo[d][1,2,3]triazole 6 from 4c. | ||
The rationale for the mechanism depicted in Scheme 2 was built on the following observations and computations: (i) apparent first order kobs = 3.1, 2.0, 1.8 and 1.4 × 10−5 s−1 were determined in methanol, acetonitrile–water, acetonitrile and dichloromethane, respectively (Fig. 3); (ii) the presence of a large excess of oxygen (bubbled through the solution) did not impact on the rate of disappearance; (iii) rather high one-electron redox potentials E° were computed and (iv) isodesmic heat (ΔHiso) for radical exchange, radical stabilization energies (RSE)25 as well as homolytic bond dissociation energies (BDE) for compounds 4c, 4d and 3c were computed (Tables 1–2).
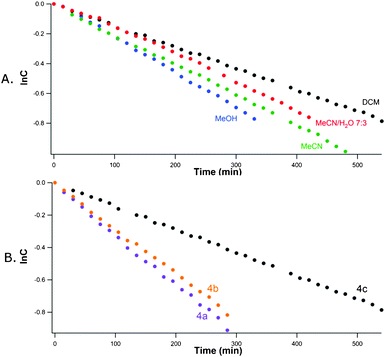 | ||
| Fig. 3 (A.) Apparent (kobs) first order disappearance of 4c in different solvents (top) and of compounds 4a–c in dichloromethane (bottom). (B.) The kinetics for the disappearance of compounds 4a–c were determined by UV-spectrophotometry at 20 °C in different solvents by following the decrease of the typical nitroso signal (λmax = 655.7 nm, 10 mg mL−1). | ||
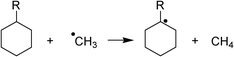
| R | ΔHiso (kcal mol−1) | RSE (kcal mol−1) |
|---|---|---|
| Me | −10.8 | 10.8 |
| Cl | −10.2 | 10.2 |
| Ph | −19.3 | 19.3 |
| 2Bt | −30.9 | 30.9 |
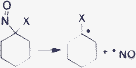
| X | BDE (kcal mol−1) |
|---|---|
| 2Bt (4c) | 22.4 |
| CN (4d) | 19.8 |
| Cl (3c) | 30.6 |
The kinetics display only moderate dependency on solvent polarity which suggested implication of radical species rather than charged intermediates. An additional set of experiments using different concentrations (5 and 20 mg mL−1) for compound 4c in acetonitrile gave different half times (t1/2 = 266.6 and 495.1 min, respectively). This observation is consistent with the formation of an inactive reservoir of nitroso compound 4c as its azodioxy dimer at higher concentration. The second observation, i.e. the apparent lack of impact of an excess of oxygen in the reaction mixture, reinforced the mechanism proposed in Scheme 2. As per Fig. 2, the steric hindrance of the backbone on αC has a profound impact on the reactivity (t1/2 = 223.6 (4a), 247.6 (4b) and 495.1(4c) min).
The one-electron redox potential E° was computed in methanol using standard procedures (see free energy cycles in the Supporting Information†).26a,bE° (MeOH) = 5.5, 6.4 and 6.2 V for 4c, 4d and 3c, respectively (E° = 6.5 V for the reference MeNO) indicated that these compounds are quite resistant to oxidation and require strong oxidizers, in agreement with previously reported data.27a–c
RSE emphasized that the 2Bt substituent stabilizes a radical much better than a phenyl does (Table 1). Further proof was obtained by computing homolytic bond dissociation energies (BDE) for the release of nitric oxide from compounds 4c, 4d and 3c (see Supporting Information† for heterolytic bond dissociation energies). These revealed that the homolytic bond rupture for the benzotriazoyl compound was very close to the value obtained for the reference 4d (Table 2),8c emphasizing its propensity to release the radical intermediate 5 (Scheme 2). The release of NO was not detected by GC-MS; subsequent rapid oxidation of the side product NO by atmospheric oxygen led to nitrogen dioxide,28a which very likely underwent recombination with the stabilized radical 5.28a–d Compound 6, the structure of which has been unambiguously determined by X-ray diffraction, was exclusively formed under aerobic conditions.29
3 Conclusion
