Immobilized functional ionic liquids: efficient, green, and reusable catalysts
Hu
Li
,
Pinaki S.
Bhadury
,
Baoan
Song
* and
Song
Yang
*
Center for Research and Development of Fine Chemicals, State Key Laboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Guizhou University, Guiyang, 550025, P. R. China. E-mail: fcc.basong@gzu.edu.cn; jhzx.msm@gmail.com; Fax: +86-851-829-2170; Tel: +86-851-829-2171
First published on 25th September 2012
Abstract
Ionic liquids (ILs) which are made up of cationic and anionic components can be designed to possess a definite set of properties. In this context, the term “designer solvents” has been used to demonstrate the potential of these environment-friendly ionic liquids in chemical reactions. Since these liquids are able to dissolve several transition metal complexes, they have often been employed in recent times in several catalytic reactions to enhance reaction rates and selectivities. The concept of “immobilized” liquids has been derived from supported liquid phase catalysts; the immobilization process transferring the desired catalytic properties of the liquids to solid catalysts could combine the advantages of ILs with those of heterogeneous support materials and various catalytically functional groups or active species. The immobilized functional ionic liquids (IFILs) capable to restrict many of the negative effects of the conventional ILs have successfully been used in various potent catalytic areas affording high catalytic activity. In this review, structural characteristics, properties and preparation of immobilized functional ionic liquids have been described and the results are compared with those of traditional ILs. Special emphasis has been paid to comprehend the mechanism of various catalytic processes using immobilized ionic liquids functionalized by different groups.
 Hu Li | Hu Li was born in Xiaogan, Hubei province, P. R. of China, in 1986. He graduated in chemical biology (National Science Base) and received his B.Sc. degree from Hubei University in 2010. Currently, he is working at Guizhou University for his M.Sc./Ph.D. degree on the catalytic conversion of biomass resources into renewable chemicals and fuels under the supervision of Prof. Pinaki S. Bhadury and Prof. Song Yang. |
 Pinaki S. Bhadury | Dr Pinaki S. Bhadury was born in India, in 1963. He obtained his M.Sc. and M.Tech. degrees from premier Indian institutes IIT Kharagpur (1987) and IIT Kanpur (1989) respectively. He then joined Def. Res and Dev. Org. (DRDO, Min. of Def.) in India where he worked for about 14 years as a Gr. A. Gaz. Scientist. While working in DRDO, he also obtained his doctoral degree awarded by Jiwaji University, Gwalior. He then moved to Dartmouth College, USA to work with renowned Prof. David M. Lemal as a Research Associate in 2004–2005. Since early 2006, he has been working as a Foreign Expert and Professor at Guizhou University, P. R. China. |
 Baoan Song | Dr Baoan Song is a professor at the Center for Research and Development of Fine Chemicals, Guizhou University, Guiyang, P. R. China. He received a B.Sc. degree at Guizhou University, got his M.Sc. degree in Shenyang Institute of Chemical Engineering, Ministry of Chemical Industry, Shenyang, P. R. China, and obtained his Ph.D. degree in Nanjing Agricultural University, Nanjing, P. R. China. For more than 20 years he has been involved in the synthetic applications of heterocyclic molecules in pesticides and catalysis, being an author or coauthor of more than 110 papers and reviews. Currently, Prof. B. A. Song is Director of the State Key Laboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, and Director of the Key Laboratory of Green Pesticide and Bioengineering, Ministry of Education. |
 Song Yang | Dr Song Yang was born in 1974 in Yangzhou, P. R. China. He received a B.Sc. degree at East China University of Science and Technology in 1995 in Shanghai, P. R. China. He then obtained his M.Sc. and Ph.D. degrees at Guizhou University, Guiyang, P. R. China, where he had been working on the synthesis of novel green pesticides (with Prof. B. A. Song). Since Nov. 2005, he is a professor in the Center for Research and Development of Fine Chemicals, Guizhou University. He has authored 12 patents and more than 90 scientific papers in international journals. |
1 Introduction
Ionic liquids (ILs) which are (at least partially organic) considered as salts with a melting point below 100 °C, have attracted considerable interest during the past decade due to their unique properties, such as low vapor pressure, wide liquid range, good conductivity and large electrochemical window.1–8 The homogeneous catalytic reactions using ILs usually present the advantages of high catalytic activity and good selectivity. Nevertheless, their widespread application in catalytic processes is still restricted by several practical problems which have been illustrated by several authors.9–11 One key drawback of homogeneous catalytic reactions is the use of a large amount of ILs as solvents or catalysts which inevitably generate lots of waste materials rendering their disposal extremely difficult and making the overall process unacceptable from environmental viewpoint. In addition, these operations are expensive despite the fact that many ILs are commercially available today. Furthermore, slow substrate diffusion owing to high viscosity of ILs often causes the reaction to proceed in the inter-phase or in the diffusion layer of the catalyst affecting the productivity of the process since a large part of the catalytically active species is not able to take part in the reaction. Again, further difficulties arise while separating organic products from the ionic phase by treatment with conventional solvents. Sometimes the process leads to the extraction of ILs and the catalyst. In addition, pure ILs, meant to minimize the effects of impurities in ILs for catalytic reactions, are very difficult to obtain. Thus, due to aforementioned shortcomings, despite showing great potential in laboratory scale, homogeneous catalysis using ILs has not revealed significant promise in large-scale industrial catalytic applications.Heterogeneous catalysis having advantages of easy separation, reusability, lower catalyst loading and corrosion effects, and the ability to provide practical conveniences in a continuous system have aroused great attentions in various catalytic processes. The concept of “immobilized” or “heterogenised” liquids is well-known from supported liquid phase catalysts.12 The immobilization process aiming to transfer the desired catalytic properties of the liquids to a solid catalyst could combine the advantages of ILs (non volatility, high solvent capacity, etc.) with those of heterogeneous support materials and catalytically functional groups or active species (such as Brønsted acidic groups and metals). When an appropriate amount of ionic liquid is immobilized on a porous solid support material with or without covalent bonds, the formation of ionic liquid layers on the carrier may act as an inert reaction phase to dissolve or combine various homogeneous or heterogeneous catalysts. Although such immobilized functional ionic liquid-phase catalysts appear as solids, the active species dissolved or combined in the ionic liquid phase on the support maintain the attractive features of ionic liquid homogeneous catalysts such as high specificity and dispersion of molecular entities. In addition, compared with biphasic reaction systems, large amounts of ionic liquid which is costly and may affect the economic viability of a potential process are not required in the catalytic systems of immobilized functional ionic liquids (IFILs).
Supported ionic liquid phase (SILP) materials, combining ILs with heterogeneous support materials, are a recent concept where a film of ionic liquid is immobilized on a solid phase. Many reviews have addressed the interactions between ionic liquids and solid supports, and the use of supported ILs in catalysis in different ways, offering to the interested readers a very suitable introduction to properties and applications thereof.13,14 However, various functional groups or active species (usually acting as catalysts) immobilized by different solid carriers through ILs, which may greatly affect or enhance the catalytic activity in many areas of catalysis, are ignored or seldom mentioned. In this review, catalysis by immobilized ionic liquids functionalized with different groups or species are emphasized. In addition, structural features, properties, methods of preparation and selected applications of IFILs are briefly described.
2 Structure, properties, and preparation of immobilized functional ionic liquids
ILs are typically composed of an organic cation (ammonium, phosphonium, pyridinium, imidazolium, guanidinium, pyrrolidinium and many other exotic cations) with a variety of substituents (generally derived from linear alkyl chains) associated either with an inorganic or organic anion. The physicochemical properties of ILs are influenced by both their cationic and their anionic moieties.15,16 Based on their differences in physicochemical properties, ILs could broadly be divided under four heads: (a) aprotic ionic liquids, the majority of ionic liquids, in which the cations are organic molecular ions; (b) protic ionic liquids, formed by the simple transfer of a proton from pure Brønsted acid to pure Brønsted base; (c) inorganic ionic liquids, in both aprotic and protic forms such as Br3ClP+, Cl3S+, ClSO2NH3; (d) solvate (chelate) ionic liquids, forming a largely unstudied class of ILs that needs to be recognized.17 Combinations of different possible cations and anions or functionalization of the alkyl chain(s) normally present on cations could result in a large number of ionic liquids with different properties (acidic, basic, neutral, coordinating and polymerizable ILs).18,19On the other hand, the IFILs generally consist of three different parts,20i.e. the porous support (such as alumina, silica and active carbon), the ILs (always a thin layer on the support surface), and the functional or active groups acting as catalysts or co-catalysts (Brønsted acidic groups, nanoparticles, and metal complexes etc.). Ionic liquids can be immobilized and functionalized in different ways, which can be classified by the interaction between the ionic liquid or its components (anions or cations) and the support material or functional group (or active species) (Fig. 1).
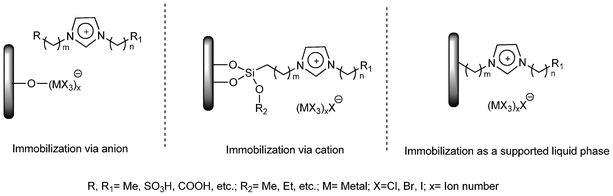 | ||
| Fig. 1 Immobilization of ionic liquids via the anion, the cation or as a supported liquid phase. | ||
The generated IFILs can have easily tunable acidity due to the availability of a vast range of support materials with their characteristic features such as surface area and pore width. Meanwhile, the incorporation of different functional or active groups into the ILs could largely change or enhance the acidity or catalytic activity of IFILs. Furthermore, by changing the length of side-chains of the inorganic cation, the hydrophilicity or hydrophobicity of the surface can be enhanced.21 In comparison to the pure ionic liquids, immobilized ionic liquids offer the additional features that have been discussed in the previous part.
To the best of our knowledge, three different synthetic pathways have been developed for the preparation of the IFIL materials.13,22 The pre- or post-functionalized ILs can be bound to a surface either by covalent bonds between silanol groups and the anion or the cation of the liquid, or without covalent bonds in the form of supported liquid phases. The general synthetic methodologies are outlined below.
Preparation of immobilised functional ionic liquids via active anion groups
This synthetic route is usually called incipient wetness method or immersion method (Scheme 1). By using this method, a solid support material was impregnated with a pre-formed functional ionic liquid containing active group (such as AlCl4−), and the anion group is usually covalently anchored to the support surface via chloroaluminate-based anion, which could further interact with the cation of IL to achieve the aim of immobilisation. The confinement of the ionic liquid to the surface is due to the formation of covalent bonds between the aluminum atoms and the surface OH groups.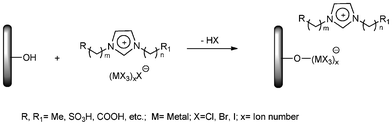 | ||
| Scheme 1 Immobilization of ILs via the anion. | ||
Preparation of immobilized functional ionic liquids via cation groups
As to the method mentioned above, the leaching of the ILs and metal-complex catalyst seems unavoidable, in particular under rigorous reaction conditions. To overcome this drawback, the grafting method or covalent anchoring method has been established for the preparation of the IFILs. In this approach, instead of adding the preformed ionic liquid to the solid support, an organic anchor group was covalently attached to the surface and was used as the cation in the formation of the ionic liquid. And a thin film of ILs can form on the surface of the support, which may be beneficial for further introducing catalytically active species to functionalize the immobilized ILs. The typical synthetic method is described in Scheme 2.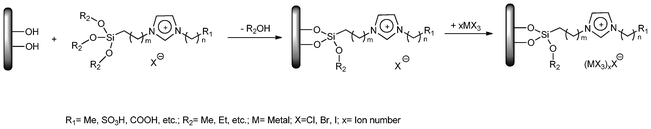 | ||
| Scheme 2 Immobilization of ILs via the cation. | ||
Preparation of immobilized functional ionic liquids via solid supports
Although the leaching of catalytically active species can be avoided in the second method, the chemical bonding of the dialkyl imidazolium cation to a solid surface may limit the degrees of freedom of the dialkyl imidazolium cation and even change the physicochemical properties of the ILs. Another approach via sol–gel synthesis, named physical confinement or encapsulation method, has received much attention in recent IL-based catalysis because of its attractive feactures.23,24 In this method, the ILs, either with or without the catalysts, can be physically confined or encapsulated into the pores of solid supports (Scheme 3). In contrast to the earlier studies, the ionic liquids used here were non-acidic and did not undergo reactions with the support.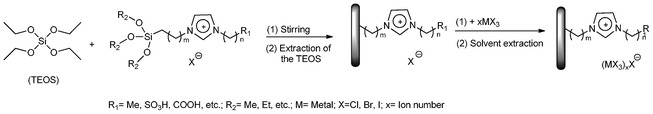 | ||
| Scheme 3 Immobilization of ILs as a supported liquid phase. | ||
Based on these three general synthetic methods, several changes, including the types of support materials, functional groups or active species, and ILs, could be made to satisfy the widespread applications of IFILs in various areas of catalysis. In the following parts, different kinds of reactions catalyzed by these modified IFILs will be presented.
3 Catalysis by immobilized ionic liquids
3.1 CO2 cycloaddition reactions
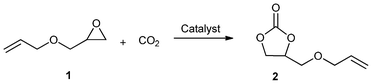
Carbon dioxide (CO2) is the most abundant waste produced by human activities and one of the most important greenhouse gases. Easy dissolution of CO2 in ILs helps to study and execute any reaction that requires this gas as a reactant.25,26 Thus, the use of ionic liquids has been found attractive for the synthesis of cyclic carbonates via CO2 cycloaddition reactions. In recent years, in an attempt to achieve excellent yields and selectivity for the production of carbonates, many efforts have been made to prepare immobilized ionic liquids (IILs).27–29 Some selected results for the synthesis of cyclic carbonates (2) from the cycloaddition of CO2 on epoxides (1) in the presence of these catalysts are listed in Table 1.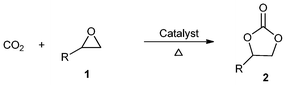
| Entry | R | Temp. (K) | Time | Catalyst | (Yield, %) [Selectivity, %] | Ref. |
|---|---|---|---|---|---|---|
| 1 | H | 383 | 7 h | IIL 1 | (97.4) | 34 |
| 2 | C6H5O | 383 | 3 h | IIL 1 | (96.4) | 34 |
| 3 | ClCH2 | 383 | 3 h | IIL 1 | (95.8) | 34 |
| 4 | C3H5OCH2 | 383 | 10 h | IIL 2 | [98.1] | 35 |
| 5 | C3H5OCH2 | 383 | 6 h | IIL 2 | [99.3] | 36 |
| 6 | C3H5OCH2 | 383 | 6 h | IIL 3 | (93.6) [94.8] | 40 |
| 7 | C6H5 | 413 | 12 h | IIL 4 | (93) | 44 |
| 8 | ClCH2 | 413 | 4 h | IIL 4 | (99) | 44 |
| 9 | CH3 | 413 | 4 h | IIL 4 | (99) | 44 |
| 10 | C6H5O | 413 | 4 h | IIL 4 | (95) | 44 |
| 11 | ClCH2 | 413 | 3 h | IIL 5 | (100) [100] | 59 |
| 12 | CH3 | 413 | 3 h | IIL 5 | (99.6) [100] | 59 |
| 13 | C6H5O | 413 | 6 h | IIL 6 | [100] | 61 |
| 14 | CH3 | 393 | 3 h | IIL 7 | [>99] | 63 |
| 15 | CH3 | 393 | 4 h | IIL 8 | (96) [>99] | 64 |
Polymeric materials are very attractive supports of catalysts as a result of some unusual advantages.30–33 In 2007, Han and his group34 reported a supported IL on a highly cross-linked polymer matrix to prepare an active and insoluble catalyst. The route to synthesize the polymer-supported IL (IIL 1) is shown in Scheme 4. 3-Butyl-1-vinylimidazolium chloride ([VBIM]Cl, 5), first prepared from 1-vinylimidazole (3) and 1-chlorobutane (4), was copolymerized with the cross-linker divinylbenzene (DVB) to afford a highly cross-linked PSIL (IIL 1), in which [VBIM]Cl (5) was covalently anchored on DVB-cross-linked polymer matrix. When IIL 1 was applied for the cycloaddition of CO2 on epoxides, it was observed that the catalyst was very active, selective, and stable, and could be easily separated from the products and reused. In addition, this catalyst was suitable for all the substrates used in their work, and up to 97.4% yield of carbonate was obtained.
