Organochalcogen ligands and their palladium(II) complexes: Synthesis to catalytic activity for Heck coupling
Arun
Kumar
,
Gyandshwar
Kumar Rao
and
Ajai
K Singh
*
Department of Chemistry, Indian Institute of Technology Delhi, New Delhi-110016, India. E-mail: aksingh@chemistry.iitd.ac.in; ajai57@hotmail.com; Fax: +91 11 26581102; Tel: +91 11 26591379
First published on 2nd October 2012
Abstract
Heck cross coupling reactions have been catalyzed with several palladium(II)-complexes of organochalcogen ligands. The complexes applied in molecular and tethered forms appear to be dispensers of catalytically active species, but this aspect has not been thoroughly looked at in all cases. The thermal stability, air and moisture insensitivity and ease of handling and synthesis have made them viable alternatives to phosphine/carbene based Pd-catalysts. This review covers developments from the design of organochalcogen ligands and their palladium(II)-complexes to the applications of these complexes in catalyzing Heck reactions.
 Arun Kumar | Dr Arun Kumar was born in 1980 in Ghaziabad, India. He did his MSc in chemistry at C. C. S. University, Meerut and PhD in inorganic chemistry at the Chemistry Department, Indian Institute of Technology Delhi, in 2009, under the supervision of Prof. A. K. Singh. His thesis was on chalcogenated Schiff bases. He is presently working as a Research Associate (CSIR) in the same department and handling research projects on the catalysis of C–C coupling reactions and nano-science. |
 Gyandshwar Kumar Rao | G. K. Rao, born in 1983 in Deoria (India), studied at St. Andrews College, Gorakhpur University, Gorakhpur (India) for his BSc and MSc degrees. Currently he is pursuing research on “Palladium Catalysts for C–C coupling Reaction” for a PhD degree in the Chemistry Department, Indian Institute of Technology Delhi, under the mentorship of Prof. A. K. Singh. |
 Ajai Kumar Singh | Dr Ajai Kumar Singh is presently a Professor (Higher Academic Grade) in the Department of Chemistry, Indian Institute of Technology Delhi, New Delhi, which he joined in 1982 as a faculty member. He was Head of the Department from Sep 2009 to Aug 2012. A PhD from University of Delhi (1977), got Post Doctoral training in the research group of Professor W. R. McWhinnie, Aston University (U. K.). His research interests are organochalcogen chemistry, organometallics and catalysis. He has nearly 180 research papers in internationally reputed journals and has mentored 30 plus graduates and post doctoral workers. Presently his research group has more than a dozen members. He is an honorary member of the Science Faculty of University of Delhi. He was President of the Indian Council of Chemists (Inorganic Section) in 2011. |
1. Introduction
The olefination of aryl halides (eqn (1)), commonly called the Heck reaction1–3 is a process generally catalyzed by palladium derivatives. This reaction, discovered independently by Heck and Mizoroki,4 is among the most important organic reactions catalyzed by palladium species. It produces chemicals that are important for the synthesis of pharmaceuticals, natural products5,6 and fine chemicals.7 In recent years excellent reviews2,3,8,9 on the Heck reaction have appeared. The mechanistic approaches plausible for this C–C cross-coupling reaction have also been discussed at length in these reviews.2,3,8–11 At high temperatures and in a reducing environment the complexes decompose transiently to the homogeneous Pd(0) phase [colloids, nano-particles (NPs) or clusters],11 which are believed to be the pre-catalyst and form heterogeneous Pd(0), which most likely catalyzes Heck coupling via stable surface defect atoms.10b It appears that the role of ligands around Pd in high temperature Heck reactions is to control the release of Pd(0) species.11 The ligand free Heck coupling catalyzed with Pd(OAc)210a,11 can be cited to support it. The in situ investigations of species involved in the Heck catalytic reactions by FT-NIR12 and quick scanning EXAFS13 have further strengthened the hypothesis regarding the key role of Pd(0) colloids/nano-particles or clusters in Heck's catalytic process. The predictions regarding the activity of ligands and solvents in homogeneous Heck reactions, and the selection of homogeneous catalysts for cross coupling, have also been addressed by computational methods.14,15| Ar–X + CH2 = CH–R → Ar–CH = CH–R + HX | (1) |
The outstanding performance giving palladium based catalytic systems for the Heck reaction has been designed using sterically bulky and electron-rich phosphine ligands2,3 or phosphine mimics such as N-heterocyclic nucleophilic carbenes (NHCs).16,17 Other phosphorous compounds (coordinating through P)2,3 used for this purpose include phosphites (mono- or polydentate) and phosphinites. Palladacycles formed by ligands containing both phosphorus and nitrogen donors are also among the efficient catalysts for this coupling reaction.18–20 Among the diverse aryl or heteroaryl halides, (for the coupling of which the Heck reaction is important), aryl chlorides are the cheapest but relatively difficult to couple. The activity of aryl halides generally follows the order ArI > ArBr > ArCl. It is not that Pd(II)-complexes of phosphorous as well as other donors do not show any catalytic activity21–29 for the coupling of ArCl. Several such complexes are active for them also but with low efficiency. However, a large number of conventional catalytic species for Heck coupling, which are generally phosphorous donor based, are moisture and air-sensitive. The strong donor properties of chalcogens have motivated many research workers to design a number of Pd(II)-complexes of organochalcogen ligands for catalyzing30 the Heck C–C coupling reaction. Such complexes have been found to be much more stable in air and moisture in comparison to Pd-complexes of phosphorous donors. In fact, as catalysts for the Heck reaction, they not only show rivalry with their respective phosphorus analogues but, in many cases, outperform them for the same aryl halide. Thus it was thought worthwhile to take stock of the catalytic activities of palladium(II) complexes of organochalcogen ligands for Heck coupling and perform a critical evaluation of them, as no such attempt has been made so far. The present review is therefore focussed on this subject. An overview of ligands and complexes relevant for Heck reactions is also presented.