In this communication, we have documented the synthesis, properties and reactivity of a new class of reagents, namely α-benzotriazoyl nitroso compounds. α-Benzotriazoyl nitroso compounds were conveniently obtained via the oxidation of the corresponding oximes in the presence of lead tetraacetate and a large excess of benzotriazole. The benzotriazole is anchored to the nitroso-substrate exclusively at the 2N position. Despite comparable activation barriers, α-benzotriazoyl nitroso compounds were not reactive towards dienes, in contrast with their α-chloro and α-acetoxy analogs. An unexpected oxidation – not observed for other α-EWG analogs – was found to occur in solution leading to the exclusive formation of α-benzotriazoyl nitro compounds. The mechanism most likely involves a rate-determining homolytic cleavage of the parent nitroso, releasing NO which is readily oxidized in solution, followed by subsequent recombination to yield the nitro analog.Acknowledgements
This work was supported by the Research Foundation-Flanders (FWO-Vlaanderen; fellowship to JCMM). JCMM acknowledges the ICT Department of Ghent University for access to HPC facilities. The authors acknowledge the Kenan Foundation (University of Florida) and the King Abdulaziz University (Saudi Arabia) for their financial support.References
- (a) B. S. Bodnar and M. J. Miller, Angew. Chem., Int. Ed., 2011, 50, 5630–5647 Search PubMed; (b) H. Yamamoto and M. Kawasaki, Bull. Chem. Soc. Jpn., 2007, 80, 595–607 CrossRef CAS; (c) Y. Yamamoto and H. Yamamoto, Eur. J. Org. Chem., 2006, 2031–2043 CrossRef CAS; (d) S. Iwasa, A. Fakhruddin and H. Nishiyama, Mini-Rev. Org. Chem., 2005, 2, 157–175 Search PubMed; (e) H. Yamamoto and N. Momiyama, Chem. Commun., 2005, 3514–3525 RSC.
- (a) D. Chaiyaveij, L. Cleary, A. S. Batsanov, T. B. Marder, K. J. Shea and A. Whiting, Org. Lett., 2011, 13, 3442–3445 CrossRef CAS; (b) J. M. de los Santos, R. Ignacio, G. Rubiales, D. Aparicio and F. Palacios, J. Org. Chem., 2011, 76, 6715–6725 CrossRef CAS; (c) T. A. Wencewicz, B. Yang, J. R. Rudloff, A. G. Oliver and M. J. Miller, J. Med. Chem., 2011, 54, 6843–6858 Search PubMed.
- (a) A. Yanagisawa, T. Fujinami, Y. Oyokawa, T. Sugita and K. Yoshida, Org. Lett., 2012, 14, 2434–2437 Search PubMed; (b) V. Krchnak, J. Zajícek, P. A. Miller and M. J. Miller, J. Org. Chem., 2011, 76, 10249–10253 Search PubMed; (c) C. P. Frazier, J. R. Engelking and J. Read de Alaniz, J. Am. Chem. Soc., 2011, 133, 10430–10433 CrossRef CAS.
- (a) K. Shen, X. Liu, G. Wang, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4684–4688 CAS; (b) M. Nielsen, D. Worgull, T. Zweifel, B. Gschwend, S. Bertelsen and K. A. Jørgensen, Chem. Commun., 2011, 47, 632–649 RSC.
- S. Murru, A. A. Gallo and R. S. Srivastava, ACS Catal., 2011, 1, 29–31 Search PubMed.
- (a) P. Sancibrao, D. Karila, C. Kouklovsky and G. Vincent, J. Org. Chem., 2010, 75, 4333–4336 Search PubMed; (b) P.-H. Lu, C.-S. Yang, B. Devendar and C.-C. Liao, Org. Lett., 2010, 12, 2642–2645 CrossRef CAS; (c) G. R. Stephenson, A. M. Balfe, D. L. Hughes and R. D. Kelsey, Tetrahedron Lett., 2010, 51, 6806–6809 Search PubMed; (d) W. Lin, K. G. Virga, K.-H. Kim, J. Zajicek, D. Mendel and M. J. Miller, J. Org. Chem., 2009, 74, 5941–5946 Search PubMed; (e) C. Kibayashi, Chem. Pharm. Bull., 2005, 53, 1375–1386 CrossRef CAS; (f) A. Hall, P. D. Bailey, D. C. Rees, G. M. Rosair and R. H. Wightman, J. Chem. Soc., Perkin Trans. 1, 2000, 329–343 RSC; (g) T. Faitg, J. Soulié, J.-Y. Lallemand and L. Ricard, Tetrahedron: Asymmetry, 1999, 10, 2165–2174 CrossRef CAS; (h) A. Hall, P. D. Bailey, D. C. Rees and R. H. Wightman, Chem. Commun., 1998, 2251–2252 RSC; (i) W. Oppolzer, O. Tamura, G. Sundarababu and M. Signer, J. Am. Chem. Soc., 1992, 114, 5900–5902 CrossRef CAS; (j) W. Oppolzer and O. Tamura, Tetrahedron Lett., 1990, 31, 991–994 CrossRef CAS; (k) H. Iida, Y. Watanabe and C. Kibayashi, Tetrahedron Lett., 1984, 25, 5091–5094 CrossRef CAS; (l) N. J. Leonard, A. J. Playtis, F. Skoog and R. Y. Schmitz, J. Am. Chem. Soc., 1971, 93, 3056–3058 CrossRef CAS; (m) B. Belleau and Y.-K. Au-Young, J. Am. Chem. Soc., 1963, 85, 64–71 CrossRef CAS.