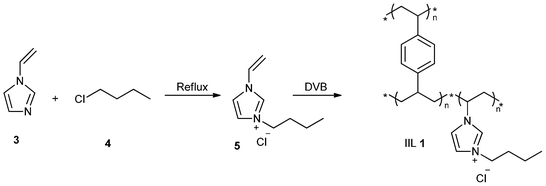 | ||
| Scheme 4 Synthesis of cross-linked-polymer-supported ionic liquid (IIL 1) . | ||
Udayakumar and co-workers35 found that ILs on ordered mesoporous materials are very useful and effective for chemical fixations of CO2, which is a vital step in the production of cyclic carbonates. In their work, trialkylamine via quaternization of hybrid MCM-41 was ionically connected to the surface of supported materials through an appropriate linker which made the active sites available towards the center of pores, leading to the trialkylamine immobilized ionic liquids (IIL 2) with high chemical and thermal stability and high catalytic activity for the synthesis of cyclic carbonates by utilizing CO2 (up to 98.1% selectivity). Then, they proposed a two-step mechanism for the overall reaction (Scheme 5), (i) an reversible reaction between oxirane (1) and immobilized ionic liquid (IIL 2) to form an intermediate complex (6), (ii) an irreversible reaction between intermediate complex (6) and carbon dioxide to form cyclic carbonate (2) and IIL 2.
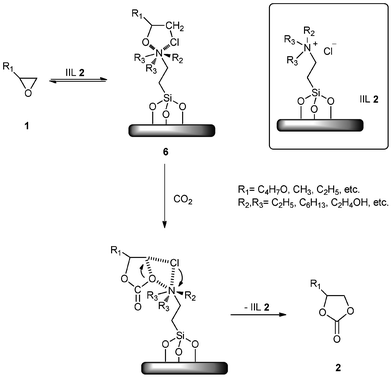 | ||
| Scheme 5 The mechanism for the formation of carbonate (2) through the cyclic intermediate (6) using IIL 2. | ||
Afterwards, Udayakumar et al.36 further examined these hybrid mesoporous materials for the CO2 insertion reaction, and found that the activity of the immobilized ionic liquids (IIL 2) was maintained after recycling and was higher in the IILs containing amines with a lower alkyl chain length (up to 99.3% selectivity). To determine the reaction kinetics between an oxirane and carbon dioxide using a quaternary onium salt catalyst, Park et al.37 proposed a model to first understand the reaction mechanism, and found that CO2 was absorbed into the heterogeneous system of the glycidyl methacrylate (GMA) solution and dispersed with solid particles of the catalyst. Moreover, a linear dependence of the reaction rate constants on the solubility parameter of the solvent was observed.
Although, immobilization of ionic liquids onto MS41 has yielded excellent activities for CO2 cycloaddition reactions, the use of expensive pore-directing agents and large amounts of organic solvents to remove the templates made these catalysts impractical for commercial scale-up. As a result, the ordering of the mesopores often decreased and these problems could be avoided through use of low-cost and highly efficient methods for synthesizing organic-functionalized amorphous silica. The hydrothermal stability of amorphous silica with rigid cross-linking skeletons could also be improved when it was synthesized under highly acidic conditions.38,39
Based on these considerations, Udayakumar and co-workers,40 in 2009, focused on this reaction in the presence of imidazole-based IILs as catalysts and/or solvents that were prepared via a simple co-condensation through direct hydrolysis of a 1-(triethoxysilylpropyl)-3-n-alkyl-imidazolium halide and tetraethyl orthosilicate (TEOS) under strong, acidic conditions without the addition of any structure-directing agents. The obtained IILs (IIL 3), with their general structure shown in Fig. 2, were used to catalyze CO2 cycloaddition reaction to afford carbonates in up to 93.6% yield.
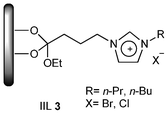 | ||
| Fig. 2 The general structure of imidazole-based IIL 3. | ||
Magnetic nanoparticles (MNPs) have recently appeared as a new type of catalyst support because of their good stability, easy synthetic route, capability for functionalization and facile separation by magnetic forces and high surface area associated with low toxicity and price.41–43 Luo and his group44 prepared three magnetic nanoparticle immobilized IL catalysts (IIL 4), following the procedure shown in Scheme 6. Magnetite nanoparticle (Fe3O4, MNP) which was easily prepared by a co-precipitation method and protected with a layer of silica to prevent aggregation, was chosen as the support. The resulting SiO2–MNP (10) was treated with ionic liquids (9) prepared by quaternazition of N-alkyl imidazole (7) with 3-chloropropyl-trimethoxysilane (8) in refluxing CHCl3 to afford the IIL 4. When used to catalyze the CO2 cycloaddition reaction, pure carbonates from CO2 and various epoxides were obtained quantitatively or in high yields.
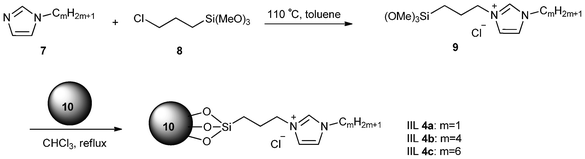 | ||
| Scheme 6 Preparation of IIL 4a–c catalysts. | ||
Layered materials, simultaneously immobilizing ILs on the surface and within the galleries of layered materials to control the release of ILs, could be another group of ideal candidates for the immobilization of ILs. Layered α-zirconium phosphate (α-ZrP), Zr(HPO4)2·H2O, is a particularly ideal candidate as a support to immobilize ILs because of its high ion-exchange capacity, highly ordered structure, ease of synthesis, and ease of crystallization and size control.45–47 However, direct intercalation of ILs into the gallery of α-ZrP in aqueous solution has met with failure.48 In order to solve the problem, Sun and co-workers49 successfully intercalated ILs into the layered α-ZrP via a facile mechanochemical reaction. This IIL was evaluated in catalytic application of CO2 cycloaddition affording up to 55.1% yield.
Most recently, Sun et al.50 perceived the concept that θ-ZrP with increased interlayer distance might act as a more ideal host for intercalation reactions since large species were difficult to directly intercalate into α-ZrP. In the light of this consideration, they again successfully intercalated ILs into the θ-ZrP layered structures in an aqueous dispersion which could readily be adopted by industrial chemical processes. Similarly, the immobilized 1-butyl-3-methylimidazolium chloride (BMIMCl) was evaluated as a catalyst for CO2 fixation and a slightly higher yield of 78.9% was obtained, compared with α-ZrP immobilized ILs.
Cross-linked polymeric nanoparticles (CLPNs) have received increasing attention in both theoretical and applied research fields due to their potential applications as nanocarriers for catalysts, drug carriers, etc.51–53 Until now, several methods have been used to prepare these materials, such as emulsion polymerization, dispersion polymerization and self-assembly.54–56 However, there are still some disadvantages in current approaches used to prepare stable polymeric nanoparticles e.g. the use of surfactants and speciality reagents requiring manipulation and in certain cases pregeneration of block copolymer57,58 Xiong and his group59 explored a facile one-step route for the synthesis of CLPNs (IIL 5) via conventional radical copolymerization of IL monomer 4-vinylbenzyl-tributylphosphorous chloride (PIL, 11) with ethylene glycol dimethacrylate (EGDMA, 12) using azobisisobutyronitrile (AIBN, 13) as initiator (Scheme 7), which was considered as a universal approach for the preparation of IL-based CLPNs. A series of CLPNs (IIL 5) were thus prepared with different molar ratio of EGDMA to PIL in different solvents (such as methanol, ethanol, iso-propanol, and acetone). It was found that these catalysts (IIL 5) presented excellent activity and selectivity in the cycloaddition reaction of CO2 to epoxides with maximum yield of 100% and selectivity of almost 100%.
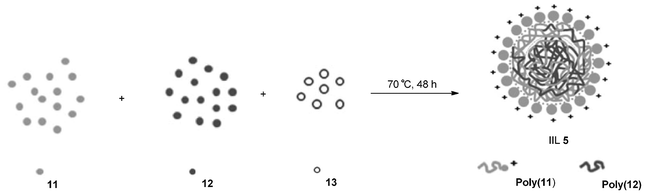 | ||
| Scheme 7 Synthesis of cross-linked polymeric nanoparticles (IIL 5). | ||
Since IL-mediated organic synthesis under microwave irradiation has been found extremely effective,60 a rapid, selective, and solventless synthesis of cyclic carbonate from phenyl glycidyl ether and CO2 was designed by Park et al.61 through microwave heating using supported IL catalysts (IIL 6, Fig. 3). It was also found that among various silica supports including commercial and amorphous silica, SBA-15, and MCM-41, IL catalysts immobilized on silica supports with relatively large pore diameters (greater than 4 nm) exhibited better catalytic activity (up to 100% selectivity).
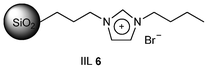 | ||
| Fig. 3 Structure of silica supported ionic liquid (IIL 6). | ||
Recently, Park and coworkers62 further investigated the effect of microwave power on the catalytic performance. In this study, they successfully immobilized tetraoctyl ammonium chloride (TOAC) ionic liquid onto K+-montmorillonite (K10) clay through the adsorption of TOAC onto K10 which showed good catalytic activity without the use of any solvent in the synthesis of cyclic carbonate from allyl glycidyl carbonate and carbon dioxide. In comparison with conventional heating batch operation, microwave assisted reaction was more rapid and selective with a very short reaction time of 20 min at low CO2 pressure (<1.34 MPa).
Solid supports seem to have significant effect on the catalytic activity of IILs. The situation demands development of stable and efficient support materials for catalytic reactions. Till date, some attempts have been made to achieve this objective. Recently, He et al.63 developed a series of polyethylene glycol (PEG)-functionalized basic ionic liquids (IIL 7a–h, Fig. 4), which showed excellent efficiency for converting CO2 into organic carbonates under mild conditions (up to >99% yield and selectivity). This is presumably attributed to simultaneous activation of epoxide assisted by hydrogen bonding and that of CO2 by the ether linkage in the PEG backbone or through the formation of carbamate species with the secondary amino group in the IL cation. Zhang and co-workers64 explored a simple and efficient chitosan (CS) supported 1-ethyl-3-methyl imidazolium bromide catalyst (IIL 8) for the synthesis of cyclic carbonates from epoxides and CO2 without metal co-catalyst and co-solvent. They envisaged a model in which CS might play bi-functional roles in promoting the reaction; the hydrogen bond-assisted ring-opening of epoxide and the nucleophilic tertiary nitrogen-induced activation of CO2 have been proposed (Scheme 8).
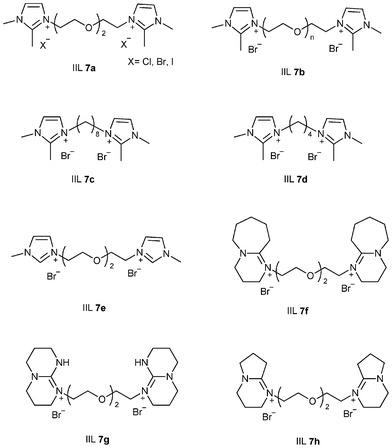 | ||
| Fig. 4 PEG-functionalized ionic liquids (IIL 7a–h). | ||
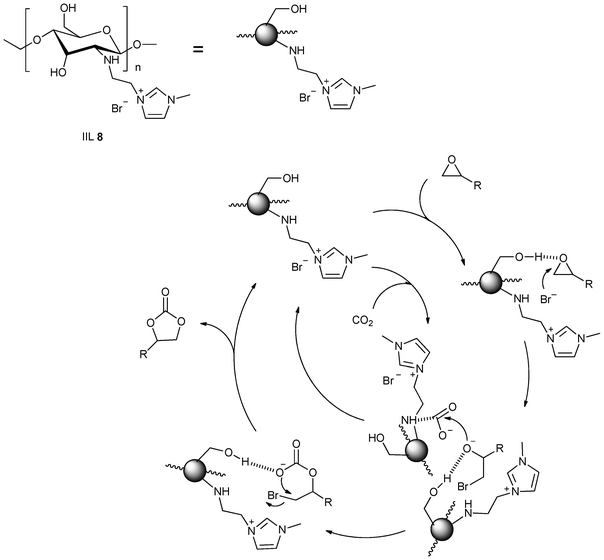 | ||
| Scheme 8 The proposed mechanism for IIL 8 catalyzed CO2 cycloaddition reaction. | ||
3.2 Transesterification reactions
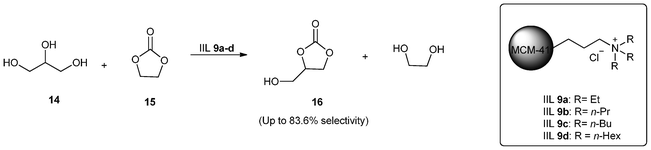
Substituted carbonates have attracted much attention in recent years due to low toxicity, good biodegradability, high boiling point, and versatility as reagent and solvent.65,66 In this regard, a number of synthetic processes such as oxidative carbonylation and other direct synthetic routes have been reported.67,68 Unfortunately, these methods usually suffer from low production rate and yield and often requires employment of corrosion resistant reactors. The transesterification reactions, therefore, offer an excellent green chemical process69 and many homogeneous and heterogeneous catalysts have been developed for this purpose.70,71 Nevertheless, with some of these catalysts, both activity and selectivity were low and several efforts were made to enhance the performance for the synthesis of substituted carbonates using IILs.In 2010, Park and co-workers72 reported the synthesis of glycerol carbonate (GC, 16) by carbonation of glycerol (14) in the presence of ethylene carbonate (EC, 15) catalyzed by the IIL 9 at moderate temperature (Scheme 9). The employed catalysts IIL 9a–d were previously prepared by immobilization of triethylamine (TEA), tri-n-propylamine (TPA), tri-n-butylamine (TBA) and tri-n-hexylamine (THA) onto MCM-41 via condensation method followed by solvent extraction and quaternization. The IILs on MCM-41 showed good catalytic activity for this transesterification reaction without using any solvent (up to 83.6% selectivity) and could be easily recovered and reused without any considerable loss of its initial activity.
In the same year, Park and his group73 demonstrated synthesis of dimethyl carbonate (DMC, 18) from EC (15) and methanol (17) via transesterification reaction using immobilized imidazolium salt as ionic liquid on amorphous silica (IIL 10) as the catalyst which was prepared by a simple co-condensation through direct hydrolysis of 1-(triethoxysilylpropyl)-3-n-alkyl-imidazolium bromide and TEOS under strong acidic conditions without the addition of any structure-directing agent. Under optimal conditions, the immobilized ionic liquid on amorphous silica showed good catalytic activity in the synthesis of DMC from EC and methanol affording 53.9% yield (Scheme 10).
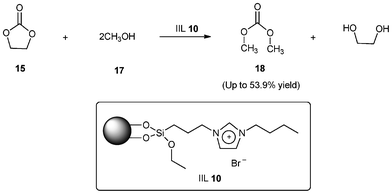 | ||
| Scheme 10 Synthesis of DMC (18) from EC (15) and methanol (17) catalyzed by IIL 10. | ||
Afterwards, Park et al.74 turned to utilize mesoporous molecular sieves CP-MS41 as solid support and quaternary ammonium salt as IL instead of amorphous silica and imidazolium-based IL, respectively. The formed immobilized ILs (IIL 11a–d, Fig. 5) also showed good catalytic activity in the synthesis of DMC from EC and methanol without using any solvent (up to 82.2% yield).