2. Organochalcogen ligands in Pd-catalyst design
2.1 General
The chemistry of organosulfur ligands has been explored more extensively than that of organo-selenium and -tellurium ligands, in which interest has built up largely in the last 3–4 decades. Some excellent reviews31–35 covering several aspects of selenides/tellurides and organoselenium/tellurium compounds have been published. The possibilities of using metal complexes of organochalcogen ligands as single source precursors of semiconductors (e.g. II–VI semiconductor) have contributed to strengthening interest in them. We have focussed here only on those organochalcogen ligands which have been used to design palladium based catalysts for the Heck reaction. Such ligands have been described in three groups: (i) pincer type, (ii) chalcogenated Schiff bases and (iii) chalcogenoethers. These ligands are not commercially available and therefore an overview of the synthetic approaches used for them is given here.2.2 Pincer ligands
The pincer ligand (Fig. 1) can make two stable chelate rings with one metal centre. Such a ligand is referred as a (Y,Z,Y′) pincer. In a large number of cases, Z is C or N and Y = Y′. Varying Y/Y′ may result in many such ligands. In chalcogen donor based pincers, Y or Y′ can be SR, SeR or TeR. The side arms stabilize the complexes of pincer ligands and are reported to affect the electronic properties of the metal center. The interesting changes in catalytic properties36–38 have been reported when the Pd–C bond is replaced with the Pd–Si or Pd–P bond (i.e. Z = Si or P). Similarly, when Z = N,39–42 and it is part of an aromatic system, changes in the activity of the resulting Pd(II)–pincer complex occur. The unsymmetrical pincer ligands, having two different donors in the side arms, have also been reported to vary somewhat catalytic activities of the resulting Pd-complexes.43,44 However, the effect of the ligand is not believed to be direct in most cases. Most likely it affects the availability of the Pd(0) species recognized as the real drivers of catalysis. The most commonly employed pincer ligands have an aryl backbone and symmetrical side arms. One obvious reason for using symmetrical pincer ligands in designing complexes suitable for catalysis is the relative ease of synthesis of such species. The pincer ligands presented here have S or Se donor sites, as success has eluded45 all attempts made so far to synthesize Te based pincers. On synthesis, properties and various applications of complexes of pincer ligands reviews have been published earlier,46–53 but their coverage of organochalcogen pincers is very low. | ||
| Fig. 1 Architecture of a pincer ligand. | ||
Chart 1 shows the chalcogenated pincer ligands that are significant in the context of the Heck coupling reaction. The symmetric (S,C,S) type pincer ligands are generally synthesized via nucleophilic substitution reactions of 1,3-bis(bromomethyl)benzene or related compounds with a suitable ArSH or RSH. The reaction is generally carried out either in DMF or in ethanol in the presence of some inorganic base (i.e. Na2CO3, K2CO3, EtONa or NaOH) at 70–80 °C.54 A benzene–water mixture has also been used as a solvent system in the presence of the phase-transfer catalyst ADOGEN–464.55,56 Generally, thiophenol derivatives are used as the source precursors of sulphur. However, L2 has been synthesized (Scheme 1) using S8 as a sulphur source precursor.57
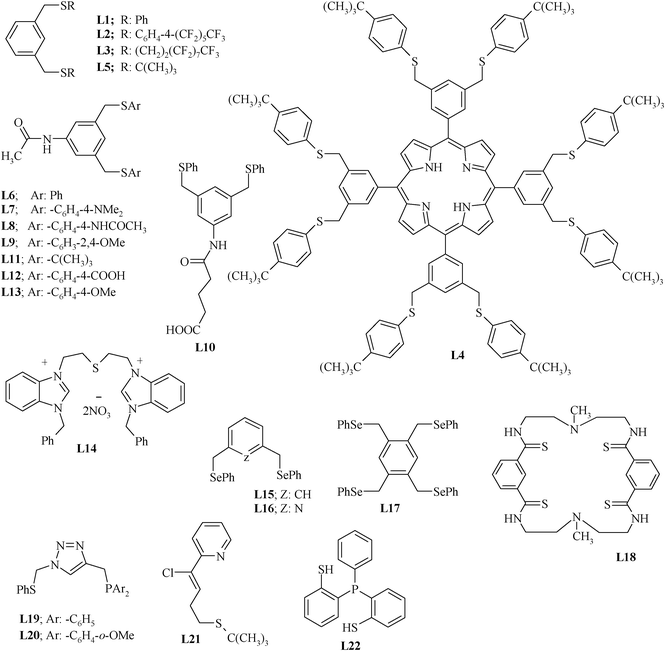 | ||
| Chart 1 Pincer ligands. | ||
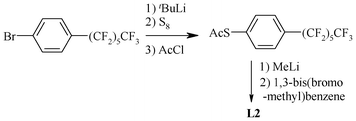 | ||
| Scheme 1 Synthesis of a pincer ligand containing a fluorous alkyl chain. | ||
The fluorous (S,C,S) pincer ligand 1,3-C6H4[CH2SCH2CH2 (CF2)7CF3]2 (L3) has been synthesized58 (Scheme 2) using 1,3-bis(bromomethyl)benzene as well as dithiol 1,3-C6H4(CH2SH)2. The latter is reacted with ICH2CH2(CH2)7CF3 in the presence of Na2CO3, K2CO3 or NaOEt (70–78 °C). L4 (Scheme 3), which is suitable for the syntheses of metallopincer–metalloporphyrin hybrid systems,59 has been considered an interesting and novel entry into catalyst modulation because of two beliefs: (i) the electronic properties of a porphyrin can be fine-tuned by the introduction of a metal into its cavity and (ii) the (S,C,S) pincer, being a monoanionic, potentially tridentate and chemically robust ligand, is suitable for synthesizing novel peripherally metalated porphyrins.59,60
 | ||
| Scheme 2 Synthesis of fluorous ligand with C10 alkyl chain. | ||
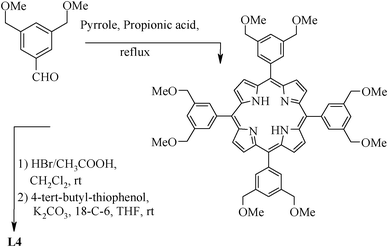 | ||
| Scheme 3 Synthesis of tetrakis pincer with porphyrin core. | ||
The reaction of an appropriate halo compound with RSNa (R = alkyl or aryl) without using an external base also results in a pincer ligand. For example, L529 is readily formed by the reaction of sodium t-butyl sulphide (NaStBu) with 1,3-bis(bromomethyl)-benzene in a benzene–water two-phase system using a phase transfer catalyst.56
The presence of substituents on the aromatic ring at a position meta to the pincer arm may have an effect on the CPh–H activation during the palladation process. Similarly, substituents at the other phenyl ring attached to chalcogen donor atoms, particularly at a position para to it, also tune the electronic effects. Thus, several substituted (S,C,S) ligands (L6–L13) given in Chart 1 have been designed (Scheme 4).61 The precursor dichloro compound, N-acetyl-1,3-bis(chloromethyl)aniline, required for their synthesis, has been prepared by a multistep route from an appropriate dicarboxylic derivative (5-aminoisophthalic acid).54 The dihalo precursor is reacted with thiophenol, N,N-dimethylaminothiophenol, p-acetamidothiophenol, 2,4-dimethoxythiophenol, 4-tert-butylbenzene-1-thiol, 4-mercapto benzoic acid and 4-mercaptoanisole in order to obtain L6–L9 and L11–L13, respectively. The Na2S can also be used as a source precursor of sulphur in designing a thio group containing pincer ligands, as in case of the (C,S,C) pincer ligand L14 (Scheme 5).62
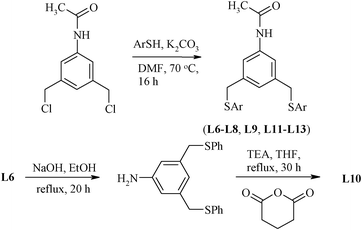 | ||
| Scheme 4 Synthesis of (S,C,S) pincer having a substituent para to C. | ||
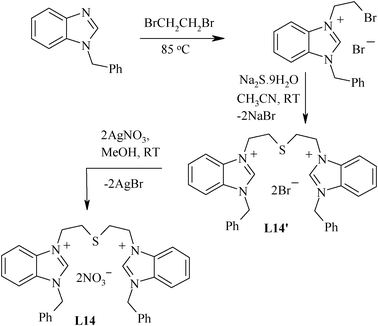 | ||
| Scheme 5 Synthesis of benzimidazolium salt type pincer. | ||
The synthesis of symmetric selenium containing pincer ligands viz (Se,C,Se) pincer/bis-pincer and (Se,N,Se) pincer involves the route of selenylation30 of an appropriate organic halo compound with diphenyl diselenide under reductive conditions (Scheme 6).30,63,64
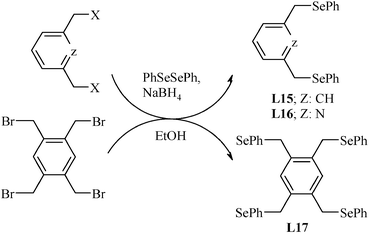 | ||
| Scheme 6 Selenylation via in situ reduction of diphenyl diselenide. | ||
The ditopic pincer ligand L18, which is also macrocyclic in nature and suitable to design a double ditopic pincer-palladium complex, has been prepared by methodology summarized in Scheme 7.65 In this case Lawesson's reagent is used to incorporate the sulphur donor into the ligand.