- (a) J.-C. Monbaliu, G. Dive, J. Marchand-Brynaert and D. Peeters, THEOCHEM, 2010, 959, 49–54 Search PubMed; (b) J. Streith and A. Defoin, Synthesis, 1994, 11, 1107–1117 Search PubMed.
- (a) M. E. Shoman, J. F. DuMond, T. S. Isbell, J. H. Crawford, A. Brandon, J. Honovar, D. A. Vitturi, C. R. White, R. P. Patel and S. B. King, J. Med. Chem., 2011, 54, 1059–1070 Search PubMed; (b) J. F. DuMond and S. B. King, Antioxid. Redox Signaling, 2011, 14, 1637–1648 Search PubMed; (c) H. Chakrapani, M. D. Bartberger and E. J. Toone, J. Org. Chem., 2009, 74, 1450–1453 Search PubMed; (d) X. Sha, T. S. Isbell, R. P. Patel, C. S. Day and S. B. King, J. Am. Chem. Soc., 2006, 128, 9687–9692 CrossRef CAS; (e) A. Di Stilo, C. Medana, B. Ferrarotti, A. L. Gasco, D. Ghigo, A. Bosia, P. A. Martorana and A. Gasco, Pharmacol. Res., 2000, 41, 469–474 Search PubMed.
- R. Bou-Moreno, S. Luengo-Arratta and W. B. Motherwell, Tetrahedron Lett., 2011, 52, 2097–2099 Search PubMed.
- (a) G. Calvet, S. C. Coote, N. Blanchard and C. Kouklovsky, Tetrahedron, 2010, 66, 2969–2980 Search PubMed; (b) H. Li, D. Gori, C. Kouklovsky and G. Vincent, Tetrahedron: Asymmetry, 2010, 21, 1507–1510 Search PubMed; (c) G. Calvet, M. Dussaussois, N. Blanchard and C. Kouklovsky, Org. Lett., 2004, 6, 2449–2451 CrossRef CAS.
- V. Gouverneur and L. Ghosez, Tetrahedron, 1996, 52, 7585–7598 CrossRef CAS.
- (a) J.-C. M. Monbaliu, F. K. Hansen, L. K. Beagle, M. J. Panzner, P. J. Steel, E. Todadze, C. V. Stevens and A. R. Katritzky, Chem.–Eur. J., 2012, 18, 2632–2638 Search PubMed; (b) A. R. Katritzky, P. Angrish and E. Todadze, Synlett, 2009, 2392–2411 CrossRef CAS; (c) A. R. Katritzky and B. V. Rogovoy, Chem.–Eur. J., 2003, 9, 4586–4593 CrossRef CAS; (d) A. R. Katritzky, X. Lan, J. Z. Yang and O. V. Denisko, Chem. Rev., 1998, 98, 409–548 CrossRef CAS.
- (a) N. Stellenboom, R. Hunter and M. R. Caira, Tetrahedron, 2010, 66, 3228–3241 Search PubMed; (b) R. Hunter, M. Caira and N. Stellenboom, J. Org. Chem., 2006, 71, 8268–8271 CrossRef CAS; (c) K. Ganapathy, R. Gurumurthy, N. Mohan and G. Sivagnanam, Monatsh. Chem., 1987, 118, 583–587 Search PubMed; (d) C. W. Rees and R. C. Storr, J. Chem. Soc. (C), 1969, 1474–1477 RSC.