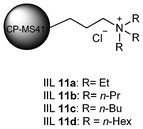 | ||
| Fig. 5 CP-MS41 immobilized ILs (IIL 11a–d). | ||
3.3 Oxidation reactions
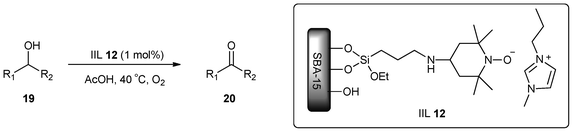
The oxidation of alcohols with molecular oxygen in the presence of metal-catalysts attracted a great deal of attention in recent years.75–78 To address the importance of metal-free aerobic oxidation of alcohols, Karimi and his group79 reported a durable and recyclable catalyst system consisting of SBA-15 supported TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl)/NaNO2/n-Bu4NBr for the aerobic oxidation of alcohols in acetic acid. However, despite the high catalytic performance and recyclability of this system, their studies showed that the method was not suitable for acid-sensitive substrates and, more importantly, the oxidation of allylic alcohols to α,β-unsaturated carbonyl compounds could not be achieved using this protocol.Recently, Karimi and Badreh80 discovered that mesochannels of SBA-15-functionalized TEMPO filled with 1-methyl-3-butylimidazolium bromide (BMIMBr) ionic liquid could result in an efficient and recyclable catalyst system (IIL 12) for the aerobic oxidation of various types of alcohols (19) to carbonyl compounds (20) with improved selectivity under metal-free conditions. In particular, they found that the catalyst IIL 12 showed excellent performance and activity in the oxidation of allylic alcohols. The yields were obtained in the range of 89–99%, and some selected results are shown in Table 2.
| Entry | R 1 | R 2 | Time (h) | Yield (%) |
|---|---|---|---|---|
| 1 | C6H5 | H | 3.5 | >99 |
| 2 | 4-Me–C6H4 | H | 3 | 96 |
| 3 | 4-MeO–C6H4 | H | 3.5 | >99 |
| 4 | 3-NO2–C6H4 | H | 3.5 | 95 |
| 5 | 4-NO2–C6H4 | H | 3.5 | 98 |
| 6 | 3-Cl–C6H4 | H | 3.5 | 99 |
| 7 | 3-Br–C6H4 | H | 3.5 | 96 |
| 8 | 4-Br–C6H4 | H | 3.5 | >99 |
| 9 | 2,4-Cl–C6H3 | H | 3.5 | 94 |
| 10 | 2-Naphthyl | H | 3.5 | >99 |
| 11 | 4-MeS–C6H4 | H | 6 | >99 |
| 12 | 3-Pyridyl | H | 6 | 93 |
| 13 | 2-Furyl | H | 3 | 98 |
| 14 | C6H5 | Me | 6 | 92 |
| 15 | C6H5 | Et | 6 | 91 |
| 16 | C6H5 | C6H5 | 9 | 89 |
| 17 | Ph-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH CH |
H | 15 | 99 |
| 18 | Ph-CHCH |
C6H5 | 17 | 99 |
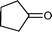
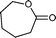
Baeyer–Villiger (BV) reaction, based on the oxidation of ketones to esters or lactones, is one of the most important reactions in organic chemistry, possessing a large range of possible applications such as the synthesis of antibiotics, steroids, pheromones, and monomers for polymerization.81 A considerable amount of research has been conducted to develop new methodologies for BV reaction.82 In this context, Chrobok and co-workers83 explored a novel approach involving the application of 68% hydrogen peroxide as an oxidant and a silica material immobilized ionic liquid with HSO4− as the counteranion to prepare the catalyst (IIL 13, Fig. 6). In their work, cyclic ketones were readily oxidized to their corresponding lactones in high isolated yields (up to 91%) under mild conditions within a short time (Table 3).
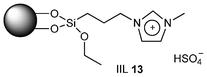 | ||
| Fig. 6 Silica material immobilized ionic liquid IIL 13. | ||
| Entry | Ketone | Lactone | Time | Yield (%) |
|---|---|---|---|---|
| 1 |
|
|
5 h | 91 |
| 2 |
|
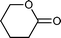
|
8 h | 71 |
| 3 |
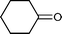
|
|
15 h | 60 |
| 4 |
|
|
10 h | 89 |
| 5 |
|
|
10 h | 88 |
| 6 |
|
|
15 h | 75 |
| 7 |

|
|
20 h | 74 |
| 8 |
|

|
20 h | 60 |
3.4 Other reactions
Various solid supported immobilized ionic liquids have also been used in many other reactions. For example, IILs could act as acidic catalysts for the alkylation of aromatic compounds with dodecene,84 in Friedel–Crafts acylation of aromatics,85 for the diazotization of aniline derivatives and subsequent synthesis of azo dyes,86 in the nucleophilic substitution reactions (such as halogenation, acetoxylation, N-acylation and azidation),87–91 aldol condensation reactions,92,93 Biginelli reaction,94 and Knoevenagel condensation reaction.95 Some modified IILs could serve as nitrosonium sources for electrophilic aromatic nitrosation96 and catalyze dealkylation and disproportionation for dodecylbenzene transformations.97 IILs also severed as efficient and recyclable heterogeneous catalysts for the dehydration of fructose to 5-hydroxymethylfurfural, which improved performances over zeolites and strong acid ion exchange resin catalysts.984 Catalysis by immobilized ionic liquids functionalized with Brønsted acidic groups
Brønsted acidic ILs used in some acid-catalyzed processes have aroused considerable interest since they offer the advantages of both solid and mineral acids.99 In comparison with other ILs, Brønsted acidic ILs generally show higher stability towards air and water. Moreover, the existence of both Brønsted acidic functional groups and acidic counter anions can enhance their acidities.100,101 For these reasons, Brønsted acidic ILs immobilized by chemical covalent bonds have recently been prepared by several groups of researchers and further applied as catalysts in various organic transformations.4.1 Catalysis by IILs functionalized with –SO3H
In 2006, Yokoyama and his group102 reported the preparation of novel 1-allylimidazolium containing acidic ionic liquids that were immobilized on modified silica gel by covalent bonds (Scheme 11). 1-Allyimidazolium containing zwitterionic salts (23) was first prepared from the reaction of 1-allyimidazole (21) with sultone (1,3-propane- or 1,4-butane sultone, 22), then treated with CF3SO3H affording Brønsted acidic ILs [(CH2)3SO3HVIM]HSO4 and [(CH2)4SO3HVIM]HSO4 (i.e.14a and 14b) which were further reacted with 3-mercaptopropyltrimethoxysilane (MPS) modified silica gel (24) to give the corresponding immobilized Brønsted acidic ILs, IIL 14a and IIL 14b.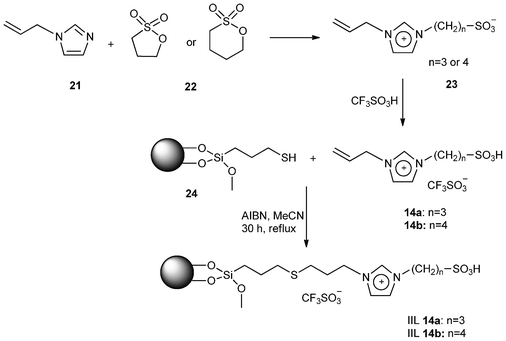 | ||
| Scheme 11 Preparation of immobilized ionic liquids, IIL 14a and IIL 14b. | ||
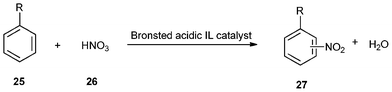
Nitration of some simple aromatic compounds (25) with 62% aqueous nitric acid (26) catalyzed by some of the IILs mentioned above could afford nitrobenzene (27) with satisfactory results (Table 4). As can be seen from the table, the two supported ionic liquids IIL 14a and IIL 14b exhibited higher activity than their non-supported counterparts 14a and 14b respectively (Entries 1–4). Their advantages as solid catalysts in separation and reusability were also observed (Entry 5).
| Entry | R | Catalyst | Conversion (%) | Product distribution (%) | ||
|---|---|---|---|---|---|---|
| ortho | meta | para | ||||
| a Result was obtained after three times recycle. b No isomer was detected. | ||||||
| 1 | H | 14a | 49.3 | Not applicable b | ||
| 2 | H | IIL 14a | 62.8 | Not applicable | ||
| 3 | H | 14b | 47.5 | Not applicable | ||
| 4 | H | IIL 14b | 61.6 | Not applicable | ||
| 5 | H | IIL 14b | 58.3a | Not applicable | ||
| 6 | Me | IIL 14b | 85.8 | 33.7 | 5.8 | 60.5 |
| 7 | Cl | IIL 14b | 10.4 | 42.7 | 1.2 | 56.1 |
| 8 | Br | IIL 14b | 22.2 | 45.8 | Trace | 54.2 |
| 9 | NO2 | IIL 14b | <1 | Not applicable | ||
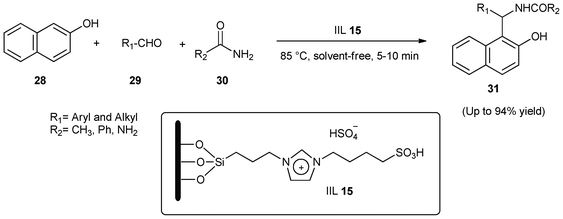
Similarly, using silica gel as the support material, an immobilized dual acidic IL catalyst (IIL 15) was prepared by Luo and co-workers103 by anchoring 3-sulfobutyl-1-(3-propyltriethoxysilane) imidazolium hydrogen sulfate onto common silica gel through covalent bonds. With this catalyst IIL 15, a simple and efficient procedure for the one-pot, three-component synthesis of amidoalkyl naphthols (31) from 2-naphthol (28), aldehydes (29) and amides (30) was further explored under solvent-free conditions (Scheme 12). It was found that a variety of aromatic aldehydes (29) with electron-donating and electron-withdrawing groups were converted to amidoalkyl naphthols (31) in excellent yields (up to 94%), and that immobilized IIL 15 showed a much higher catalytic activity, shortened the reaction time and required only mild conditions compared to other catalysts (such as p-TSA, iodine, and K3CoW12O40·3H2O).
 | ||
| Scheme 12 Synthesis of amidoalkyl naphthols catalyzed by IIL 15. | ||
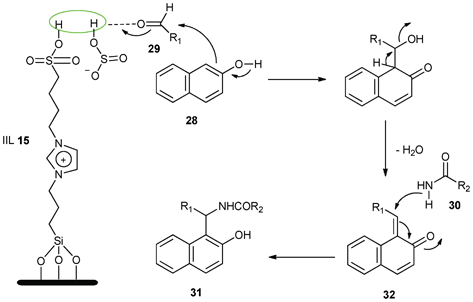
Meanwhile, a mechanism was proposed to portray the probable sequence of events (Scheme 13). They presumed that ortho-quinone methides (o-QMs, 32) were formed by the nucleophilic addition of 2-naphthol (28) to the aldehyde (29), which was assisted by IIL 15. The o-QMs (32) then reacted with amides or urea (30) via a Michael addition to afford the expected amidoalkyl naphthols (31). The immobilized IIL 15 can effectively facilitate the reaction by providing dual acidic sites to activate the aldehydes.
 | ||
| Scheme 13 A plausible mechanism for the synthesis of amidoalkyl naphthols (31) using IIL 15. | ||
Compared to the amorphous silica gel, mesoporous silica is characterized by a stable mesoporous structure, high surface area, controllable pore size and a large amount of silanol group. Based on this consideration, Guan et al.104 successfully immobilized Brønsted acidic ionic liquid [(CH2)3SO3HVIM]HSO4 (33) together with 3-mercaptopropyltrimethoxysilane (34) and AIBN onto silica gel using TEOS (35) as silica source to generate the catalyst (IIL 16) with mesoporous structure (Scheme 14).
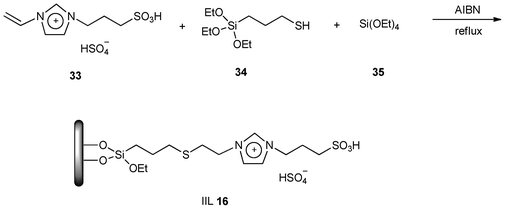 | ||
| Scheme 14 Preparation of IIL 16. | ||
In order to investigate the scope and limitation of the catalyst for esterification, different alcohols (36) and carboxylic acids (37) as the reactants were also tested (Table 5). It was found that good to excellent yields ranging from 84.1% to 99.4% with perfect selectivity (100%) for corresponding esters (38) were obtained, and satisfactory catalytic activity of 88.7% could still be kept after the catalyst was recycled for 7 times for the synthesis of n-butyl acetate.
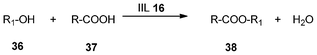
| Entry | R | R 1 | Temperature | Time | Yield (%) | Selectivity (%) |
|---|---|---|---|---|---|---|
| 1 | CH3 | n-C4H9 | 89 °C | 3 h | 99.2 | 100 |
| 2 | CH3 | CH3(CH2)5 | 95 °C | 3 h | 99.4 | 100 |
| 3 | CH3 | CH3(CH2)3(C2H5)CHCH2 | 94 °C | 3 h | 98.6 | 100 |
| 4 | CH3 | CH3(CH2)3OCH2CH2 | 93 °C | 3 h | 99.0 | 100 |
| 5 | CH3(CH2)2 | n-C4H9 | 93 °C | 3 h | 86.3 | 100 |
| 6 | CH3CH2 | S-C4H9 | 88 °C | 3 h | 92.2 | 100 |
| 7 | CH3 | C2H5 | 71 °C | 3 h | 84.1 | 100 |
| 8 | CH3CH2 | C6H5CH2 | 88 °C | 3 h | 93.4 | 100 |
| 9 | CH3 | C6H5CH2 | 90 °C | 3 h | 98.5 | 100 |
In 2011, Guan and his group105 again employed almost the same approach to immobilize Brønsted acidic ionic liquid 1-(propyl-3-sulfonate) imidazolium hydrosulfate ([(CH2)3SO3H-HIM]HSO4) onto silica-gel with TEOS as the silica source. The immobilized ionic liquid catalyst showed good catalytic performance for acetalization of different aldehydes and alcohols. Satisfactory yields of 81.0–98.5% and 100% selectivity for acetal were achieved under optimal reaction conditions. Moreover, the catalyst could be reused for 10 times without significant loss of catalytic activity, showing good reusability for the reaction.
Instead of designing covalently attached ILs onto the surface of a solid support, Skoda-Földes and co-workers106 recently prepared seven different silica-immobilized SO3H-functionalised ILs via simple physical adsorption process. It was proved that these catalysts were efficient for isobutene ologomerisaton with excellent C8 selectivity (up to 87%) and good C12+ selectivity (up to 84%), and that the catalysts bearing trifluoromethanesulfonate anion could be reused for several times without loss of activity.
Treating PS as the support material, Guan et al.107 synthesized PS-supported acidic ionic liquid (PS-CH2-[SO3H-pIM][HSO4]) catalyst (IIL 17, Scheme 15). The pre-synthesized PS-CH2Cl (39) was successively reacted with imidazole, 1,3-propane sultone, and sulfuric acid to afford PS-CH2-[HIM][Cl] (40), PS-CH2-[SO3H-pIM][Cl] (41), and the target catalyst (IIL 17) respectively. Meanwhile, an acidic ionic liquid (44) was prepared from H2SO4 acidification of 43 which was pre-synthesized from 1,3-propane sultone (42) reacting with an equal mole of imidazole.
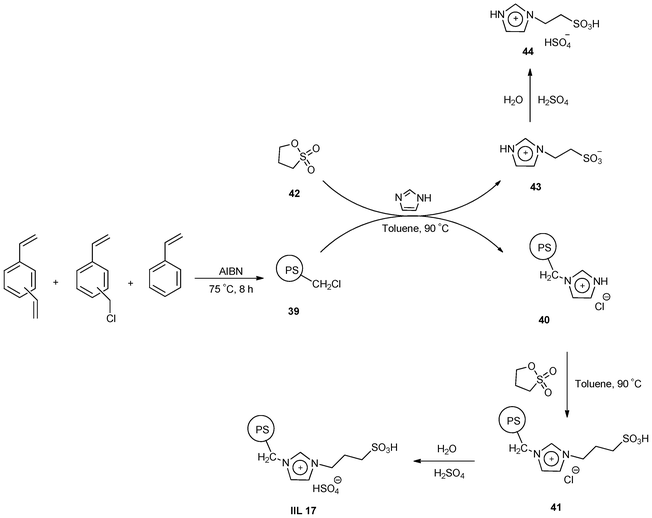 | ||
| Scheme 15 Procedures for the synthesis of PS-CH2Cl (39), acidic ionic liquid (44) and IIL 17 catalyst. | ||
To evaluate the performance of IIL 17, the catalytic activities of various catalysts for the esterification reaction of n-butyl alcohol and acetic acid were examined under the same reaction conditions (Table 6). Obviously, the yield of n-butyl acetate in the presence of IIL 17 (Table 6, Entry 4) was much higher compared to the reaction conducted with acidic ionic liquid (44, Table 5, Entry 3), the support catalyst (39, Table 6, Entry 2) and without any catalyst (Table 6, Entry 1).
| Entry | Catalyst | Active sites (mmol) | Yield (%) | TONa |
|---|---|---|---|---|
| a Turnover number as mole of product/mole of acidic ionic liquid in catalyst. | ||||
| 1 | Blank | 0 | 22.6 | — |
| 2 | 39 | 0 | 35.8 | — |
| 3 | 44 | 0.0018 | 98.2 | 43.4 |
| 4 | IIL 17 | 0.0018 | 99.1 | 43.8 |
Shan and his group108 disclosed a magnetic nano-solid acid catalyst (IIL 18) by grafting an ionic liquid onto Fe3O4 nanoparticles followed by sulfonation of phenyl groups in the ionic liquid (Scheme 16).