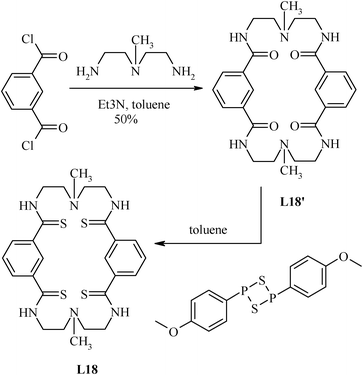 | ||
| Scheme 7 Synthesis of the polythioamide-based macrocycle. | ||
Undoubtedly, the large number of pincer ligands used for catalyst design are symmetrical, but palladium(II) complexes of the unsymmetrical ones have been not only found to be promising but also to behave somewhat differently in comparison to their symmetric counterparts while catalyzing Heck coupling. The synthesis of the unsymmetrical type of pincer ligands (Y ≠ Y′) is a considerable challenge, as the methodologies involved are usually comprised of a series of steps and separations, which commonly result in low yields.66,67 Consequently, the development of efficient and powerful methods for the rapid synthesis of a wide variety of tailor-made unsymmetrical pincer ligands has been considered as an area of high importance. The unsymmetrical pincers are made by designing both arms or by starting from a precursor in which one arm already exists. Unsymmetrical pincer ligand 4-(tert-butylthio)-1-(2-pyridinyl)-1-butyne (L21)43 has been synthesized by a route in which substitution of the mesylated hydroxyl group of the corresponding propargyl alcohol with NaSC(CH3)3 was carried out.68,69 However, using this route only a limited number of unsymmetrical pincers may be designed. An efficient strategy, which may used for the facile preparation of a novel and bigger family of tridentate (Y,C,Y′) type pincer ligands, was reported by Gandelman et al.,44 as shown in Scheme 8. This route is based on Huisgen dipolar cycloaddition of azides with alkynes to yield triazoles.70 The required azide and alkyne were prepared44 from PhS–CH2–Cl and Ar2PH–BH3, respectively. This reaction can be carried out under ambient conditions and with exclusive regioselectivity for the 1,4-disubstituted triazole product, when mediated by catalytic amounts of CuI salt.71,72 This method is to “click” two monomeric groups with coordinating atoms and functionalized with azidomethyl and propargyl units, respectively, under Sharpless conditions to provide a pincer-type ligand framework. The resulting triazole-based ligand possesses two coordinating “arms” in 1,4-positions and the relatively acidic C–H bond in between. Importantly, in contrast to possible traditional synthetic methods, (Y,C,Y′) pincer ligands are selectively obtained in such an approach, since only this covalent assembly is possible under the conditions of the “click” reaction. The application of this “click” strategy to a separately prepared series of monodentate ligand units functionalized with azido and alkynyl groups, respectively, in a systematic combinatorial manner, may result in a wide variety of triazole-based pincer ligands. A number of required azido- and alkynyl-based monomers can be easily prepared, if they are not commercially available, by simple and short protocols.44
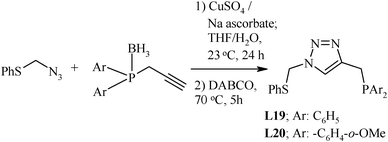 | ||
| Scheme 8 Synthesis of unsymmetrical pincer ligands. | ||
2.3 Schiff base ligands
Metal complexes of Schiff bases are well known as catalysts.73–76 Schiff bases have been prepared following the traditional template procedure as well as via a template-free synthesis.77,78 The interest in Schiff bases having sulphur, selenium or tellurium donor atoms stems from their possible role in designing a variety of catalysts, as they contain both ‘soft’ and ‘hard’ donor sites and may behave as hemilabile ligands,79,80 which allow facile reversible generation of a coordinatively unsaturated metal center.81,82Schiff bases containing a thioether donor site have been explored more than selenated and tellurated ones, known scarcely before 1980. In fact, they have picked up mostly in the last two decades only. The chalcogenated Schiff bases used to design Pd(II) catalysts for Heck coupling are summarized in Chart 2. They can be prepared either by treating chalcogenated aldehydes/ketones with amines or by treating chalcogenated amines with aldehydes or ketones. The preparation of a wide variety of chalcogenated carbonyl compounds is not easy. If a chalcogenated amine is treated with aldehyde or ketone, then a wider range of Schiff-base ligands may be designed by varying the carbonyl group containing compound.83–85
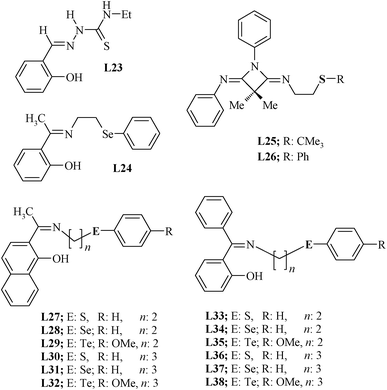 | ||
| Chart 2 Schiff base ligands. | ||
The first such ligand whose Pd(II) complex was explored for the Heck reaction was a thiosemicarbazone, L23.86 It was synthesized by direct reaction of salicylaldehyde with N-ethylthiosemicarbazide in ethanol. For the synthesis of Schiff base ligands, both generalized and specific procedures have been reported. The ligands L25 and L26 were made by a non-generalized route (Scheme 9).87 Their precursor compound was not available commercially and had to be synthesized.88 The C–N bond lengths [1.253(2)–1.257(2) Å] indicate significant double bond character and the bond angles around the C and N atoms are indicative of their sp2 hybridized nature. The ligands L27–L38 were synthesized by a general methodology in which chalcogenated amines H2N–(CH2)n–E–Ar (E = S, Se or Te) were reacted with the corresponding carbonyl group containing the compound. The chalcogenated amines are not commercially available but can be easily synthesized89–93 by reacting the appropriate halogenated alkyl amines with ArE− (E = S/Se/Te). The ArS− may be generated by reacting ArSH with NaOH and ArSe− and ArTe− (in situ) by reduction of Ar2Se2 and Ar2Te2, respectively, with NaBH4.