- T. V. Hughes, S. D. Hammond and M. P. Cava, J. Org. Chem., 1998, 63, 401–402 CrossRef CAS.
- Caution! 1-Chloro-1H-benzo[d][1,2,3]triazole (1-CBT) is a shock- and heat-sensitive material. There is little to no specific hazard associated with the synthesis of 1-CBT as reported in ref. 14. The recrystallization in boiling hexane–dichloromethane must be carried out on a small scale (<1 g) behind a blast shield. The white crystals obtained after recrystallization (mp 108.0–109.0 °C) are extremely shock sensitive. Thermogravimetric (TGA) analysis showed that 1-CBT starts decomposing at 90 °C; at 113.5 °C, 50.3% wt of the sample was lost. The heat of combustion was measured: ΔUc = 0.82 Mcal mol−1.
- (a) L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168–7175 RSC; (b) L. R. Domingo and J. A. Sáez, Org. Biomol. Chem., 2009, 7, 3576–3583 RSC; (c) L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS; (d) L. R. Domingo, J. A. Sáez and P. Pérez, Chem. Phys. Lett., 2007, 438, 341–345 CrossRef CAS; (e) M. J. Aurell, L. R. Domingo, P. Pérez and R. Contreras, Tetrahedron, 2004, 60, 11503–11509 CrossRef CAS; (f) P. Perez, L. R. Domingo, M. J. Aurell and R. Contreras, Tetrahedron, 2003, 59, 3117–3125 CrossRef CAS; (g) L. R. Domingo, P. Perez and R. Contreras, J. Org. Chem., 2003, 68, 6060–6062 CrossRef CAS; (h) L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417–4423 CrossRef CAS.
- R. R. Contreras, P. Fuentealba, M. Galvan and P. Perez, Chem. Phys. Lett., 1999, 304, 405–413 CrossRef CAS.
- (a) A. H. A. Mohammed and G. Nagendrappa, J. Chem. Sci., 2010, 122, 571–577 Search PubMed; (b) A. O. Terent'ev, I. B. Krylov, Y. N. Ogibin and G. I. Nikishin, Synthesis, 2006, 22, 3819–3824 Search PubMed.
- (a) A. K. Gupta, J. Acharya, D. Pardasani and D. K. Dubey, Tetrahedron Lett., 2007, 48, 767–770 Search PubMed; (b) V. Kumar and M. P. Kaushik, Tetrahedron Lett., 2005, 46, 8121–8123 Search PubMed; (c) A. Zaks, A. V. Yabannavar, D. R. Dodds, C. Anderson Evans, P. R. Das and R. Malchow, J. Org. Chem., 1996, 61, 8692–8695 CrossRef CAS; (d) H. Diekmann and W. Luttke, Angew. Chem., Int. Ed. Engl., 1968, 7, 387–388 Search PubMed.
-
Procedure for the synthesis of 2-(1-nitrosocyclohexyl)-2H-benzo[d][1,2,3]triazole
4c
. A solution of lead(IV) tetraacetate (4.43 g, 10.0 mmol) and 1H-benzotriazole (11.81 g, 100.0 mmol) in dry THF (100 mL) was stirred for 15 min at 0 °C. The resulting homogeneous solution was treated dropwise with a solution of cyclohexanone oxime (1.13 g, 10.0 mmol) in dry THF (10 mL) over 15 min at 0 °C. After 2 h, the solvent was removed under reduced pressure and the brown residue was triturated several times with hexanes (5 × 50 mL) containing 10% of ethyl acetate. The combined organic fractions were evaporated under reduced pressure and the greenish oily residue was purified by column chromatography over silica gel (hexanes–ethyl acetate 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 2-(1-nitrosocyclohexyl)-2H-benzo[d][1,2,3]triazole as a blue solid. Yield: 74% (1.70 g, 7.4 mmol), blue microcrystals. m.p. 125.0–127.0 °C. 1H NMR (300 MHz, CDCl3): δ = 1.34–1.51 (m, 2H), 1.55–1.67 (m, 1H), 1.67–1.80 (m, 1H), 1.94–2.06 (m, 2H), 2.65–2.86 (m, 4H), 7.36–7.45 (m, 2H), 7.84–7.93 (m, 2H) ppm. 13C NMR (75 MHz, CDCl3): δ = 21.7, 24.6, 29.2, 118.7, 127.0, 128.1, 144.9 ppm. Elemental analysis calcd (%) for C12H14N4O1: C, 