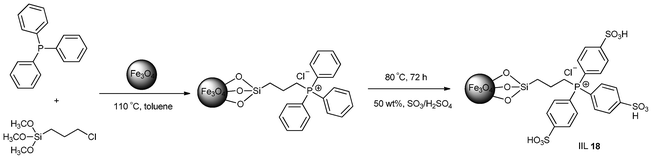 | ||
| Scheme 16 The synthesis process of IIL 18. | ||
Using this catalyst (IIL 18), acetals (47) were obtained in high yield (up to 97%) by acetalization or ketalization of different aldehydes or ketones (45) with ethylene glycol (46) (Scheme 17). It was also found that the Brønsted acidic IL (IIL 18) catalyst could be easily separated after completion of the reaction and reused without any significant reduction of catalytic activity.
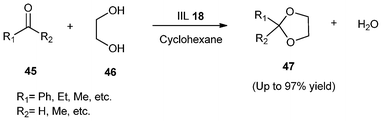 | ||
| Scheme 17 Acetalization or ketalization catalyzed by IIL 18. | ||
Driven by the unique properties of magnetic nanoparticles and the valuable potential applications of acidic ILs in catalysis,109 Luo and co-workers110 in 2012 designed a nanosized novel silica-coated Fe3O4 immobilized Brønsted acidic ionic liquid (IIL 19) and used it in a one-pot three-component condensation of dimedones (48) with 2-naphthol (28) and aldehydes (29) to prepare benzoxanthenes (49) in excellent yields (up to 94%), as shown in Scheme 18. The combination of nano-support features and soft imidazolium linkers made the active sites of the supported catalyst more free and soluble in the reaction system thereby facilitating the condensation reaction. Moreover, the catalyst could be readily separated by use of a magnetic force and reused without any significant loss of catalytic activity after six runs.
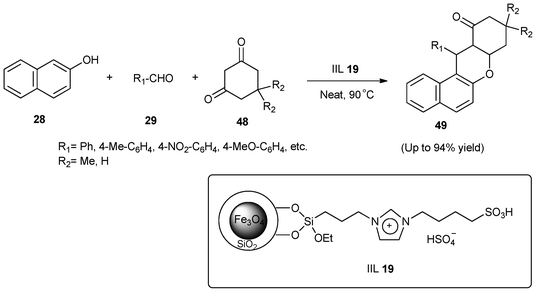 | ||
| Scheme 18 One-pot synthesis of benzoxanthenes (49) catalyzed by IIL 19. | ||
In comparison with porous inorganic supports, porous polymers usually showed adjustable hydrophobicity and good stability for various acids. Most recently, Qi et al.111 demonstrated a high temperature (200 °C) hydrothermal synthesis of functionalized strong acidic ionic liquids e.g. ordered and stable mesoporous resins (OMR-ILs). The high temperature used in the operation proved beneficial for obtaining the samples with a high degree of cross-linking resulting in their extra-ordinary thermal and mechanical stabilities. To examine the catalytic performance of OMR-ILs, the esterification of acetic acid with cyclohexanol (EAC), transesterification of tripalmitin with methanol (TTM), Peckmann reaction of resorcinol with ethyl acetoacetate (PRE), and hydration of propylene oxide (HPW) over various catalysts were carried out. It was found that the OMR-ILs samples showed much higher catalytic activities (91.2–99% conversions) than those of conventional solid acids of Amberlyst 15 (51.3–80.5% conversions), SBA-15-[C3mim][SO3CF3] (42.8–71.2% conversions) and acidic zeolites (6.5–48.6% conversions). The results in terms of yields were even comparable with that obtained with homogeneous sulfuric acid (94.5–99.1% conversions). It is worth noting that the OMR-ILs samples exhibited very good recyclability; after being recycled for five times there was no noticeable decrease in catalytic activity for these reactions.
4.2 Catalysis by IILs functionalized with –COOH
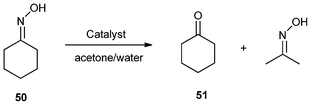
In 2004, Deng and co-workers112 synthesized a series of silica gel confined ILs (IIL 20a–j) from the corresponding pure acid-functionalized ILs (20a–j, Fig. 7) through a sol–gel process. It was found that most of the prepared immobilized Brønsted acidic ILs displayed almost the same conversions and selectivities as the pure ionic liquid catalysts for the deoximation reaction of cyclohexanone oxime (50) to cyclohexanone (51) (Table 7). In addition to easy separation and reusability, the uses of stoichiometric, reducing or oxidizing agents were also avoided in this catalytic system.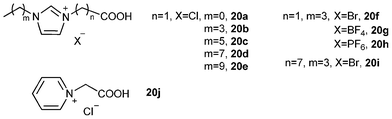 | ||
| Fig. 7 Ionic liquid with an acid group on the side chain. | ||
| Entry | Catalyst | Conversion (%) | Selectivity (%) | TON |
|---|---|---|---|---|
| a The data in brackets were results of the deoximation over pure ILs catalysts 20a–j. b The immobilised catalyst was reused for the second time. | ||||
| 1 | IIL 20a | 94.3 (94.4)a | >99 | 118 (30)a |
| 2 | IIL 20b | 92.5 (94.8) | >99 | 143 (37) |
| 3 | IIL 20c | 90.3 (92.1) | >99 | 158 (40) |
| 4 | IIL 20d | 76.9 (93.7) | >99 | 150 (46) |
| 5 | IIL 20e | 72.4 (83.8) | >99 | 155 (45) |
| 6 | IIL 20f | 93.8 (93.2) | >99 | 180 (45) |
| 7 | IIL 20g | 94.1 (94.2) | >99 | 219 (55) |
| 8 | IIL 20h | 88.2 (94.6) | >99 | 164 (49) |
| 9 | IIL 20i | 26.2 (84.6) | >99 | 65 (52) |
| 10 | IIL 20j | 92.2 (96.8) | >99 | 114 (30) |
| 11b | IIL 20f | 90.1 | >99 | 173 |
Considering the fact that incorporation of carboxylic acid moieties (strong Brønsted acid) into ILs might provide an effective alternative for improving the catalytic performance of ILs toward the cycloaddition of CO2 and epoxides, Park and his group113 prepared a range of carboxylic-acid-functionalized imidazolium-based ILs bearing different halide anions and grafted them onto a silica gel surface, forming various heterogeneous catalysts (IIL 21). For comparison, non-functionalized IL (IIL 22) was also synthesized, and the general synthetic route is shown in Scheme 19.
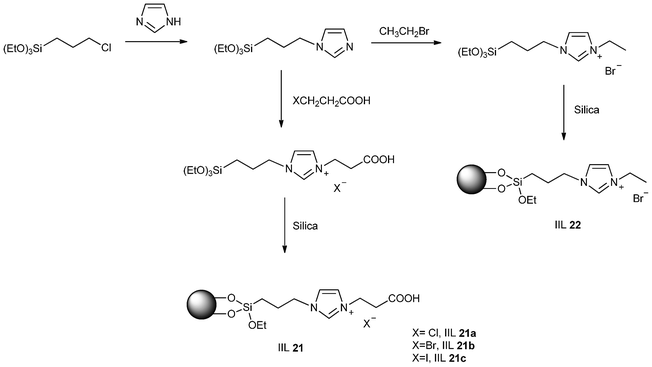 | ||
| Scheme 19 Synthesis of IIL 21a–c and IIL 22. | ||
They further investigated the effects of molecular composition (functional group and anion type) of grafted ILs on catalytic reactivity for the cycloaddition reaction of allyl glycidyl ether (1) and CO2 to cyclic carbonate 2, and realized that grafted ILs with carboxyl functional groups demonstrated better catalytic activities than grafted ILs without any functional group (Table 8). This advantage of carboxylic acid-functionalized ILs might be attributed to the –COOH group in the IL cation which can greatly accelerate the rates of the reactions and exhibits a synergistic effect with the halide anions. Furthermore, they also found that these heterogeneous catalysts can be easily separated from the products and reused.
| Entry | Catalyst | Conversion (%) | Selectivity (%) | TON |
|---|---|---|---|---|
| 1 | IIL 21a | 71 | 93 | 143 |
| 2 | IIL 21b | 73 | 96 | 156 |
| 3 | IIL 21c | 76 | 98 | 170 |
| 4 | IIL 22 | 65 | 95 | 40 |
4.3 Catalysis by IILs functionalized with –OH
Hydroxyl group is believed to facilitate the coordination of the H and O or N atoms of epoxides or aziridines through a hydrogen bond, which could result in the polarization of C–O or C–N bonds and avial anions (such as halides) to facilitate a nucleophilic attack on the sterically hindered β-carbon atom of the epoxides or aziridines.114,115 Taking into consideration these aspects, several attempts were made to explore the catalytic performance for CO2 cycloaddition reactions in the presence of IILs containing –OH as catalysts.In 2009, Zhang et al.116 reported three different polystyrene resin-supported hydroxyl-functionalized ILs, the synthetic approach is shown in Scheme 20. Imidazole (52) was first chemically immobilized on polystyrene resin (53) through covalent bonds to afford polymer 54, which was further reacted with the corresponding halide substituted alcohols to give IIL 23. It was demonstrated that the hydroxyl group in the catalyst had a synergetic effect with the halide anion. As a result, high yields (up to 99%) and excellent selectivity (up to 99%) of cyclic carbonates were achieved from corresponding epoxides and CO2 under mild conditions (2.5 MPa, 120 °C and 4 h) without any co-solvent. Among the catalysts designed, IIL 23a was found to be applicable to the cycloaddition of a variety of terminal epoxides with CO2, insuring high yields (up to 99%) and high selectivity (up to >99%) of the corresponding cyclic carbonates. In addition, the catalyst recycle test showed that the supported catalyst could be reused for as many as six times without any loss of catalytic activity.
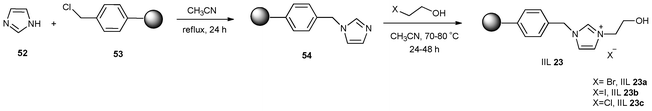 | ||
| Scheme 20 Synthesis of polymer-supported ionic liquids (IIL 23). | ||
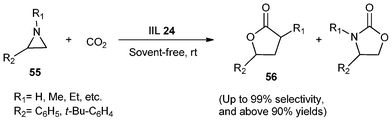
Afterwards, polymer supported diol functionalized ionic liquids were investigated by Bhanage and co-workers.117 IIL 24 was prepared by carrying out the N-alkylation reaction two times on the heterocyclic compound (Scheme 21). The polymer (54) for immobilizing diol functionalized ionic liquids was first synthesized from cross linked polystyrene resin (53) by reacting with imidazole (52), which was subsequently reacted with the corresponding halogenated diol to provide the polymer supported 1-(2,3-dihydroxyl-propyl)-imidazolium halide (IIL 24a and IIL 24b). The catalysts were then employed for coupling of CO2 to aziridines (55) to afford the desired products (56) in good yields (above 90%) and excellent stereoselectivity (up to 99%); the synthetic route is depicted in Scheme 22.
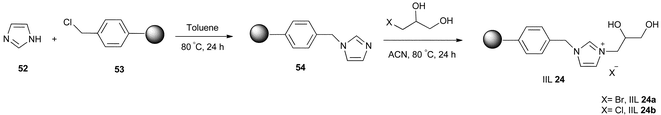 | ||
| Scheme 21 Preparation of polymer-supported diol functionalized ionic liquids (IIL 24). | ||
 | ||
| Scheme 22 Carboxylation of aziridines (55) and CO2 catalyzed by IIL 24. | ||
Most recently, using the same catalysts (IIL 24), Bhanage and his group118 developed a facile, stable, halide free, highly selective and recyclable catalytic protocol for synthesis of cyclic carbonates under solvent free conditions. In order to further prove that the vicinal hydroxyl group was favorable for the cycloaddition reaction, a co-catalyst KBr combined with 3-bromo-propane-1,2-diol was introduced for the coupling reaction of CO2 and methyloxirane, the isolated yield of corresponding product was 97% within 0.5 h which was much higher than that obtained by immobilized IL without a functional group (trace yield). The remarkable activity of these catalysts bearing diol functional groups might be attributed to the presence of vicinal hydroxyl groups which facilitated the forward reaction.
5 Catalysis by immobilized ionic liquids functionalized with metals
The very concept of the term “Supported Ionic Liquid Phase” (SILP) catalysis first emerged in 2002 when the immobilization of a transition metal catalyst in a thin film of ionic liquid immersed on a porous inorganic support was published by Mehnert et al.119 Synergy between catalytically active sites and ILs can be remarkably affected by the chemical functionalities of solid support immobilized functional ILs. This type of synergy might reach a maximum degree if immobilized ILs provide a “matched function” or an “enhancement effect” onto the grafted active sites and/or vice versa through a cooperative mechanism. In particular, metal-containing immobilized ILs represent a promising area in the multi-faced IL universe, since they combine the unique properties of ILs with the peculiar properties (magnetic, photophysical/optical or catalytic) of the incorporated metals (such as metal nanoparticles, metal ions and metal complexes). Some early reviews on catalysis by immobilized ILs functionalized with metals have summarized parts of the reported results in different ways till the end of 2009.120–123 Therefore, we intend to pay more emphasis on the research results that emerged in subsequent investigations.5.1 Hydrogenation reactions
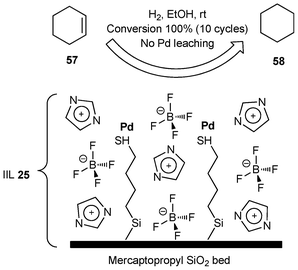
The catalytic systems, imidazolium-base ILs immobilized on metal nanoparticles using various solid supports (such as molecular sieves, montmorillonite, activated carbon fibers) have been used for different selective hydrogenation processes with unprecedented activity and stability.124–136 The combination of nanoparticles, the ILs, and solid supports showed excellent synergistic effects to enhance the activity and durability of the catalysts.Hagiwara et al.,137 in 2011, disclosed a simple and cost effective immobilization procedure without employing synthetic transformations or a large amount of expensive ionic liquid for the preparation of IL immobilized metal nanoparticle catalyst. In their work, palladium acetate was immobilized as IIL 25 in the pores of amorphous mercaptopropyl silica gel with the aid of ionic liquid [BMIM]BF4 (BMIM = 1-butyl-3-methylimidazolium). The hydrogenation of cyclohexene (57) to cyclohexane (58) was used as a benchmark reaction to investigate the catalytic performance of IIL 25 (Fig. 8). It was proved that the catalyst exhibited comparable catalytic performance to Pd/C at room temperature under atmospheric pressure, and could be recycled up to 10 times by simple decantation, while maintaining 100% conversions.
 | ||
| Fig. 8 Catalytic hydrogenation of cyclohexene by IIL 25. | ||
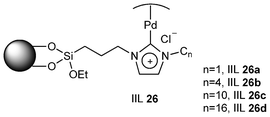
Controlling the selectivity of chemical reactions by steric hindrance is an interesting topic. Han and his group138 synthesized a series of Pd(II)-NHC IL complexes immobilized on mesoporous silica by N-heterocyclic carbene (NHC) ionic liquids (ILs) with different alkyl chain lengths (Fig. 9). The catalytic activity of the catalysts (IIL 26) for the hydrogenation of different alkenes and allyl alcohol was studied, and the results of the reactions are shown in Table 9. The selectivity of IIL 26a, IIL 26b, IIL 26c and IIL 26d for 1-propanol was 80.0%, 80.5%, 84.1% and 84.0%, respectively, which was higher than that obtained using the commercial Pd/C. It was observed that the selectivity for the hydrogenation of allyl alcohol to 1-propanol increased with the increase of the alkyl chain length of the ILs. However, the activity of IMM-1 for 1-hexene and cyclohexene was lower than that of Pd/C. This can be explained by the steric effect. It was also disclosed that the selectivity for hydrogenation of allyl alcohol catalyzed by IIL 26c increased from 84.1% to 90.2% as the pressure of CO2 changed from 0.0 to 7.0 MPa at a fixed H2 pressure of 4.0 MPa, demonstrating that CO2 can enhance the selectivity for the desired product considerably. In addition, the catalytic activity, selectivity and Pd content remained almost unchanged after being reused three times.