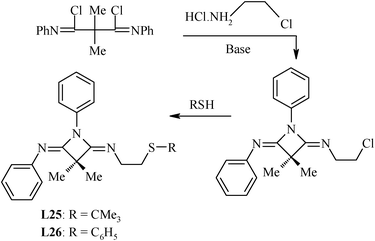 | ||
| Scheme 9 Synthesis of four-membered heterocyclic ring based diimines. | ||
The reactions of PhSe–(CH2)2–NH2 and ArTe–(CH2)2–NH2 were monitored by 77Se{1H} and 125Te{1H} NMR spectroscopy. Their 77Se{1H}/125Te{1H} NMR signals were found significantly deshielded on the formation of the corresponding Schiff base. These signals, in the case of PhSe–(CH2)3–NH2 and ArTe–(CH2)3–NH2, remain almost unchanged on the formation of Schiff bases.84,85
2.4 Chalcogenoether ligands
The chalogenoethers, which neither behave as a pincer nor have a >C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group, but have been used to design palladium(II) complexes for Heck coupling, are summarized in Chart 3. Most of these ligands can be synthesized by chalcogenylation of the appropriate organic halo-compound with suitable diaryldichalcogenide or dialkyldichalcogenide under reductive conditions, which give ArE−/RE− (E = S, Se or Te). Most of the organochalcogen precursors needed, Me2S2, Me2Se2, Me2Te2, Ph2S2, Ph2Se2 and Ph2Te2, are commercially available. Thus, L39–L43 and L46–L52 have been synthesized using this methodology.30,94,95L44 and L45 have been synthesized by the reaction of NaSCH3 and PhSLi with 2-bromo-3-(bromomethyl)pyridine.96 PhSLi was generated in situ by the reaction of BuLi and PhSH at −78 °C in THF. The functionalized diaryldiselenides, required for the synthesis of L46–L52, were not commercially available, and therefore they had to be synthesized by earlier reported procedures.97,98 Thereafter, chalcogenylation of alkyl halides (CH2Br2, BrCH2CH2Br and PhCH2Cl) with appropriate selenolate, generated by the in situ reduction of diaryldiselenide, was adopted for the synthesis of these ligands also.99
N group, but have been used to design palladium(II) complexes for Heck coupling, are summarized in Chart 3. Most of these ligands can be synthesized by chalcogenylation of the appropriate organic halo-compound with suitable diaryldichalcogenide or dialkyldichalcogenide under reductive conditions, which give ArE−/RE− (E = S, Se or Te). Most of the organochalcogen precursors needed, Me2S2, Me2Se2, Me2Te2, Ph2S2, Ph2Se2 and Ph2Te2, are commercially available. Thus, L39–L43 and L46–L52 have been synthesized using this methodology.30,94,95L44 and L45 have been synthesized by the reaction of NaSCH3 and PhSLi with 2-bromo-3-(bromomethyl)pyridine.96 PhSLi was generated in situ by the reaction of BuLi and PhSH at −78 °C in THF. The functionalized diaryldiselenides, required for the synthesis of L46–L52, were not commercially available, and therefore they had to be synthesized by earlier reported procedures.97,98 Thereafter, chalcogenylation of alkyl halides (CH2Br2, BrCH2CH2Br and PhCH2Cl) with appropriate selenolate, generated by the in situ reduction of diaryldiselenide, was adopted for the synthesis of these ligands also.99
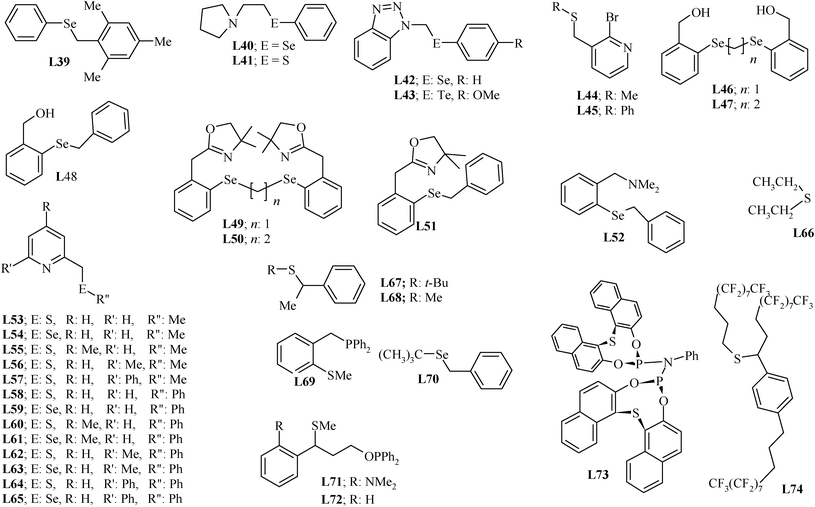 | ||
| Chart 3 Chalcogenoether ligands. | ||
Ligands L53–L65100 were obtained when PyCH2Li (generated in situ by the reaction of n-BuLi and PyCH3) was reacted directly with R′′–E–E–R′′ at low temperature.100–115 Diethylsulfide, L66, can be synthesized easily116 and is also commercially available. The synthesis of L69 involves two steps. Both the steps were carried out at low temperature (−78 °C). In first step, BrC6H4–2-(CH2PPh2) was synthesized by reacting 2-bromobenzylbromide with LiPPh2, and in the second step the resulting bromo derivative was converted to L69 by its reaction with n-BuLi and dimethyldisulfide.117
L70 has been synthesized by treating Se powder with t-BuLi and benzyl bromide.30 However, for synthesis of ligand L71, a multistep process (Scheme 10), starting from N,N-dimethyl-o-toluidine was used. The n-BuLi was used to carry out the extension of the side arm (from CH3) and introduce the PPh2 group.118ortho-Lithiation of CH3 of the precursor by n-BuLi, and subsequent reaction with dimethyl disulfide, yielded a thioether, which was again subjected to lithiation at the benzylic carbon with n-BuLi/TMEDA, and subsequently reacted with ethylene oxide to give a functionalised alcohol. The hydrogen abstraction from this alcohol using n-BuLi, and the subsequent reaction of the resulting alkoxide with PPh2Cl resulted in phosphinite ligand L71. The L72 in good yield was prepared by the same procedure using benzyl methyl sulfide (C6H5CH2–S–Me).