62.59; H, 6.13; N, 24.33; found: C, 62.20; H, 5.90; N, 24.41. Crystal data for compound 4c: blue crystal (plates), dimensions 0.4 × 0.1 × 0.04 mm, crystal system monoclinic, space group P21/c, Z = 4, a = 11.7352(5), b = 8.5659(3), c = 12.1021(4) Å, β = 110.616(2)°, V = 1138.63(7) Å3, ρ = 1.349 g cm−3, T = 100(2) K, Θmax = 30.60°, radiation Mo-Kα, λ = 0.71073 Å, 0.3 ω-scans with CCD area detector, covering a whole sphere in reciprocal space, 14435 reflections measured, 3407 unique (Rint = 0.0218), 2873 observed (I > 2σ(I)), intensities were corrected for Lorentz and polarization effects, an empirical absorption correction was applied using SADABS22 based on the Laue symmetry of the reciprocal space, m = 0.091 mm−1, Tmin = 0.6840, Tmax = 0.7461, structure solved by direct methods and refined against F2 with a full-matrix least-squares algorithm using the SHELXL-97 software package, 164 parameters refined, hydrogen
atoms were treated using appropriate riding models, goodness of fit = 1.069 for observed reflections, final residual values R1(F) = 0.0376, wR(F2) = 0.0976 for observed reflections. CCDC 883509†.
:1) to give 2-(1-nitrosocyclohexyl)-2H-benzo[d][1,2,3]triazole as a blue solid. Yield: 74% (1.70 g, 7.4 mmol), blue microcrystals. m.p. 125.0–127.0 °C. 1H NMR (300 MHz, CDCl3): δ = 1.34–1.51 (m, 2H), 1.55–1.67 (m, 1H), 1.67–1.80 (m, 1H), 1.94–2.06 (m, 2H), 2.65–2.86 (m, 4H), 7.36–7.45 (m, 2H), 7.84–7.93 (m, 2H) ppm. 13C NMR (75 MHz, CDCl3): δ = 21.7, 24.6, 29.2, 118.7, 127.0, 128.1, 144.9 ppm. Elemental analysis calcd (%) for C12H14N4O1: C, 62.59; H, 6.13; N, 24.33; found: C, 62.20; H, 5.90; N, 24.41. Crystal data for compound 4c: blue crystal (plates), dimensions 0.4 × 0.1 × 0.04 mm, crystal system monoclinic, space group P21/c, Z = 4, a = 11.7352(5), b = 8.5659(3), c = 12.1021(4) Å, β = 110.616(2)°, V = 1138.63(7) Å3, ρ = 1.349 g cm−3, T = 100(2) K, Θmax = 30.60°, radiation Mo-Kα, λ = 0.71073 Å, 0.3 ω-scans with CCD area detector, covering a whole sphere in reciprocal space, 14435 reflections measured, 3407 unique (Rint = 0.0218), 2873 observed (I > 2σ(I)), intensities were corrected for Lorentz and polarization effects, an empirical absorption correction was applied using SADABS22 based on the Laue symmetry of the reciprocal space, m = 0.091 mm−1, Tmin = 0.6840, Tmax = 0.7461, structure solved by direct methods and refined against F2 with a full-matrix least-squares algorithm using the SHELXL-97 software package, 164 parameters refined, hydrogen
atoms were treated using appropriate riding models, goodness of fit = 1.069 for observed reflections, final residual values R1(F) = 0.0376, wR(F2) = 0.0976 for observed reflections. CCDC 883509†. - The exo lone pair effect, i.e. the preferential endo approach of nitrosodienophiles in Diels–Alder reactions, has been introduced by Houk et al. (see M. A. McCarrick, Y.-dong Wu and K. N. Houk, J. Am. Chem. Soc., 1992, 114, 1499–1500 Search PubMed ). The minimization of the electrostatic repulsions between the diene and the dienophile leads to a highly favored endo approach, releasing the nitrogen lone pair of the nitroso in the exo position.
- I. V. Alabugin, K. M. Gilmore and P. W. Peterson, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 109–141 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- G. Kaupp and J. Schmeyers, J. Org. Chem., 1995, 60, 5494–5503 CrossRef CAS.