 | ||
| Fig. 9 Structure of IIL 26 with different alkyl chain lengths of the ILs. | ||
| Entry | Catalyst | Substrate | Time | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|---|
| 1 | IIL 26a | 1-Hexene | 100 min | 100 | 100 |
| 2 | Pd/C | 1-Hexene | 50 min | 100 | 100 |
| 3 | IIL 26a | Cyclohexene | 140 min | 100 | 100 |
| 4 | Pd/C | Cyclohexene | 80 min | 100 | 100 |
| 5 | IIL 26a | Allyl alcohol | 35 min | 100 | 80.0 |
| 6 | IIL 26b | Allyl alcohol | 37 min | 100 | 80.5 |
| 7 | IIL 26c | Allyl alcohol | 40 min | 100 | 84.1 |
| 8 | IIL 26d | Allyl alcohol | 41 min | 100 | 84.0 |
| 9 | Pd/C | Allyl alcohol | 40 min | 100 | 74.0 |
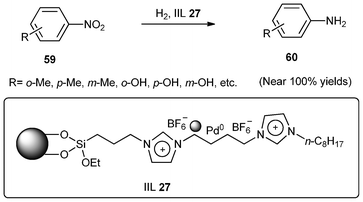
Anilines are very important intermediates for manufacturing 4,4′-methylene-dianiline, dyes, pharmaceuticals, pesticides, explosives, fibers, photographic and rubber chemicals. Wei and co-workers139 developed a heterogeneous catalytic assembly of Pd nanoparticles in-SiO2 supported ionic liquid brushes (IIL 27) for the hydrogenation of nitrobenzenes (59) to arylamines (60) (Scheme 23). This catalytic assembly showed uniform Pd particles size distribution and was proved to be highly efficient, selective, and recyclable in the hydrogenation reactions under solvent-free or neat water conditions, giving nearly 100% yields and selectivity. This approach overcame simultaneously the disadvantages of the requirement for organic solvents in the liquid hydrogenation process and of high temperature in the vapor hydrogenation process.
 | ||
| Scheme 23 Catalytic hydrogenation of nitrobenzenes (59) to arylamines (60) by IIL 27. | ||
Asymmetric hydrogenation of prochiral ketones is one of the most important methods for the synthesis of enantiomerically pure secondary alcohols, which are valuable building blocks and intermediates for the manufacture of pharmaceuticals and advanced materials.140 Furthermore, judicious selection of support material can solve the problem arising due to lack of stability of the liquid film on the support vs. mechanical removal by convection. In 2010, a chiral ruthenium complex containing a cheap achiral monophosphine [RuCl2(PPh3)2(S,S-DPEN)] (61, DPEN = 1,2-diphenylethylenediamine) was prepared and immobilized in the ionic liquid [BMIM][BF4] which was confined on the surface of mesoporous silicates, including MCM-48, MCM-41, SBA-15, and amorphous SiO2 (Fig. 10). The four immobilized catalysts (IIL 28a/61, IIL 28b/61, IIL 28c/61, and IIL 28d/61) were evaluated in the asymmetric hydrogenation of acetophenone. For comparison, the chiral Ru pre-catalyst 61 was also investigated under the same reaction conditions. It could be observed that all the heterogeneous catalysts showed comparable conversions and ee values compared to their homogeneous counterparts (Table 10, Entry 1 vs. Entries 2–5). The high catalytic activities may be attributed to the homogeneous-like properties of the active species in ionic liquid phase on the mesoporous supports. Besides acetophenone, the immobilized catalysts also showed satisfactory catalytic activity and enantioselectivity for other aromatic ketones (Table 10, Entry 6 vs. Entry 7, and Entry 8 vs. Entry 9). In particular, the SiO2-supported catalyst showed the best stability while the conversion and ee values (100% and 76%, respectively) were comparable to the homogeneous catalytic system for the fifth run.141
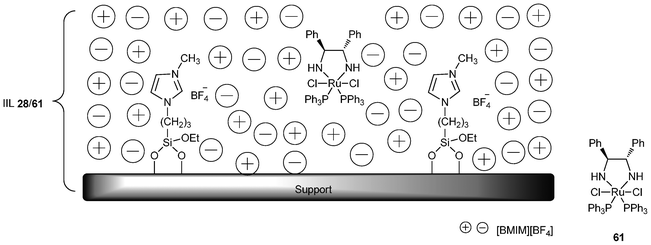 | ||
| Fig. 10 Immobilized ionic liquid catalyst IIL 28/61. | ||
| Entry | R | Catalyst | Conversion (%) | ee (%) |
|---|---|---|---|---|
| 1 | H | 61 | 100 | 78 (R) |
| 2 | H | IIL 28a/61 | 100 | 78 (R) |
| 3 | H | IIL 28b/61 | 100 | 72 (R) |
| 4 | H | IIL 28c/61 | 100 | 72 (R) |
| 5 | H | IIL 28d/61 | 100 | 77 (R) |
| 6 | Cl | 61 | 99 | 66 (R) |
| 7 | Cl | IIL 28a/61 | 99 | 64 (R) |
| 8 | F | 61 | 97 | 45 (R) |
| 9 | F | IIL 28a/61 | 98 | 48 (R) |
Wasserscheid et al.142 further extended asymmetric hydrogenation reaction using immobilised chiral ligand functionalized IL as catalyst to a continuous, gas-phase process. In the hydrogenation of methyl acetoacetate (62) to methyl hydroxybutyrate(63) using RuLBr2 in 1-ethyl-3-methylimidazolium bis(trifluoromethyl-sulfonyl)imide on silica gel 30 (IIL 29/RuLBr2), an enantiomeric excess in the range of 65–82% ee could be obtained in 100 h using a continuous stream operation (Scheme 24). Moreover, it was demonstrated that the ionic liquid film on the support was absolutely essential for catalytic activity in the system and a relatively high degree of pore filling with the ionic liquid could be beneficial. By changing the reactor from a back-mixed Berty-type to a tubular reactor, the accumulation of side-products in the immobilized functional IL catalyst could be significantly reduced and much higher catalyst stability was observed. Remarkably, this continuous gas phase hydrogenation has been achieved for a substrate of relatively low volatility by using helium as carrier gas in the reactor.
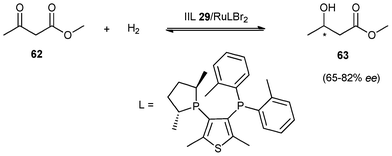 | ||
| Scheme 24 Hydrogenation of methyl acetoacetate using IIL 29/RuLBr. | ||
5.2 Friedel–Crafts reactions
Friedel–Crafts reactions are of considerable importance in the production of petroleum and pharmaceutical products. Conventional homogeneous acid catalysts (such as AlCl3, FeCl3, ZnCl2, or H2SO4) for Friedel–Crafts reactions in the liquid phase inevitably possessed some inherent shortcomings such as corrosion, difficulties in separation and/or recovery, environmental hazard, and disposal problems coming from the large amount of acidic effluents. Currently, immobilized functional ILs with excellent physicochemical properties are considered as an appropriate alternative.Hölderich et al.143 developed a process for the alkylation of benzene, toluene, naphthalene, and phenol with dodecene in the presence of immobilized 1-butyl-3-methyl-imidazolium chloride with AlCl3 (Al-IIL). The immobilized IL played an important role in significant reduction of overall cost of the process. A comparison of different supports showed that carriers based on SiO2 and Al2O3 retained higher amounts of chloroaluminate IL than ZrO2 and TiO2 carriers after the extraction, and only the silica-based supports were active in the alkylation reactions. Under optimized conditions, the alkylation of benzene using Al-IIL as catalyst resulted in 98% selectivity of monoalkylated products with 99% conversion of dodecene at 80 °C. Naphthalene and phenol exhibited lower conversions and high selectivity towards monoalkylated products.
Two different methods for preparation of immobilized Al-IIL, which might affect the catalytic activity of Friedel–Crafts type alkylation reactions, were further investigated by the same group.144 It was found that the method of incipient wetness allowed a very fast and easy immobilization, but it is limited to anions with a high affinity to oxygen. Without immobilization, no surface bonds would exist and leaching of the ionic liquid might also be expected. The second method of immobilization of ILs onto support materials via covalent bonds was found more stable and active. For the alkylation of benzene with dodecene at 80 °C in reactions catalyzed by amorphous silica and MCM-41 immobilized ILs, conversions greater than 90% with very high selectivity to the desired monoalkylated product were observed. In comparison, the reaction catalyzed by immobilized AlCl3 showed only 15.7% conversion, and a high selectivity towards the isomers of dodecene. The low conversion achieved with the pure ionic liquid as catalyst can be explained by the low solubility of the catalysts in the reaction mixture.
Yin and co-workers145 demonstrated a two-step approach for preparation of immobilized chloroferrate ionic liquid (IIL 30). The methodology involved generation of 1-trimethoxysilylpropyl-3-methylimidazolium chloride–FeCl3 (64) and its subsequent grafting onto siliceous MCM-41 (Scheme 25). In comparison with the 1-butyl-3-methylimidazolium chloride–FeCl3 (BMIMCl–FeCl3), the catalyst obtained showed much higher efficiency in Friedel–Crafts reaction between benzene and benzyl chloride. Typically, the conversions and selectivity were all achieved in 100%, and the catalyst could be recycled and reused for ten times without significant loss of catalytic activity. However, only 19.2% conversion was achieved after five recycles in the presence of BMIMCl–FeCl3.
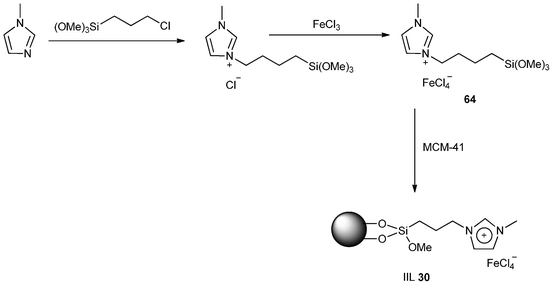 | ||
| Scheme 25 Procedures for the synthesis of immobilized chloroferrate IL (IIL 30) . | ||
Novel periodic mesoporous organosilica (PMO) materials of SBA-15 incorporating Lewis acidic chloroindate(III) ionic liquid moieties (IIL 31) were prepared via a multistep process (Scheme 26). N-(3-triethoxysilylpropyl), N(3)-(3-trimethoxysilylpropyl-4,5-dihydroimidazolium chloride (65) was first synthesized and used as bridging organosilica precursor to co-condense with tetraethoxysilane (TEOS) in the presence of P123, affording the immobilized IL (66). After removal of the template, the addition of InCl3 gave rise to PMO materials incorporating chloroindate(III) ionic liquid moieties bearing a Lewis acid group. Friedel–Crafts reaction between benzene and benzyl chloride over various catalysts was examined. It was observed that the siliceous SBA-15 showed no catalytic activity, however, the immobilization of the Lewis acidic ionic liquids with increasing amount of InCl3 led to a drastic increase in the catalytic activity ranging from 14 to 100 conversions with constant selectivity of 100%. Moreover, the immobilized functional IL catalyst could be recycled and reused six times with 100% selectivity albeit a gradual decrease in conversion (100%, 100%, 100%, 100%, 99%, and 97%). By contrast, only 76% conversion and 95% selectivity could be detected after five times in the absence of the IL, suggesting that the acidic PMO material showed better reusability than SBA-15-supported InCl3.146
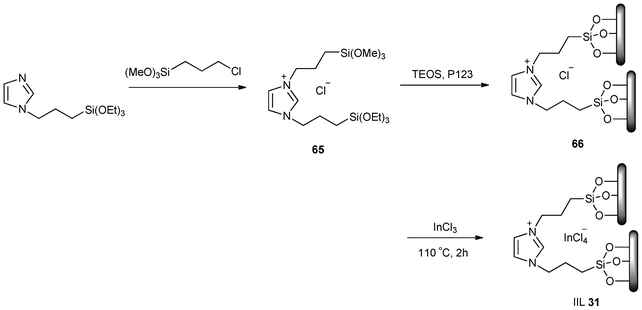 | ||
| Scheme 26 Procedure for the synthesis of immobilized chloroindate IL (IIL 31) . | ||
5.3 Carbonylation reactions
The oxidation of alcohols to the corresponding aldehydes and ketones is one of the most important functional group transformations in organic chemistry.147 In 2010, Chrobok et al.148 presented the copper-TEMPO-catalyzed aerobic oxidation of primary alcohols based on the new supported ionic liquid phase (SILP) catalysts (IIL 32). The reaction system applied in their study has been shown in Scheme 27. The ionic liquid phase (1-butyl-3-methylimidazolium octylsulfate, 1-butyl-3-methylimidazolium hexafluorophosphate, or 1-butyl-3-methylimidazolium tetrafluoroborate) together with CuCl2, which is insoluble in the organic solvent, was immobilized on the solid support (ionogel or bi-modal pore structure silica). The alcohols studied and TEMPO were dissolved in dibutyl ether as the organic solvent and the gas phase consisting of oxygen. The process gave high yields of corresponding aldehydes (up to 95%) with the application of SILP catalysts. Moreover, the catalysts were used in seven cycles without significant loss of activity. High recoveries of the catalysts were also observed.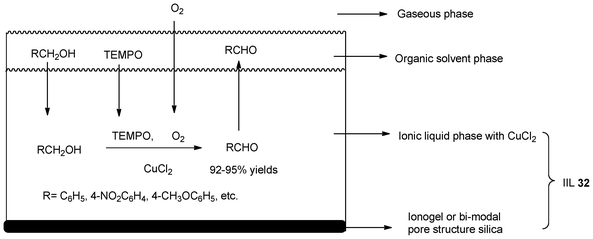 | ||
| Scheme 27 The reaction system for the aerobic oxidation of alcohols catalyzed by IIL 32. | ||
Several new catalysts (IIL 33) of copper ion-containing ionic liquids immobilized on SBA-15 were prepared by Dong and co-workers149 through grafting of 3-trimethoxysilylpropyl-pyridinium chloride (67) onto SBA-15 followed by the addition of copper salts including CuCl2, CuCl, CuBr2 to the resulting immobilized IL (68) (Scheme 28). The oxidative carbonylation of methanol to dimethyl carbonate (DMC) was investigated using these immobilized catalysts (Table 11). It was clearly shown that the immobilization of CuCl2, CuCl, and CuBr2 on ionic liquid-modified SBA-15 can remarkably improve the catalytic activity and selectivity of CuCl2, CuCl, and CuBr2 catalyst, respectively. The IIL 33c catalyst exhibited the best catalytic activity with a 17.0% conversion of methanol, 97.5% selectivity of DMC. The recycling of the IIL 33c catalyst was also examined. Although the IIL 33c catalyst deactivated rapidly during the initial three runs, it still maintained a comparable activity with the fresh CuBr2/SBA-15 catalyst after three times of recycling.
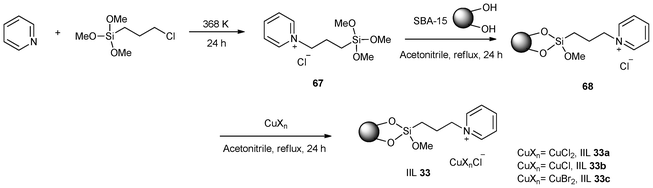 | ||
| Scheme 28 Procedures for synthesis of immobilized copper ion-containing ILs (IIL 33). | ||
| Entry | Catalyst | Conversion (%) | Selectivity (%) |
|---|---|---|---|
| 1 | CuCl2 | 10.7 | 96.4 |
| 2 | CuCl2/SBA-15 | 9.6 | 94.0 |
| 3 | IIL 33a | 9.5 | 98.4 |
| 4 | CuCl | 9.7 | 94.3 |
| 5 | IIL 33b | 7.3 | 97.6 |
| 6 | CuBr2 | 10.5 | 86.0 |
| 7 | CuCl2/SBA-15 | 8.2 | 85.3 |
| 8 | IIL 33c | 17.0 | 97.5 |
In view of its importance, hydroformylation, one of the most investigated catalytic reactions, has been a subject of recent studies in both industries and academia. Cole-Hamilton et al.150 prepared a supported ionic liquid phase catalyst (IIL 34) from [PrMIM][Ph2P(3-C6H4SO3)] (PrMIM = 1-propyl-3-methylimidazolium), [Rh(CO)2(acac)][OctMIM]NTf2 (acac = 2,4-pentanedione, OctMIM = 1-n-octyl-3-methylimidazolium, Tf = CF3SO2) and microporous silica for the continuous flow hydroformylation of 1-octene (69) to aldehydes (70) in the presence of compressed CO2 (Scheme 29). Under optimized conditions, the catalyst was shown to be stable over at least 40 h of continuous catalysis with a steady state turnover frequency (TOF, mol product (mol Rh)−1) of 500 h−1 at low Rh leaching (0.2 ppm). The conversion of 1-octene (69) was 42 ± 2% throughout the reaction and the ratios of linear and branched aldehydes (nonanal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2-methyloctanal, 70a
:2-methyloctanal, 70a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :70b) were ca. 3, similar to that obtained using exactly the same catalyst in conventional organic solvents.