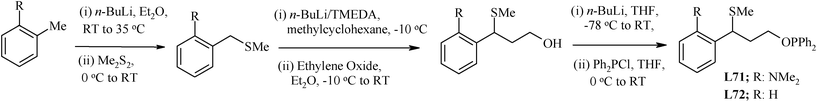 | ||
| Scheme 10 Synthesis of hemilabile sulfur-containing phosphinite ligands. | ||
L73 was synthesized in good yield by treatment of PhN(PCl2)2 with two equivalents of bis(2-hydroxy-1-naphthyl)sulfide, prepared by the reaction of SCl2 with 2-naphthol in the presence of Et3N and a catalytic amount of DMAP (4-N,N-dimethylaminopyridine) or a straightforward reaction of aniline with two equivalents of an appropriate cyclic chlorophosphite of thiobis(2,2′-naphthol), {–OC10H6(μ–S)C10H6O–}PCl, in the presence of an excess of Et3N at −15 °C.119–121
The synthesis of L74, a chiral and fluorous thioether,122 has been started from commercially available p-iodobenzaldehyde. In the first step of its synthesis, this aromatic aldehyde undergoes high-yield Wittig reactions with the readily available phosphonium salt CF3(CF2)7CH2CH2 PPh3+I−123–126 to give an alkene, which reacts with i-PrMgCl to effect an iodine/magnesium exchange. The subsequent addition of fluorous aldehyde CF3(CF2)7CH2CH2CHO results in the benzylic alcohol. Thiols are known to condense with benzylic alcohols in the presence of Lewis acids.127 Thus, fluorous thiol CF3(CF2)7CH2CH2CH2SH,128,129 synthesized by using thiourea and the fluorous iodide CF3(CF2)7CH2CH2CH2I126 was condensed with the fluorous benzylic alcohol (Scheme 11) to give the triply fluoro-pony-tailed chiral fluorous thioether L74. This synthetic route has also been found suitable for other fluorous thiols.130
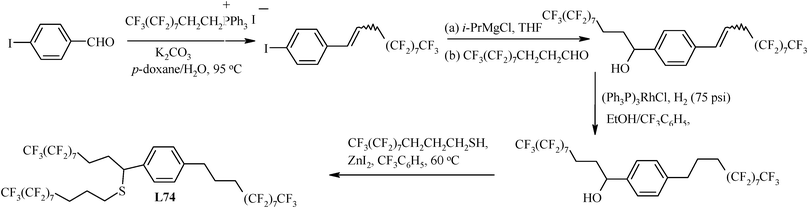 | ||
| Scheme 11 Synthesis of the triply pony-tailed chiral fluorous thioether ligand. | ||
3. Palladium(II)-organochalcogen ligand complexes and Heck coupling
Palladium(II) complexes of organochalcogen ligands, explored for Heck coupling, are not commercially available. Therefore, it is essential to have an overview of the complexes first. This will also help in understanding the relationship between the catalytic activity and the molecular architecture (if any). The Pd(II) complexes explored for the Heck coupling are listed in Charts 4–7. The significant aspects of their preparation are summarized below. Their characterization has been carried out spectroscopically and by X-ray crystallography whenever single crystals could be grown.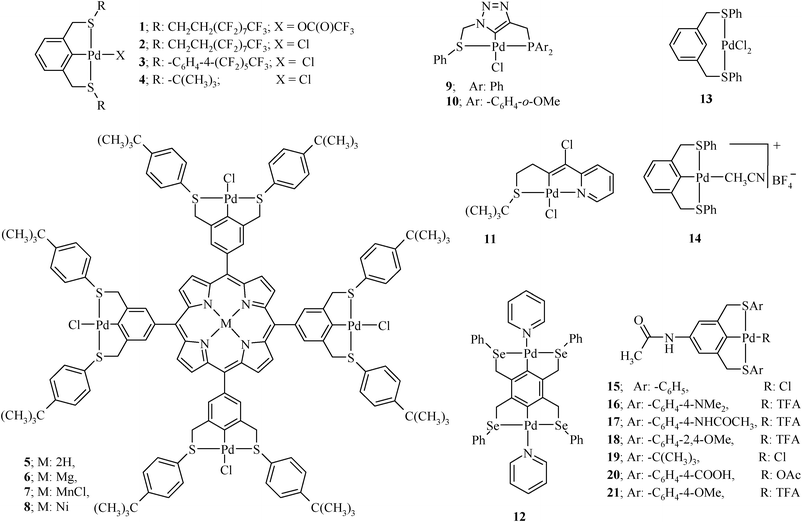 | ||
| Chart 4 Palladium complexes of pincer ligands. | ||
 | ||
| Chart 5 Non-palladacyclic pincer complexes. | ||
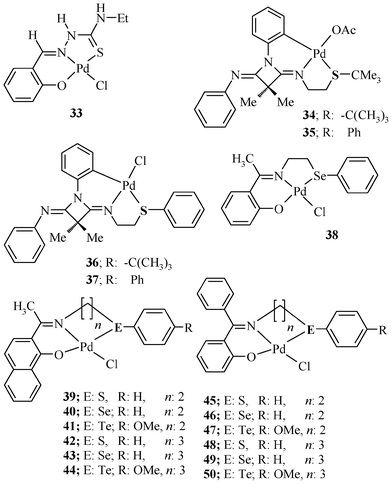 | ||
| Chart 6 Palladium(II) complexes of Schiff base ligands. | ||
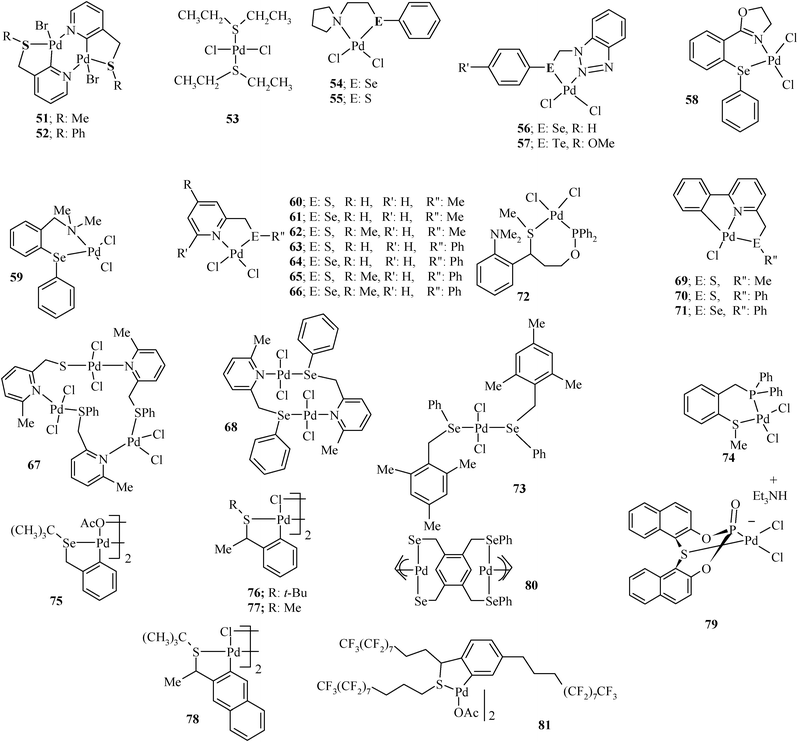 | ||
| Chart 7 Palladium(II) complexes of chalcogenoether ligands. | ||
3.1 Complexes of pincer ligands
The complexes of palladium(II) with pincer type organochalcogen ligands have got much attention as catalysts for Heck coupling, in spite of the fact that sometimes their synthesis is not easy and desired modifications in the ligand architecture are also difficult. However, the complexes of some of the (S,C,S), (Se,C,Se) and (Se,N,Se) types of pincer ligands are easy to prepare using some of the commonly used palladium precursors for this purpose, such as Pd[OC(O)CF3]2, PdCl2(PhCN)2, [Pd(CH3CN)4][BF4]2 or Na2PdCl4.The C–H activation/cyclometallation is a major strategy for the preparation of palladium pincer complexes,131,132 which is dependent on the electronic character and bulkiness of the side arms of the ligand (i.e. donor atoms Y/Y′). The C–H activation is carried out using Pd(II) species (sometimes in the presence of a base) and is usually dependent on the ancillary ligands present on the metal. Using salts such as PdCl2(PhCN)2, Pd(OCOCF3)2,133 and Na2PdCl4 in the presence of CH3COONa,29,55,56 or [Pd(CH3CN)4](BF4)264,134,135 and PdCl2(TMEDA)44 in the presence of NEt3, an efficient C–H activation can be achieved. All these salts are commercially available but expensive. However, [Pd(CH3CN)4](BF4)2136,137 can be easily generated in situ134 prior to the C–H activation process. The optimum conditions for a complexation reaction depends on the Pd(II) species as well as the pincer ligand. Some examples are as follows. The reaction of the fluorous (S,C,S) pincer ligand L3 and Pd[OC(O)CF3]2 and PdCl2(PhCN)2 at 80 °C directly leads to the Pd complexes 1 and 2 (Chart 4), respectively.58 The Pd complex 3 has been synthesized by heating the corresponding ligand L2 with [PdCl2(PhCN)2] in a mixture of acetonitrile–benzotrifluoride (BTF) for 48 h.57 Refluxing the mixture of Na2PdCl4 and CH3COONa with ligand L5 in EtOH for 25 min results in complex 4.29,55,56 Unsymmetric pincer ligands L19 and L20 on reaction with [PdCl2(TMEDA)] (TMEDA = tetramethylethylene-diamine) in DMF in the presence of NEt3 for 12 h at 70 °C gave complexes 9 and 10, respectively.44