- M. L. Coote, C. Y. Lin, A. L. J. Beckwith and A. A. Zavitsas, Phys. Chem. Chem. Phys., 2010, 12, 9597–9610 RSC.
- See for example: (a) N. Shao, X.-G. Sun, S. Dai and D.-en Jiang, J. Phys. Chem. B, 2012, 116, 3235–3238 CrossRef CAS; (b) N. Shao, X.-G. Sun, S. Dai and D.-en Jiang, J. Phys. Chem. A, 2011, 115, 12120–12125 Search PubMed.
- (a) Y. Ogata and T. Akada, Tetrahedron, 1972, 28, 15–19 CrossRef CAS; (b) T. G. Bonner and R. A. Hancock, J. Chem. Soc. (B), 1970, 519–524 RSC; (c) K. M. Ibne-Rasa, C. G. Lauro and E. J. O, J. Am. Chem. Soc., 1963, 85, 1165–1167 CrossRef CAS.
- (a) P. Astolfi, P. Carloni, E. Damiani, L. Greci, M. Marini, C. Rizzoli and P. Stipa, Eur. J. Org. Chem., 2008, 3279–3285 Search PubMed; (b) B. G. Gowenlock, B. King, J. Pfab and M. Witanowski, J. Chem. Soc., Perkin Trans. 2, 1998, 483–485 RSC; (c) B. G. Gowenlock, J. Pfab and V. M. Young, J. Chem. Soc., Perkin Trans. 2, 1997, 1793–1798 RSC; (d) G. L. Squadrito, F. R. Fronczek, S. F. Watkins, D. F. Church and W. A. Pryor, J. Org. Chem., 1990, 55, 4322–4326 Search PubMed.
-
Procedure for the synthesis of 2-(1-nitrocyclohexyl)-2H-benzo[d][1,2,3]triazole
6
. A solution of 2-(1-nitrosocyclohexyl)-2H-benzo[d][1,2,3]triazole 4c (1.15 g, 5.0 mmol) in 50 mL dry methanol was stirred for 15 h in an open flask. The solvent was evaporated and the resulting white solid was recrystallized from a 10:1 hexanes–dichloromethane mixture. Yield: 92% (1.21 g, 4.9 mmol), white microcrystals. m.p. 143.0–145.0 °C. 1H (300 MHz, CDCl3): 1.30–1.72 (m, 4H), 1.86–1.99 (m, 2H), 2.72 (dt, J = 13.1, 13.1, 4.0 Hz, 1H), 3.44–3.44 (m, 2H), 7.41–7.49 (m, 2H), 7.86–7.95 (m, 2H) ppm. 13C NMR (75 MHz, CDCl3): δ = 22.6, 24.0, 34.1, 105.1, 119.1, 128.1, 145.0 ppm. Crystal data for compound 6: white crystal (rods), dimensions 0.55 × 0.29 × 0.26 mm, crystal system orthorhombic, space group P212121, Z = 4, a = 5.9246(9), b = 11.7114(19), c = 16.898(3) Å, β = 90.00°, V = 1172.5(3) Å3, ρ = 1.395 g cm−3, T = 100(2) K, Θmax = 31.73°, radiation Mo-Kα, λ = 0.71073 Å, 0.3 ω-scans with CCD area detector, covering a whole sphere in reciprocal space, 8626 reflections measured, 2124 unique (Rint = 0.0554), 2045 observed (I > 2σ(I)), intensities were corrected for Lorentz and polarization effects, an empirical absorption correction was applied using SADABS22 based on the Laue symmetry of the reciprocal space, m = 0.099 mm−1, Tmin = 0.6747, Tmax = 0.7463, structure solved by direct methods and refined against F2 with a full-matrix least-squares algorithm using the SHELXL-97 software package, 163 parameters refined, hydrogen atoms were treated using appropriate riding models, goodness of fit = 1.071 for observed reflections, final residual values R1(F) = 0.0346, wR(F2) = 0.0888 for observed reflections. CCDC 883510†.
Footnote |
| † Electronic Supplementary Information (ESI) available: Detailed computational procedures, cartesian coordinates for the isolated transitions states, kinetic data, 1H and 13C NMR for compounds 4c and 6 and thermogravimetric analysis of 1-CBT. CCDC 883509-883510. See DOI: 10.1039/c2ra21311g/ |
| This journal is © The Royal Society of Chemistry 2012 |