:70b) were ca. 3, similar to that obtained using exactly the same catalyst in conventional organic solvents.
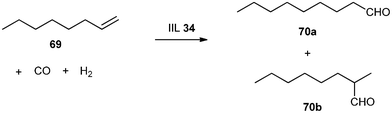 | ||
| Scheme 29 The hydroformylation of 1-octene (69) catalyzed by IIL 34. | ||
Recently, Bell and co-workers151 investigated the effects of parameters such as ligand composition, ligand:rhodium ratio, ionic liquid composition, ionic liquid loading, and temperature of silica pretreatment on the activity, selectivity, and stability of Rh-based supported ionic liquid phase catalysts in catalytic hydroformylation of propene. Highest activity (TOF of 84 h−1, n-butanal:iso-butanal of 12) was achieved with sulfoxantphos (SX) as the ligand and [BMIM][OctSO4] (Oct = 1-n-octyl) as the IL supported on silica pretreated at 100 °C by using an SX:Rh ratio of 10 and IL loading corresponding to α = 0.2.
5.4 Suzuki and Heck reactions
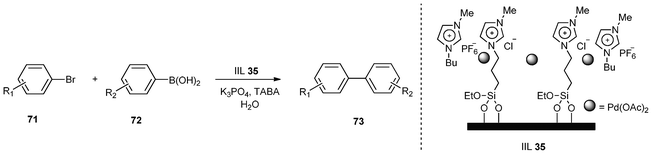
Suzuki coupling of aryl halides with arylboronic acids is a powerful reaction for the construction of biaryl units which are valuable compounds used in production of fine and bulk chemicals such as pharmaceuticals, agrochemicals and natural products.152 Usually, these cross-coupling reactions were catalyzed by palladium complexes in the presence of various phosphine ligands under homogeneous conditions. Therefore, one of the most challenging topics in this area is the development of more appropriate heterogeneous systems for practical applications.Palladium acetate immobilized in thin ionic liquid layers on the mesopore wall of hierarchical MFI zeolite was demonstrated by Jin and co-workers, which was tested as a heterogeneous catalyst (IIL 35) for Suzuki coupling of aryl bromides (71) with arylboronic acids (72) in water (Table 12). In order to enhance the reactivity of the reaction, a phase transfer agent, tetrabutylammonium bromide (TBAB) was added. Under optimized conditions, high catalytic activity was observed, affording the corresponding biphenyl compounds (73) in excellent isolated yields. In addition to the high activity, the catalyst (IIL 35) could be recycled five times without significant loss of catalytic activity.153
| Entry | R 1 | R 2 | Time | Yield (%) |
|---|---|---|---|---|
| 1 | H | 4-Me | 0.5 h | 94 |
| 2 | 4-MeO | 4-Me | 0.5 h | 92 |
| 3 | 4-Me | 4-Me | 1.0 h | 95 |
| 4 | 2-Me | 4-Me | 0.5 h | 91 |
| 5 | H | 4-MeO | 0.5 h | 94 |
| 6 | 4-MeO | 4-MeO | 0.5 h | 91 |
| 7 | 4-Me | 4-MeO | 1.0 h | 91 |
| 8 | 2-Me | 4-MeO | 0.5 h | 93 |
| 9 | H | 4-Cl | 0.5 h | 95 |
| 10 | 4-MeO | 4-Cl | 0.5 h | 93 |
| 11 | 4-MeO | 2-Cl | 0.5 h | 90 |
| 12 | 4-Me | 2-Cl | 2.0 h | 85 |
| 13 | 2-Me | 2-Cl | 1.0 h | 87 |
| 14 | H | 3-NO2 | 0.5 h | 96 |
| 15 | 4-MeO | 3-NO2 | 0.5 h | 96 |
| 16 | 4-Me | 3-NO2 | 0.5 h | 92 |
| 17 | 4-Me | 3-NO2 | 0.5 h | 92 |
Song and his group154 synthesized a co-polymer composed of di-OH functional ionic liquid 3-(2,3-dihydroxypropyl)-1-vinylimidazolium chloride with styrene which was applied in the immobilization of Pd nanoparticles. The prepared catalyst showed efficient catalytic activity in Suzuki coupling reactions for a wide range of aryl iodides and bromides with 0.05 mol% Pd at 70 °C in water–ethanol solution under air. Both the electron rich and electron deficient aryl iodides gave the products with good to excellent yields (96–98%) within 1 h; the sterically demanding aryl iodide also afforded good yields (up to 84%). Moreover, the catalyst could be recycled and reused at least five times without significant decline in the catalytic activity.
Wei et al.155 developed a series of ionic liquid brush immobilized PdEDTA catalysts (IIL 36) for coupling of arylboronic acids with aryl iodides and bromides in water under aerobic conditions. Compared to the analogous catalysts with n-butyl, benzyl, or n-dodecyl as the terminal alkyl group of IL (Fig. 11), the catalyst with octyl group was more efficient in balancing critical hydrophobic and hydrophilic parameters and hence applied for the Suzuki reaction. In order to enhance the solvent power of the immobilized ionic liquid (IIL 36b), water-insoluble aryl halides (71) and arylboronic acids (72) were efficiently transformed to the desired products (73) in excellent yields of 88–99% (Table 13). It is particularly noteworthy that all of the reactions were carried out in neat water avoiding the need for phase transfer catalyst, organic co-solvent and inert gas.
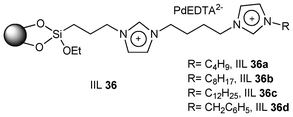 | ||
| Fig. 11 Catalysts with different terminal alkyl groups in ILs. | ||
| Entry | X | R 1 | R 2 | Time | Yield (%) |
|---|---|---|---|---|---|
| 1 | Br | H | 4-MeO | 7 h | 94 |
| 2 | Br | 4-MeO | 4-MeO | 6 h | 94 |
| 3 | Br | 3-NO2 | 4-MeO | 5 h | 99 |
| 4 | I | 4-NO2 | 4-MeO | 6 h | 99 |
| 5 | Br | H | 4-CF3 | 6 h | 98 |
| 6 | Br | 4-Ac | 4-CF3 | 7 h | 99 |
| 7 | Br | 4-MeO | 4-CF3 | 8 h | 93 |
| 8 | I | 4-MeO | 4-CF3 | 6 h | 99 |
| 9 | Br | H | 4-CO2H | 6 h | 88 |
| 10 | Br | 4-Ac | 4-CO2H | 6 h | 90 |
| 11 | Br | 4-MeO | 4-CO2H | 7 h | 91 |
SBA-15 functionalized palladium complex partially confined with 1-butyl-3-methylimidazolium hexafluorophosphate ionic liquid (IIL 37) was demonstrated to be very efficient for the Suzuki coupling reaction of aryl halides (71) including aryl chlorides and heteroaryl halides with different aryl boronic acids (72) under aqueous conditions without any organic co-solvent (Scheme 30). The yields of the resulting products (73) were obtained in the range of 84–100%, and the catalyst could be recycled and reused up to four times without considerable loss of its reactivity. It was also found that almost similar yields of cross-coupling products could be obtained in open air as compared to the same transformation performed under an argon atmosphere.156
 | ||
| Scheme 30 Suzuki coupling reaction of aryl halides with aryl boronic acids using IIL 37. | ||
The palladium catalyzed Heck reaction is one of the most widely used catalytic carbon-carbon bond forming tools in organic synthesis. The reaction has been used to generate alkenes, dienes, and other unsaturated structures which are important intermediates in the production of fine chemicals.157 Recently, Mesoporous SBA-15 was again used as a solid support to develop a heterogeneous catalyst in which the Pd–porphyrin functionalized ionic liquid was tightly locked in the channels of mesoporous SBA-15 through ion-pair electrostatic interaction between imidazolium-cationic and Pd–porphyrin-anionic moieties. It was found that the materials prepared through post-synthesis maintained better mesophase structure of SBA-15 than those prepared through one-pot sol–gel procedure. The Heck reaction of aryl iodides (or aryl bromides) (74) and ethyl acrylate (75) catalyzed by IIL 38 was studied (Scheme 31). Most of the cross-coupling products (76) were obtained in excellent yields (up to 100%). Particularly, this catalytic system could be recovered and recycled 9 times without SBA-15 structural collapse, Pd species leaching, or obvious activity loss.158
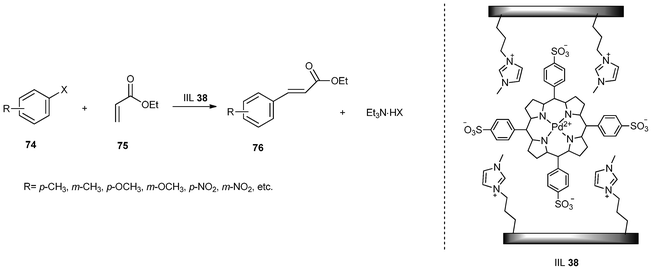 | ||
| Scheme 31 Heck reaction of aryl halides with ethyl acrylate catalyzed by IIL 38. | ||
Han et al.159 employed the cross-linked polymer polydivinylbenzene (PDVB) and grafted it with 1-aminoethyl-3-vinylimidazolium bromide ([VAIM]Br) as a support to immobilize palladium nanoparticles. The catalytic performance of the copolymer-supported Pd nanoparticles (IIL 39) for the Heck arylation of olefins (77) with different aryl iodides (71) was demonstrated under solvent-free conditions (Table 14). The coupling reaction of both electron-deficient and electron-rich iodobenzenes with olefins proceeded to afford the desired products (78) in high yields (up to 98%) using the catalyst (Table 14, Entry 1–8). Due to the insoluble nature of the cross-linked copolymer and the coordination force between the amine group and the palladium nanoparticles, the catalyst was very stable and could be easily separated from the products and reused without significant loss of catalytic activity (Table 14, Entry 9–11).
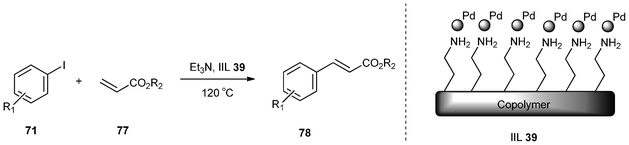
| Entry | R 1 | R 2 | Time | Yield (%) |
|---|---|---|---|---|
| a Entries 9, 10 and 11 are the results of reusing the catalyst for the second, third and fourth times under the reaction conditions of entry 1. | ||||
| 1 | H | Me | 4 h | 97 |
| 2 | H | Et | 5 h | 94 |
| 3 | H | Bu | 6 h | 95 |
| 4 | 4-F | Me | 3 h | 93 |
| 5 | 4-Cl | Me | 2 h | 94 |
| 6 | 4-OH | Me | 1 h | 98 |
| 7 | 4-OMe | Me | 6 h | 96 |
| 8 | 4-Me | Me | 4 h | 93 |
| 9a | H | Me | 4 h | 95 |
| 10a | H | Me | 4 h | 95 |
| 11a | H | Me | 4 h | 94 |
5.5 Other reactions
Hydroamination, a C–N bond forming process, offers a highly attractive pathway for the synthesis of nitrogen containing molecules that are utilized as building blocks in industrial applications. Efforts were made to prepare catalysts with sufficient activity and long-term stability for this type of reaction. For example, immobilization of organometallic complexes on thin supported films of ionic liquid was used to generate a new class of hydroamination catalysts (IIL 40).160,161 High activities for the addition of aniline (79) to styrene (80) were obtained with these bi-functional systems resulting in the Markovnikoff product (81) under kinetically controlled conditions and mainly the anti-Markovnikoff product (82) under the thermodynamic regime (Scheme 32).
To further improve the catalytic performance of imidazolium-based IL catalysts for the cycloaddition reactions of CO2 and epoxides, various metal chlorides were incorporated into silica-grafted 1-methyl-3-[(triethoxysilyl)propyl] imidazolium chloride to obtain heterogeneous catalysts. For example, Park et al.162 introduced CoCl2, NiCl2, CuCl2, ZnCl2, and MnCl2 into the catalytic system and found that the incorporation of such metal ions significantly enhanced the catalytic reactivity of the silica-grafted ILs towards cycloaddition reactions of CO2 and epoxides in solvent-free conditions. Relatively higher selectivity (90–96%) was achieved in the synthesis of allyl glycidyl carbonate (AGC) via a cycloaddition reaction of CO2 and allyl glycidyl ether (AGE) catalyzed by immobilized ILs functionalized with metal ions, compared with the corresponding reaction using pure immobilized IL (66% selectivity).
H2O2-based catalytic epoxidation has received much attention from an economic and environmental perspective.163 For instance, immobilized ILs linked to metals have been designed to improve the catalytic activity for the oxidation of allylic alcohols and alkenes to corresponding aldehydes.164,165 With the aid of the IL, both hydrophobic reactant and the hydrophilic reactant were accessible to the active sites. This has significantly helped to achieve promising results in catalytic reactions.
Recently, many other types of interesting organic transformations such as asymmetric Strecker hydrocyanation of aldimines for the preparation of chiral α-aminonitriles,166 selective oxidation of sulfide to sulfoxide or sulfone167 and reduction of 4-nitrophenol to 4-aminophenol168 have also been executed using the type of catalysts mentioned above.
6 Catalysis by immobilized ionic liquids functionalized with other groups
Not only acidic ILs, but also basic ILs have been immobilized by some researchers. As an illustration, Hardacre and his group169 reported a typical Knoevenagel reaction mediated by basic ionic liquid (BIL) immobilized on silica. The silica was suspended in an organic solvent and as a biphasic system; the cations (BIL 1–3) are shown in Fig. 12 and in each case bis{(trifluoromethyl)sulfonyl}imide ([NTf2]−) was employed as the counter ion. By increasing the distance between the ammonium head group and Hünig's base, the activity of the catalyst was found to increase. Moreover, excellent recyclability of the catalyst for over five times without any loss of catalytic activity or leaching of the ionic liquid into the extractant phase was observed. In addition to tethered ammonium ionic liquids, the ILs with protonated amine groups were also prepared for the Knoevenagel reaction. The mesoporous silica (MCM-41 and SBA-15) supported basic IL catalysts showed much higher catalytic performance and better recyclability as compared with the homogeneous counter-part, N-(3-aminopropyl), N(3)-(3-triethoxysilylpropyl)-4,5-dihydroimidazolium bromide hydrobromide.170 | ||
| Fig. 12 Structure of the cations of basic ionic liquids (BILs). | ||
7 Other applications of immobilized functional ILs
The electrolytes and electrodes, employed in the fields of biosensors, fuel/solar cells, etc., are crucial to the performance of electrocatalysis or electrochemical reaction, and ionic liquids comprising of metal-modified electrolytes and electrodes were found to exhibit increased thermal stability, conductivity, electrocatalytic activity, and cycleability over a long period. For these reasons, many attempts have been made to solidify the ionic liquids into solid-state electrolytes and electrodes e.g. by copolymerization with an ionic liquid monomer, impregnation into an organic polymer, confinement within inorganic gels, solidification using low-molecular-weight gelating agents, and gelation with nanoparticles.171–181The immobilization of enzymes or proteins onto supported ionic liquids have been carried out to improve the stability and activate the enzymes or proteins and to develop environment-friendly alternative solvents for various biocatalysis, leading to greener and highly efficient bioprocesses.182–184 For example, Liu and co-workers185 synthesized room temperature ionic liquid-decorated mesoporous SBA-15 for papain immobilization. In the casein hydrolysis, the papain immobilized on SBA-15 via ionic liquid (1-methyl-3-[(triethoxysilyl)propyl] imidazolium chloride) showed a higher specific activity than that on SBA-15, implying that the ionic liquid was beneficial to improve the activity of the immobilized papain.