The synthesis of complex 11 involves reaction of the methanolic solution of Li2PdCl4 with unsymmetrical pincer ligand L21.43 The reaction of ligand L18 with two equivalents of K2PdCl4 in DMSO in the presence of the base gives the complex ditopic double pincer palladacycle, 25 (Fig. 2), at room temperature.65
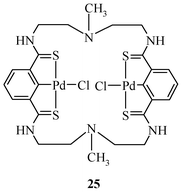 | ||
| Fig. 2 Ditopic pincer palladacycle. | ||
Bis-pincer palladium complex 12 has been synthesized64 from L17 using C–H activation with [Pd(CH3CN)4][BF4]2 in the presence of pyridine, which is required in the case of some other pincer ligands also.
Sometimes in an attempt to prepare the Pd(II) complex of a pincer ligand, the Pd–C bond is not formed. For example, the reaction of L1 {a (S,C,S) pincer} with [Pd(PhCN)2Cl2] in refluxing acetonitrile results in a complex of PdCl2 in which the ligand coordinates with its two sulphur donor sites (13 in Chart 4). The pretreatment of [Pd(PhCN)2Cl2] with an excess of AgBF4 facilitates the formation of the Pd–C bond (14).54
The quantitative yields of the pure tridentate pincer ligand–Pd(II) complexes were obtained when a substituent in the ligand at a position para to C going to bond with Pd was present. Treatment of the 5-acetamido-(S,C,S) ligands (L6–L13) with [Pd(PhCN)2Cl2] or Pd[OC(O)CF3]2 in refluxing MeCN easily yielded complexes 15–21 (Chart 4).61
The C–H functionalization by “transcyclometallation”138 gives pincer-Pd(II) complex 22 (Scheme 12).30 The ancillary ligand Cl on palladium can easily be exchanged with other halides, pyridines, phosphines etc. in order to fine-tune the electron density on the metal centre of the pincer complex.
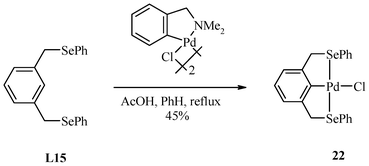 | ||
| Scheme 12 Transcyclometallation used for the synthesis of 22. | ||
For pincer ligands having nitrogen, sulphur or phosphorous as a central donor atom, the direct reaction of the ligand with Na2PdCl4 or K2PdCl4 gives a pincer ligand–Pd(II) complex, as shown in Scheme 13 for the first pincer (Se,N,Se) ligand (L16).63
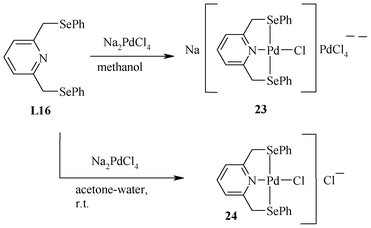 | ||
| Scheme 13 Synthesis of Pd(II) complexes of the (Se,N,Se) pincer. | ||
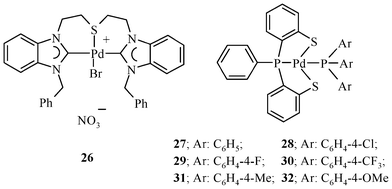
A (C,S,C) pincer-type ligand L14, on reaction with an equimolar amount of Pd(OAc)2, gives a stable complex 26 (Chart 5) only in the presence of a stabilizing anionic ligand like Br, which is added in the form of KBr.62 The trans-[PdCl2(PAr3)2] has also been used to synthesize pincer–Pd(II) complexes (27–32, Chart 5) of L22.139–142 The NEt3 is added to deprotonate the SH group.
3.2 Complexes of Schiff base ligands
Palladium(II)-chalcogenated Schiff base complexes (Chart 6, 33–50) explored for Heck coupling have been synthesized by the reaction of Na2PdCl4,83–85 K2PdCl4, Li2PdCl4 or Pd(OAc)2 with appropriate ligands at room temperature. Generally the reactions were carried out in a water–acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture and stoichiometric complexes [PdCl(L–H)] were isolated. They were authenticated by crystal structures83–85 in some cases. The ligands coordinating in a monoanionic tridentate (E, N, O−) mode form a six membered chelate ring through O− and N (azomethine) and a five/six membered ring with E and N.
:1) mixture and stoichiometric complexes [PdCl(L–H)] were isolated. They were authenticated by crystal structures83–85 in some cases. The ligands coordinating in a monoanionic tridentate (E, N, O−) mode form a six membered chelate ring through O− and N (azomethine) and a five/six membered ring with E and N.