Apart from catalysis, immobilized ionic liquids are playing an increasingly important role in separation science. A variety of chemicals including gases (e.g. CO2, N2, SO2),186–190 metals (e.g. Cr4+, Hg2+),191,192 and organic compounds (e.g. thiophene, dioxins, protein)193–197 have been separated with enhanced selectivity. Thus, Ihara et al.198 prepared a multi-mode poly(ionic liquid)-grafted silica stationary phase for analytical studies in HPLC from the ionic liquid monomer (1-(2-acryloyloxyundecyl)-3-methyl-imidazolium bromide) –it was subsequently polymerized on the silica via surface-initiated radical chain-transfer reaction. Compared with commonly used octadecyl silica (ODS) columns, the modified stationary phase showed considerably higher molecular-planarity and selectivity towards polycyclic aromatic hydrocarbon (PAH) isomers, additional anion-exchange ability and hydrophilic interaction in liquid chromatography (HILIC) mode. Moreover, immobilized ionic liquids can also serve as sorbents in liquid chromatography199 with high-selectivity, thermally stable GC stationary phases200 and as recyclable ligands cooperated with L-proline in asymmetric catalytic aldol condensations.201–203
Additionally, immobilized ionic liquids have been applied for preparation of various types of materials. For instance, Fletcher and co-workers204 reported a series of emulsions synthesized from silica nanoparticle stabilized ionic liquids, showing excellent stability with respect to coalescence and Ostwald ripening; Li et al.205 developed a stable poly(ionic liquid) brush coated electrospun membranes, the surface properties of the membrane were reversibly altered between hydrophilic to hydrophobic with counter anion exchange, which helped pores to withdraw or expel solvent molecules (H2O) thereby controlling the transport of probe molecules through the membrane.
8 Summary
As is clear from this review, immobilized functional ionic liquids in catalysis combines the advantages of homogeneous and heterogeneous catalysis such as high activity and selectivity, the ease of product separation and recyclability. Besides the successful immobilization of homogeneous catalysts, the use of immobilized functional ILs in catalysis also significantly lowers the requirement for the amount of ILs while retaining the efficiency of catalytic reaction pretty well. In summary, suitably functionalized ILs play a central role for the generation of immobilized catalysts with high activity and stability and have been successfully employed to execute useful organic transformations. However, some limitations remain and significant research still needs to be conducted in future to understand the basic kinetics and detailed mechanistic studies for specific transformations, the precise interaction of the ionic liquid with the support, the support's influence on activity and selectivity of the catalyst, and the leaching of the catalyst or the ionic liquid.Acknowledgements
This work was financially supported by the International Science & Technology Cooperation Program of China (2010DFB60840), Key Science and Technology Project of Guizhou Province (No. 20076004), Social Development S&T Program (No. SZ-[2009] 3011), and the National Key Technology R&D Program (2006BAD07A12).References
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 366–3692 Search PubMed.
- F. Jutz, J.-M. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322–353 CrossRef CAS.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS.
- M. Haumann and A. Riisager, Chem. Rev., 2008, 108, 1474–1497 CrossRef CAS.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- K. Binnemans, Chem. Rev., 2007, 107, 2592–2614 CrossRef CAS.
- F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS.
- C. Chiappe and M. Malvaldi, Phys. Chem. Chem. Phys., 2010, 12, 11191–11196 RSC.
- A. Riisager, R. Fehrmann, M. Haumann and P. Wasserscheid, Top. Catal., 2006, 40, 91–102 CrossRef.
- C. V. Doorslaer, J. Wahlen, P. Mertens, K. Binnemans and D. D. Vos, Dalton Trans., 2010, 39, 8377–8390 RSC.
- I. Tóth, I. Guo and B. E. Hanson, J. Mol. Catal. A: Chem., 1997, 116, 217–229 CrossRef CAS.
- C. P. Mehnert, Chem.–Eur. J., 2005, 11, 50–56 CrossRef.
- Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- T. Welton, P. Wasserscheid, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- C. Chiappe and D. Pieraccini, J. Phys. Org. Chem., 2005, 18, 275–297 CrossRef CAS.
- C. A. Angell, Y. Ansari and Z. Zhao, Faraday Discuss., 2012, 154, 9–27 RSC.
- S. Lee, Chem. Commun., 2006, 1049–1063 RSC.
- J. H. Davis Jr, Chem. Lett., 2004, 33, 1072–1077 CrossRef.
- M. H. Valkenberg, C. deCastro and W. F. Hölderich, Green Chem., 2002, 4, 88–93 RSC.
- X. X. Han and D. W. Armstrong, Acc. Chem. Res., 2007, 40, 1079–1086 CrossRef CAS.
- Q.-H. Zhang, S.-G. Zhang and Y.-Q. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- A. Safavi, N. Maleki, N. Iranpoor, H. Firouzabadi, A. R. Banazadeh, R. Azadi and F. Sedaghati, Chem. Commun., 2008, 6155–6157 RSC.
- M.-A. Néouze, J. L. Bideau, P. Gaveau, S. Bellayer and A. Vioux, Chem. Mater., 2006, 18, 3931–3936 CrossRef CAS.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef.
- J. L. Anthony, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2002, 106, 7315–7320 CrossRef CAS.
- L. Han, S.-W. Park and D.-W. Park, Energy Environ. Sci., 2009, 2, 1286–1292 RSC.
- L.-F. Xiao, F.-W. Li, J.-J. Peng and C.-G. Xia, J. Mol. Catal. A: Chem., 2006, 253, 265–269 CrossRef CAS.
- J. Sun, S. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS.
- X. Q. Yu, J. S. Huang, W. Y. Yu and C. M. Che, J. Am. Chem. Soc., 2000, 122, 5337–5342 CrossRef CAS.
- R. Akiyama and S. Kobayashi, Angew. Chem., 2002, 114, 2714–2716 CrossRef.
- M. Takeuchi, R. Akiyama and S. Kobayashi, J. Am. Chem. Soc., 2005, 127, 13096–13097 CrossRef CAS.
- C. W. Tsang, B. Baharloo, D. Riendl, M. Yam and D. P. Gates, Angew. Chem., 2004, 116, 5800–5803 CrossRef.
- Y. Xie, Z.-F. Zhang, T. Jiang, J.-L. He, B.-X. Han, T.-B. Wu and K.-L. Ding, Angew. Chem., 2007, 119, 7393–7396 CrossRef.
- S. Udayakumar, S.-W. Park, D.-W. Park and B.-S. Choi, Catal. Commun., 2008, 9, 1563–1570 CrossRef CAS.
- S. Udayakumar, Y.-S. Son, M.-K. Lee, S.-W. Park and D.-W. Park, Appl. Catal., A, 2008, 347, 192–199 CrossRef CAS.
- K.-S. Hwang, Y.-S. Son, S.-W. Park, D.-W. Park and K. J. Oh, J. Ind. Eng. Chem., 2009, 15, 854–859 Search PubMed.
- E. Cano-Serrano, G. Blanco-Brieva, J. M. Campos-Martin and J. L. G. Fierro, Langmuir, 2003, 19, 7621–7627 CrossRef CAS.
- K. Shimizu, E. Hayashi, T. Hatamachi, T. Kodama and Y. Kitayama, Tetrahedron Lett., 2004, 45, 5135–5138 Search PubMed.
- S. Udayakumar, V. Raman, H.-L. Shim and D.-W. Park, Appl. Catal., A, 2009, 368, 97–104 CrossRef CAS.
- A.-H. Lu, W. Schmidt, N. Matoussevitch, H. Bönnemann, B. Spliethoff, B. Tesche, E. Bill, W. Kiefer and F. Schuth, Angew. Chem., Int. Ed., 2004, 43, 4303–4306 CrossRef CAS.
- H. M. R. Gardimalla, D. Mandal, P. D. Stevens, M. Yen and Y. Gao, Chem. Commun., 2005, 4432–4434 RSC.
- R. Abu-Reziq, H. Alper, D. Wang and M. L. Post, J. Am. Chem. Soc., 2006, 128, 5279–5282 CrossRef.
- X.-X. Zheng, S.-Z. Luo, L. Zhang and J.-P. Cheng, Green Chem., 2009, 11, 455–458 RSC.
- L. Sun, W. J. Boo, R. L. Browning, H.-J. Sue and A. Clearfield, Chem. Mater., 2005, 17, 5606–5609 CrossRef CAS.
- G. Alberti, Acc. Chem. Res., 1978, 11, 163–170 CrossRef CAS.
- W. J. Boo, L. Sun, J. Liu, A. Clearfield and H.-J. Sue, J. Phys. Chem. C, 2007, 111, 10377–10381 CrossRef CAS.
- H. Wang, M. Zou, N. Li and K. Li, J. Mater. Sci., 2007, 42, 7738–7744 CrossRef CAS.
- H. Hu, J. C. Martin, M. Xiao, C. S. Southworth, Y.-Z. Meng and L.-Y. Sun, J. Phys. Chem. C, 2011, 115, 5509–5514 CrossRef CAS.
- H. Hu, J. C. Martin, M. Zhang, C. S. Southworth, M. Xiao, Y.-Z. Meng and L.-Y. Sun, RSC Adv., 2012, 2, 3810–3815 RSC.
- R. Savic, L. B. Luo, A. Eisenberg and D. Maysinger, Science, 2003, 300, 615–618 CrossRef CAS.
- W. Meier, Chem. Soc. Rev., 2000, 29, 295–303 RSC.
- T. Higuchi, A. Tajima, K. Motoyoshi, H. Yabu and M. Shimomura, Angew. Chem., Int. Ed., 2009, 48, 5125–5128 CrossRef CAS.
- K. Min, H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 10521–10526 CrossRef CAS.
- J. Yuan and M. Antonietti, Macromolecules, 2011, 44, 744–750 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS.
- S. Liu and S. Armes, J. Am. Chem. Soc., 2001, 123, 9910–9911 CrossRef CAS.
- K. Min, H. Gao, J. A. Yoon, W. Wu, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2009, 42, 1597–1603 CrossRef CAS.
- Y.-B. Xiong, Y.-J. Wang, H. Wang and R.-M. Wang, Polym. Chem., 2011, 2, 2306–2315 RSC.
- S. Caddick, Tetrahedron, 1995, 51, 10403–10432 CrossRef.
- M. M. Dharman, H.-J. Choi, D.-W. Kim and D.-W. Park, Catal. Today, 2011, 164, 544–547 CrossRef CAS.
- D.-W. Kim, D.-K. Kim, M.-I. Kim and D.-W. Park, Catal. Today, 2012, 185, 217–223 Search PubMed.
- Z.-Z. Yang, Y.-N. Zhao, L.-N. He, J. Gao and Z.-S. Yin, Green Chem., 2012, 14, 519–527 RSC.
- J. Sun, J.-Q. Wang, W.-G. Cheng, J.-X. Zhang, X.-H. Li, S.-J. Zhang and Y.-B. She, Green Chem., 2012, 14, 654–660 RSC.
- Y. Ono, Appl. Catal., A, 1997, 155, 133–166 CrossRef CAS.
- M. Aresta, A. Dibenedetto, F. Nocito and C. Ferragina, J. Catal., 2009, 268, 106–114 CrossRef CAS.
- K. Tomishige and K. Kunimori, Appl. Catal., A, 2002, 237, 103–109 CrossRef CAS.
- T. Sakakura, J. C. Choi, Y. Satio and T. Sako, Polyhedron, 2000, 19, 573–576 CrossRef CAS.
- Y. Fang and W. Xiao, Sep. Purif. Technol., 2004, 34, 255–263 CrossRef CAS.
- T. Tatsumi, Y. Watanabe and K. A. Koyano, Chem. Commun., 1996, 2281–2282 RSC.
- B. S. Ahn, B. G. Lee, H. S. Kim and M. S. Han, React. Kinet. Catal. Lett., 2001, 73, 33–38 CrossRef CAS.
- H.-J. Cho, H.-M. Kwon, J. Tharun and D.-W. Park, J. Ind. Eng. Chem., 2010, 16, 679–683 Search PubMed.
- D.-W. Kim, C.-W. Kim, J.-C. Koh and D.-W. Park, J. Ind. Eng. Chem., 2010, 16, 474–478 Search PubMed.
- D.-W. Kim, D.-O. Lim, D.-H. Cho, J.-C. Koh and D.-W. Park, Catal. Today, 2011, 164, 556–560 Search PubMed.
- F. Rajabi and B. Karimi, J. Mol. Catal. A: Chem., 2005, 232, 95–99 CrossRef CAS.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666 CrossRef CAS.
- J.-E. Bäckvall, Modern Oxidation Methods, Wiley-VCH, Weinheim, 2004 Search PubMed.
- Z. Hou, N. Theyssen and W. Leitner, Green Chem., 2007, 9, 127–132 RSC.
- B. Karimi, A. Biglari, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2007, 46, 7210–7213 CrossRef CAS.
- B. Karimi and E. Badreh, Org. Biomol. Chem., 2011, 9, 4194–4198 RSC.
- G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4123 CrossRef.
- S. Baj, A. Chrobok and R. Słupska, Green Chem., 2009, 11, 279–282 RSC.
- A. Chrobok, S. Baj, W. Pudło and A. Jarzębski, Appl. Catal., A, 2009, 366, 22–28 CrossRef CAS.
- C. DeCastro, E. Sauvage, M. H. Valkenberg and W. F. Hölderich, J. Catal., 2000, 196, 86–94 CrossRef CAS.
- M. H. Valkenberg, C. deCastro and W. F. Hölderich, Appl. Catal., A, 2001, 215, 185–190 CrossRef CAS.
- H. Valizadeh, M. Amiri and F. Hosseinzadeh, Dyes Pigm., 2012, 92, 1308–1313 CrossRef CAS.
- D. W. Kim, D. J. Hong, K. S. Jang and D. Y. Chi, Adv. Synth. Catal., 2006, 348, 1719–1727 CrossRef CAS.
- S. M. Coman, M. Florea, V. I. Parvulescu, V. David, A. Medvedovici, D. D. Vos, P. A. Jacobs, G. Poncelet and P. Grange, J. Catal., 2007, 249, 359–369 CrossRef CAS.
- D. W. Kim, H.-J. Jeong, S. T. Lim, M.-H. Sohn and D. Y. Chi, Tetrahedron, 2008, 64, 4209–4214 CrossRef CAS.
- S. S. Shinde, B. S. Lee and D. Y. Chi, Tetrahedron Lett., 2008, 49, 4245–4248 Search PubMed.
- D. W. Kim and D. Y. Chi, Angew. Chem., Int. Ed., 2004, 43, 483–485 CrossRef CAS.
- S. Abelló, F. Medina, X. Rodríguez, Y. Cesteros, P. Salagre, J. E. Sueiras and D. T. B. Coq, Chem. Commun., 2004, 1096–1097 RSC.
- X. Zhang, W.-S. Zhao, C.-K. Qu, L.-L. Yang and Y.-C. Cui, Tetrahedron: Asymmetry, 2012, 23, 468–473 Search PubMed.
- Z.-T. Wang, S.-C. Wang and L.-W. Xu, Helv. Chim. Acta, 2005, 88, 986–989 CrossRef CAS.
- Y.-L. Xu, C.-S. Sun and G.-H. Liu, Advanced Materials Research, 2012, 347–353, 1944–1948 Search PubMed.
- H. Valizadeh, M. Amiri and A. Shomali, C. R. Chim., 2011, 14, 1103–1108 Search PubMed.
- A. L. Petre, W. F. Hoelderich and M. L. Gorbaty, Appl. Catal., A, 2009, 363, 100–108 CrossRef CAS.
- K. B. Sidhpuria, A. L. Daniel da Silva, T. Trindade and J. A. P. Coutinho, Green Chem., 2011, 13, 340–349 RSC.
- J. S. Wilkes, J. Mol. Catal. A: Chem., 2004, 214, 11–17 CrossRef CAS.
- X. M. Liu, M. Liu, X. W. Guo and J. X. Zhou, Catal. Commun., 2008, 9, 1–7 CrossRef CAS.
- Y. Y. Wang, X. Gong, Z. Wang and L. Y. Dai, J. Mol. Catal. A: Chem., 2010, 322, 7–16 CrossRef CAS.
- K. Qiao, H. Hagiwara and C. Yokoyama, J. Mol. Catal. A: Chem., 2006, 246, 65–69 CrossRef CAS.
- Q. Zhang, J. Luo and Y.-Y. Wei, Green Chem., 2010, 12, 2246–2254 RSC.
- J.-M. Miao, H. Wan and G.-F. Guan, Catal. Commun., 2011, 12, 353–356 CrossRef CAS.
- J.-M. Miao, H. Wan, Y.-B. Shao, G.-F. Guan and B. Xu, J. Mol. Catal. A: Chem., 2011, 348, 77–82 Search PubMed.
- C. Fehér, E. Kriván, J. Hancsók and R. Skoda-Földes, Green Chem., 2012, 14, 403–409 RSC.
- Z.-J. Xu, H. Wan, J.-M. Miao, M.-J. Han, C. Yang and G.-F. Guan, J. Mol. Catal. A: Chem., 2010, 332, 152–157 CrossRef CAS.