For selenium and tellurium containing derivatives, the 77Se{1H} and 125Te{1H} NMR spectra were found to be interesting. In the 77Se{1H} NMR spectra of those complexes in which one chelate ring formed around the central metal atom is five membered and the other is six membered, the signals show high deshielding (of the order of 158 ppm) relative to those of the corresponding free ligands.83–85 When both of these rings are six membered, the deshielding is reduced drastically (of the order of only 7–8 ppm) and in the case of 49, the signal shows shielding (16 ppm) rather than deshielding.85 A similar trend is observed in the case of tellurium analogues. In the 125Te{1H} NMR spectra of 41 and 47, the signals are deshielded by 298 and 308 ppm, respectively, relative to those of the corresponding free ligands, but in the case of 44 and 50, the magnitude of deshielding is only 24 and 11 ppm, respectively.84,85
3.3 Complexes of chalcogenoether ligands
The palladium complexes of chalcogenoethers used for Heck coupling are summarized in Chart 7. They include palladacycles (51, 52, 69–71, 75–78 and 81) as well as coordination compounds (53–68, 72–74, 79 and 80).A simple and efficient way to synthesize palladacycles is based on the oxidative addition of a Pd(0) species, such as Pd(dba)2, to the carbon–halogen bond of the pincer pro-ligand. For example, bimetallic Pd-complexes 51 and 52 (which convert easily into monomeric species) can be prepared96 by this route as yellow solids.
Li2PdCl4,116 Na2PdCl4,94,95 PdCl2(CH3CN)2,99,100 PdCl2(C6H5CN)2,73,118 PdCl2(COD)117,119 and Pd(OAc)229,30,56,122 are the other palladium precursors used for the synthesis of Pd-complexes (Chart 7) of this type of chalcogenated ligands. The air and moisture stable PdCl2(SEt2)2 complex was prepared in almost quantitative yield by the direct reaction of lithium tetrachloropalladate(II) with the ligand.116 Sodium tetrachloropalladate(II) (Na2PdCl4) has been used in the synthesis of 54–57.94,95 Generally the reaction of Na2PdCl4 with an appropriate ligand gives the complex within 30 min at room temperature.94,95
Complexes 58 and 59 were readily obtained on reacting [PdCl2(CH3CN)2] with an appropriate ligand (L51 and L52, respectively) in dry dichloromethane at room temperature within 2 h.99 For complexation reactions of [PdCl2(CH3CN)2] with the ligands, acetonitrile was found to be a most suitable solvent. For example, complexes 60–71 were obtained on the reaction of [PdCl2(CH3CN)2] with the corresponding ligands in acetonitrile for 12 h at room temperature.100 The treatment of [PdCl2(PhCN)2] in dichloromethane with the ligand L71 gives complex 72 in high yield.118 Similarly for synthesis of complex 73, [PdCl2(PhCN)2] was reacted with the selenide ligand in acetonitrile.30 Palladium(II) acetate, another precursor employed, forms complexes 76 and 77 on reaction with ligands L67 and L68, respectively.29,56 The [Pd(COD)Cl2] also forms complexes readily and is a good Pd(II) precursor. The 1:2 reaction of L73 with [Pd(COD)Cl2] in the presence of H2O and Et3N in THF at room temperature gives anionic complex 79.119 The effect of the palladium precursor on the coordination mode of the ligand and the type of resulting complex has been observed in the case of 1,2,4,5-tetrakis(phenylselenomethyl)-benzene (L17), which forms a non-palladacyclic complex 80 with two seven membered chelate rings on reaction with [Pd(η3–C3H5)(μ-Cl)]2, and a bis-pincer palladium complex 12 on reaction with [Pd(CH3CN)4][BF4]2.64 In the 77Se{1H} NMR spectrum of L17, a signal appears at 361.2 ppm. It shifts to higher frequency by ∼33 ppm on the formation of complex 80 and by ∼35 ppm on the formation of 12. The difference between the shifts shown by the two complexes is insignificant with respect to the variation in size of the chelate rings and donor atoms. The synthesis of some of the complexes of Chart 7 requires a higher reaction temperature. The palladacycle 75 of selenium ligand L70 has been synthesized by its reaction with Pd(OAc)2 in benzene at 70 °C.30 Similarly, palladacycle 81 has been synthesized by the reaction of L74 with Pd(OAc)2 in AcOH at 95 °C.122 The complex 74 has been synthesized by the refluxing ligand with [PdCl2(COD)] in acetone for 2 h.117
3.4 Catalytic performances of Pd-complexes taken in molecular form for Heck coupling
Undoubtedly all Pd(II)-complexes of organochalcogen ligands explored for Heck coupling are not equally efficient. However some of them definitely outperform phosphine complexes. Hybrid organochalcogen ligands have been generally used to design Pd(II) complexes for the Heck reaction. However, only a few ligands having a P donor atom in combination with chalcogen44,117,118 have been explored for this purpose. The common observations regarding palladium(II) complexes of organochalcogen ligands investigated for Heck coupling are that they show (i) relatively good stability in air and moisture than phosphine and carbene complexes, (ii) better TON values for reactions of aryl iodides and activated aryl bromides in comparison to those of aryl chlorides and (iii) good catalytic activity for several unactivated bromides and activated chlorides, but at high temperature in the range of 130–180 °C and often in the presence of additional reagents, which probably help in stabilizing small clusters of Pd(0) in solution (believed to be invariably formed at higher temperatures) and prevent their further agglomeration to inactive species.29,43,100,116 The most popular of such stabilizing reagents are quaternary ammonium halides.143–147The palladacycles and Pd(II) complexes of pincer type organochalcogen ligands generally show good catalytic activity. Among the complexes of pincer type organochalcogen ligands found promising for Heck coupling, complex 22, having a (Se,C,Se)-pincer ligand, is unique as experimental conditions may be so tuned that it shows very high catalytic activity for the Heck coupling of a variety of aryl bromides including activated, deactivated, and heterocyclic ones with n-butyl acrylate. However, aryl chlorides were found to be almost inactive under similar conditions. Thus with complex 22, TON of the order 2.4 × 105 (70 h) has been achieved for the coupling of PhBr with n-butyl acrylate in DMA. It is interesting to note that the TON of 22 is almost twice that of TON (133000 in 63 h) obtained when Milstein's (P,C,P) pincer palladium complex is used for bromobenzene–methylacrylate coupling in N-methyl pyrrolidone.148 The catalytic performances reported for complex 22 also appear to be better than those of 12, a homodinuclear Pd(II) complex of a bis-pincer (Se,C,Se) ligand.64 However, 12 required in higher mol% than 22 for comparable conversion completes the reaction in less time. The sulphur analogues of 22, i.e.429 and 15,54 the Pd-complexes of (S,C,S)-type pincer ligands are shown to be active for the coupling of aryl iodides with methyl acrylate, but the reported data on them is insufficient for any conclusive comparison with 22.30 However, 4 catalyzes the arylation of methyl acrylate and styrene with C6H5I efficiently (TON up to 48500 and TOF 9.4 to 31.2 min−1) in the presence of NEt3 as a base.29