- P. Wang, A.-G. Kong, W.-J. Wang, H.-Y. Zhu and Y.-K. Shan, Catal. Lett., 2010, 135, 159–164 CrossRef CAS.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J. M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS.
- Q. Zhang, H. Su, J. Luo and Y.-Y. Wei, Green Chem., 2012, 14, 201–208 RSC.
- F.-J. Liu, S.-F. Zuo, W.-P. Kong and C.-Z. Qi, Green Chem., 2012, 14, 1342–1349 RSC.
- D.-M. Li, F. Shi, S. Guo and Y.-Q. Deng, Tetrahedron Lett., 2004, 45, 265–268 CrossRef CAS.
- L.-N. Han, H.-J. Choi, S.-J. Choi, B.-Y. Liu and D.-W. Park, Green Chem., 2011, 13, 1023–1028 RSC.
- A. L. Zhu, T. Jiang, B. X. Han, J. C. Zhang, Y. Xie and X. M. Ma, Green Chem., 2007, 9, 169–172 RSC.
- Y. X. Zhou, S. Q. Hu, X. M. Ma, S. G. Liang, T. Jiang and B. X. Han, J. Mol. Catal. A: Chem., 2008, 284, 52–57 CrossRef CAS.
- J. Sun, W.-G. Cheng, W. Fan, Y.-H. Wang, Z.-Y. Meng and S.-J. Zhang, Catal. Today, 2009, 148, 361–367 CrossRef CAS.
- R. A. Watile, D. B. Bagal, K. M. Deshmukh, K. P. Dhake and B. M. Bhanage, J. Mol. Catal. A: Chem., 2011, 351, 196–203 CrossRef CAS.
- R. A. Watile, K. M. Deshmukh, K. P. Dhake and B. M. Bhanage, Catal. Sci. Technol., 2012, 2, 1051–1055 RSC.
- C. P. Mehnert, R. A. Cook, N. C. Dispenziere and M. Afeworki, J. Am. Chem. Soc., 2002, 124, 12932–12933 CrossRef CAS.
- Y. Gu and G. Li, Adv. Synth. Catal., 2009, 351, 817–847 CrossRef.
- P. Tundo and A. Perosa, Chem. Soc. Rev., 2007, 36, 532–550 RSC.
- C. Van Doorslaer, J. Wahlen, P. Mertens, K. Binnemans and D. De Vos, Dalton Trans., 2010, 39, 8377–8390 RSC.
- S. Werner, M. Haumann and P. Wasserscheid, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 203–230 Search PubMed.
- C. P. Mehnert, E. J. Mozeleski and R. A. Cook, Chem. Commun., 2002, 3010–3011 RSC.
- A. Wolfson, I. F. J. Vankelecom and P. A. Jacobs, Tetrahedron Lett., 2003, 44, 1195–1198 CrossRef CAS.
- J. Huang, T. Jiang, H.-X. Gao, B.-X. Han, Z.-M. Liu, W.-Z. Wu, Y.-H. Chang and G.-Y. Zhao, Angew. Chem., 2004, 116, 1421–1423 CrossRef.
- J.-P. Mikkola, P. Virtanen, H. Karhu, T. Salmi and D. Y. Murzin, Green Chem., 2006, 8, 197–205 RSC.
- S.-D. Miao, Z.-M. Liu, B.-X. Han, J. Huang, Z.-Y. Sun, J.-L. Zhang and T. Jiang, Angew. Chem., Int. Ed., 2006, 45, 266–269 CrossRef CAS.
- P. Virtanen, H. Karhu, K. Kordas and J.-P. Mikkola, Chem. Eng. Sci., 2007, 62, 3660–3671 CrossRef CAS.
- M. Ruta, I. Yuranov, P. J. Dyson, G. Laurenczy and L. Kiwi-Minsker, J. Catal., 2007, 247, 269–276 CrossRef CAS.
- M. A. Gelesky, S. S. X. Chiaro, F. A. Pavan, J. H. Z. dos Santos and J. Dupont, Dalton Trans., 2007, 5549–5553 RSC.
- R. Abu-Reziq, D. Wang, M. Post and H. Alper, Adv. Synth. Catal., 2007, 349, 2145–2150 CrossRef CAS.
- Y. Kume, K. Qiao, D. Tomida and C. Yokoyama, Catal. Commun., 2008, 9, 369–375 CrossRef CAS.
- L. Rodríguez-Pérez, E. Teuma, A. Falqui, M. Gómez and P. Serp, Chem. Commun., 2008, 4201–4203 RSC.
- Y. S. Chun, J. Y. Shin, C. E. Song and S.-G. Lee, Chem. Commun., 2008, 942–944 RSC.
- A. G. Panda, S. R. Jagtap, N. S. Nandurkar and B. M. Bhanage, Ind. Eng. Chem. Res., 2008, 47, 969–972 CrossRef CAS.
- H. Hagiwara, T. Nakamura, T. Hoshi and T. Suzuki, Green Chem., 2011, 13, 1133–1137 RSC.
- G. Liu, M.-G. Hou, T.-B. Wu, T. Jiang, H.-L. Fan, G.-Y. Yang and B.-X. Han, Phys. Chem. Chem. Phys., 2011, 13, 2062–2068 RSC.
- J. Li, X.-Y. Shi, Y.-Y. Bi, J.-F. Wei and Z.-G. Chen, ACS Catal., 2011, 1, 657–664 Search PubMed.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS.
- L.-L. Lou, Y.-L. Dong, K. Yu, S. Jiang, Y. Song, S. Cao and S.-X. Liu, J. Mol. Catal. A: Chem., 2010, 333, 20–27 Search PubMed.
- E. Öchsner, M. J. Schneider, C. Meyer, M. Haumann and P. Wasserscheid, Appl. Catal., A, 2011, 399, 35–41 CrossRef.
- C. DeCastro, E. Sauvage and M. H. Valkenberg, and W. F. Hölderich, J. Catal., 2000, 196, 86–94 Search PubMed.
- M. H. Valkenberg, C. deCastro and W. F. Hölderich, Top. Catal., 2001, 14, 139–144.
- G.-J. Wang, N.-Y. Yu, L. Peng, R. Tan, H.-H. Zhao, D.-H. Yin, H.-Y. Qiu, Z.-H. Fu and D.-L. Yin, Catal. Lett., 2008, 123, 252–258 CrossRef CAS.
- H.-H. Zhao, N.-Y. Yu, J.-Q. Wang, D.-Y. Zhuang, Y. Ding, R. Tan and D.-H. Yin, Microporous Mesoporous Mater., 2009, 122, 240–246 Search PubMed.
- G. Tojo, M. Fernandez, Oxidation of Alcohols to Aldehydes and Ketones, Springer, USA, 2006 Search PubMed.
- A. Chrobok, S. Baj, W. Pudło and A. Jarzębski, Appl. Catal., A, 2010, 389, 179–185 Search PubMed.
- H. Wang, B. Wang, C.-L. Liu and W.-S. Dong, Microporous Mesoporous Mater., 2010, 134, 51–57 Search PubMed.
- U. Hintermair, Z.-X. Gong, A. Serbanovic, M. J. Muldoon, C. C. Santini and D. J. Cole-Hamilton, Dalton Trans., 2010, 39, 8501–8510 RSC.
- S. Shylesh, D. Hanna, S. Werner and A. T. Bell, ACS Catal., 2012, 2, 487–493 Search PubMed.
- N. T. S. Phan, M. V. D. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- M.-J. Jin, A. Taher, H.-J. Kang, M. Choi and R. Ryoo, Green Chem., 2009, 11, 309–313 RSC.
- J. Wang, G. Song and Y. Peng, Tetrahedron Lett., 2011, 52, 1477–1480 CrossRef CAS.
- J.-F. Wei, J. Jiao, J.-J. Feng, J. Lv, X.-R. Zhang, X.-Y. Shi and Z.-G. Chen, J. Org. Chem., 2009, 74, 6283–6286 CrossRef CAS.
- B. Karimi and A. Zamani, Org. Biomol. Chem., 2012, 10, 4531–4536 RSC.
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS.
- J. Zhang, G.-F. Zhao, Z. Popović, Y. Lu and Y. Liu, Mater. Res. Bull., 2010, 45, 1648–1653 Search PubMed.
- G. Liu, M. Hou, J. Song, T. Jiang, H. Fan, Z. Zhang and B. Han, Green Chem., 2010, 12, 65–69 RSC.
- O. Jimenez, T. E. Müller, C. Sievers, A. Spirkl and J. A. Lercher, Chem. Commun., 2006, 2974–2976 RSC.
- C. Sievers, O. Jiménez, R. Knapp, X. Lin, T. E. Müller, A. Türler, B. Wierczinski and J. A. Lercher, J. Mol. Catal. A: Chem., 2008, 279, 187–199 CrossRef CAS.
- L. Han, M.-S. Park, S.-J. Choi, Y.-J. Kim, S.-M. Lee and D.-W. Park, Catal. Lett., 2012, 142, 259–266 Search PubMed.
- B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2473 CrossRef CAS.
- G. Liu, M. Hou, J. Song, Z. Zhang, T. Wu and B. Han, J. Mol. Catal. A: Chem., 2010, 316, 90–94 Search PubMed.
- S. Doherty, J. G. Knight, J. R. Ellison, D. Weekes, R. W. Harrington, C. Hardacre and H. Manyar, Green Chem., 2012, 14, 925–929 RSC.
- B. Karimi, A. Maleki, D. Elhamifar, J. H. Clark and A. J. Hunt, Chem. Commun., 2010, 46, 6947–6949 RSC.
- X. Shi, X. Han, W. Ma, J. Wei, J. Li, Q. Zhang and Z. Chen, J. Mol. Catal. A: Chem., 2011, 34, 57–62 Search PubMed.
- J. Yuan, S. Wunder, F. Warmuth and Y. Lu, Polymer, 2012, 53, 43–49 Search PubMed.
- C. Paun, J. Barklie, P. Goodrich, H.Q.N. Gunaratne, A. McKeown, V.I. Pârvulescu and C. Hardacre, J. Mol. Catal. A: Chem., 2007, 269, 64–71 CrossRef CAS.
- H. Zhao, N. Yu, Y. Ding, R. Tan, C. Liu, D. Yin, H. Qiu and D. Yin, Microporous Mesoporous Mater., 2010, 136, 10–17 CrossRef CAS.
- M. Kosmulski, M. Szafran, C. Saneluta and K. Marczewska-Boczkowska, J. Colloid Interface Sci., 2005, 291, 214–217 CrossRef CAS.
- S. Shimano, H. Zhou and I. Honma, Chem. Mater., 2007, 19, 5216–5221 CrossRef CAS.
- Y. Zhang and J. Zheng, Electrochem. Commun., 2008, 10, 1400–1403 Search PubMed.
- K.-F. Wang, F.-F. Jian and R.-R. Zhuang, Dalton Trans., 2009, 4532–4537 RSC.
- A. Garcia-Bernabe, V. Compan, M. I. Burguete, E. García-Verdugo, N. Karbass, S. V. Luis and E. Riande, J. Phys. Chem. C, 2010, 114, 7030–7037 CrossRef CAS.
- Q.-L. Sheng, J.-B. Zheng, X.-D. Shang-Guan, W.-H. Lin, Y.-Y. Li and R.-X. Liu, Electrochim. Acta, 2010, 55, 3185–3191 CrossRef CAS.
- L. Qiu, B. Liu, Y. Peng and F. Yan, Chem. Commun., 2011, 47, 2934–2936 RSC.
- J.-S. Lee, T. Lee, H.-K. Song, J. Cho and B.-S. Kim, Energy Environ. Sci., 2011, 4, 4148–4154 RSC.
- J. Chai, F.-H. Li, Y.-W. Hu, Q.-X. Zhang, D.-X. Han and L. Niu, J. Mater. Chem., 2011, 21, 17922–17929 RSC.
- Y. Zhang, X. Sun and N. Jia, Sens. Actuators, B, 2011, 157, 527–532 CrossRef CAS.
- A. Eguizábal, J. Lemus, M. Urbiztondo, A. M. Moschovi, S. Ntais, J. Soler and M. P. Pina, J. Power Sources, 2011, 196, 4314–4323 CrossRef CAS.
- P. Lozano, E. García-Verdugo, N. Karbass, K. Montague, T. D. Diego, M. I. Burguete and S. V. Luis, Green Chem., 2010, 12, 1803–1810 RSC.
- H. Vila-Real, A. J. Alfaia, J. N. Rosa, P. M. P. Gois, M. E. Rosa, A. R. T. Calado and M. H. Ribeiro, J. Biotechnol., 2011, 152, 147–158 CrossRef CAS.
- J. Li, L. Liu, F. Xiao, Z. Gui, R. Yan, F. Zhao, L. Hu and B. Zeng, J. Electroanal. Chem., 2008, 613, 51–57 Search PubMed.
- W. Bian, B. Yan, N. Shi, F. Qiu, L.-L. Lou, B. Qi and S. Liu, Mater. Sci. Eng., C, 2012, 32, 364–368 Search PubMed.
- D.-H. Kim, I.-H. Baek, S.-U. Hong and H.-K. Lee, J. Membr. Sci., 2011, 372, 346–354 CrossRef CAS.
- J. J. Close, K. Farmer, S. S. Moganty and R. E. Baltus, J. Membr. Sci., 2012, 390–391, 201–210 Search PubMed.
- Y. Gu and T. P. Lodge, Macromolecules, 2011, 44, 1732–1736 CrossRef CAS.
- X.-B. Hu, Y.-X. Li, K. Huang, S.-L. Ma, H. Yu, Y.-T. Wu and Z.-B. Zhang, Green Chem., 2012, 14, 1440–1446 RSC.
- M. Gupta, Y. Chen, Z. Hu and J. Jiang, Phys. Chem. Chem. Phys., 2012, 14, 5785–5794 RSC.
- M.-L. Chen, Y.-N. Zhao, D.-W. Zhang, Y. Tian and J.-H. Wang, J. Anal. At. Spectrom., 2010, 25, 1688–1694 RSC.
- F. Loe-Mie, G. Marchand, J. Berthier, N. Sarrut, M. Pucheault, M. Blanchard-Desce, F. Vinet and M. Vaultier, Angew. Chem., 2010, 122, 434–437 Search PubMed.
- X. Wang, H. Wan, M. Han, L. Gao and G. Guan, Ind. Eng. Chem. Res., 2012, 51, 3418–3424 Search PubMed.
- H. Meng, X.-W. Chen and J.-H. Wang, J. Mater. Chem., 2011, 21, 14857–14863 RSC.
- P. S. Kulkarni, L. A. Neves, I. M. Coelhoso, C. A. M. Afonso and J. G. Crespo, Environ. Sci. Technol., 2012, 46, 462–468 Search PubMed.
- A. P. de los Ríos, F. J. Hernández-Fernández, M. Rubio, D. Gómez and G. Víllora, Desalination, 2010, 250, 101–104 Search PubMed.
- W. Bi, M. Tian and K. H. Row, Analyst, 2012, 137, 2017–2020 RSC.
- H. Qiu, A. K. Mallik, M. Takafuji, S. Jiang and H. Ihara, Analyst, 2012, 137, 2553–2555 RSC.
- F. Yang, R. Shen, Y. Long, X. Sun, F. Tang, Q. Cai and S. Yao, J. Environ. Monit., 2011, 13, 440–445 RSC.
- J. L. Anderson and D. W. Armstrong, Anal. Chem., 2005, 77, 6453–6462 CrossRef CAS.
- M. Gruttadauria, S. Riela, P. L. Meo, F. D'Anna and R. Noto, Tetrahedron Lett., 2004, 45, 6113–6116 CrossRef CAS.
- M. Gruttadauria, S. Riela, C. Aprile, P. L. Meo, F. D. Anna and R. Noto, Adv. Synth. Catal., 2006, 348, 82–92 CrossRef CAS.
- C. Aprile, F. Giacalone, M. Gruttadauria, A. M. Marculescu, R. Noto, J. D. Revell and H. Wennemers, Green Chem., 2007, 9, 1328–1334 RSC.
- B. P. Binks, A. K. F. Dyab and P. D. I. Fletcher, Chem. Commun., 2003, 2540–2541 RSC.
- W. Jia, Y. Wu, J. Huang, Q. An, D. Xu, Y. Wu, F. Li and G. Li, J. Mater. Chem., 2010, 20, 8617–8623 RSC.
| This journal is © The Royal Society of Chemistry 2012 |