The Pd(II) complexes of fluorous (S,C,S) pincer ligands and palladacycles of fluorous ligands have been found to be very interesting for Heck coupling. Among the species 1, 2, 3 and 81 explored for the coupling of iodobenzene with methyl acrylate in solution (Table 1), the mol% needed is the lowest for 81.122 This palladacycle and 1–3 are unique as they have poor solubility or are nearly insoluble in many solvents at 20–24 °C, but become soluble in the same ones at elevated temperatures. This enables catalytic reactions in the absence of fluorous solvents. Further, their precipitation as halo-bridged species on cooling makes them recyclable, of course with a somewhat lower activity. Thus, 81 is not only efficient but can be recovered by a liquid–solid phase separation and reused. At the limiting concentration level, recovery may be difficult and the additional advantage of reusability is mitigated. However, purification of 3 prior to reuse on either a small or large scale is relatively easy.57 It is the reusability of these catalysts that makes them different.57,122 The performance of complex 81 (Table 1) is excellent as a catalyst in solution for Heck reactions of aryl iodide (TON 1.51 × 106 for C6H5I) as well as activated aryl bromide (TON 3.01 × 105 for OHC–C6H4–4-Br).122
| 1 | 2 | 3 | 81 | |
|---|---|---|---|---|
| Ar–X | Ph–I | Ph–I | Ph–I | Ph–I |
| Alkene | Methyl acrylate | Methyl acrylate | Methyl acrylate | Methyl acrylate |
| Mol% | 0.22 | 0.22 | 3.0 | 6.22 × 10−5 |
| Base | i-Pr2NEt | i-Pr2NEt | Bu3N | Et3N |
| Solvent | DMF | DMA | DMA | DMF |
| Temperature | 125–135 | 135 | 140 | 140 |
| Time | 45 min | 60 min | 30–45 min | 18 h |
| Yield (%) | 94 | 65 | 76 | 100 |
| TON | — | — | — | 1.51 × 106 |
The Pd complexes of several other (S,C,S) type pincer ligands are known since 1980,55,56 but their catalytic potential has been examined in 2004 for Heck coupling.61 Recently, such species have been used by Reinhoudt and co-workers as scaffolds to build self assembling metallodendrimers.149 The Pd complexes of (S,C,S) pincers 15–21 have the distinction of being the first chalcogenated pincer–Pd(II) complexes evaluated as catalysts in solution for the Heck coupling of a variety of alkene acceptors and aryl halides.54,61 In comparison to the analogous (P,C,P)-pincer based palladium catalysts, their preparation did not involve air sensitive phosphine synthesis.148 They were found to promote Heck reactions of aryl iodides but not of aryl bromides very effectively. For example, for coupling of activated aryl bromides, 17, 18, 19 and 21 gave TON values of the order of 350–500 only. Even for these low values, the presence of the promoter tetrabutylammonium bromide (50 mol %) with K2CO3 as a base, higher loadings of (S,C,S) pincer palladium(II) complexes (17, 18, 19 and 21) and a long reaction time were required for complete conversion of even bromobenzene to a Heck product. In the case of deactivated aryl bromides e.g. 3,5-dimethoxybromobenzene, the conversions were very low (e.g. ∼15% with 19).61
The palladium complexes (9–11)43,44 of unsymmetrical chalcogenated pincer ligands like (S,C,P), (S,C,N) etc. have been found to show moderate catalytic activity for Heck C–C coupling. For the coupling of methyl acrylate and bromobenzene in DMF in the presence of sodium carbonate at 140 °C, TON values for (S,C,P) pincer based complexes 9 and 10 were found to be 42000 and 6900, respectively.44 These values indicate that the catalytic activity of the Pd complex of (S,C,P) pincer is better than those of its (S,C,S) counterparts54 but is lower than that of the Pd–(P,C,P) pincer complex.148 The instability of the Pd–(S,C,P) pincer complexes is not an issue of great concern, as the same turnover frequencies are observed for these catalysts after a reaction time of 24 and 48 h.44 The potential of Pd-complex (11) of an unsymmetrical (S,C,N) pincer43 as a Heck catalyst has been probed in the coupling of n-butyl acrylate with 2- and 4-bromotoluene and 4-bromoanisole at 150 °C, using DMA as the solvent and NaOAc as the base in the presence of N(n-Bu)4Br as a salt additive.43 The yields (9–20%) of the cross coupled products as well as the TON values (900–2000) indicate that the (S,C,N) pincer based palladacycle 11 cannot be considered efficient, even for catalyzing the Heck coupling of aryl bromides.
The catalytic activity of ditopic double pincer complex 25 for Heck coupling has been explored for 4-iodotoluene only. It is reported that the catalyst survives in oxygen and at high temperature (165 °C) and gives good a turnover number in a reaction time of 48 h (up to 94300).65 The survival at 165 °C is somewhat surprising. Although for ArI these TON values do not fall in the range of most efficient catalysts known at present, the catalyst is shown to have the advantage of reusability as it works without any evidence of lowering the activity, at least for 10 cycles. However, full characterization of the catalyst before reuse has not been attempted.
The Pd(II) complexes (23, 24, 26–32)62,63,139 of chalcogenated pincer ligands that are not palladacycles (i.e. no C–Pd bond) have been investigated for Heck coupling but found to show lower catalytic efficiency than that of 22, a palladacycle with a (Se,C,Se) pincer.30 The catalytic activities of 23 and 24, Pd(II) complexes with a (Se,N,Se) pincer, have been tested at a concentration of 1 × 10−3 mol% using n-butylamine as a base.63 The 23 having two palladium centers shows a higher catalytic activity than 24. Although the complex of 23 has two Pd per molecule, its TON is not double the value shown by 24. TON and TOF values depend on the reaction conditions (solvent, base, temperature and time). Thus, comparisons of the results of different research groups are not straightforward. In comparison to the Pd complex of the (Se,C,Se) pincer ligand, which is considered an excellent catalyst,30 TON values achieved by employing 23 and 24 within 24 h reactions look promising. For example, for coupling of bromobenzene with n-butylacrylate, the use of 23 and 24 (concentration 1 × 10−3 mol%) gives TON values of 43000 and 40000, respectively, in 24 h whereas the value of TON for the same Heck reaction catalyzed with 22 [Pd(II) complex of (Se,C,Se) pincer], known to be among the best catalysts, is ∼2.4 × 105 in 70 h (catalyst loading 3.5 × 10−4 mol%).30 The Pd(II) complexes (27–32) of (S,P,S) pincers139 catalyze the Heck coupling reaction of styrene with bromobenzene. At 1.73 × 10−1 mol% loading of the catalyst in N,N′-dimethylformamide (DMF) and in the presence of Cs2CO3 as the base, only 65–75% conversion in 4 h at 160 °C could be achieved. The Pd-complex 26 of the benzimidazole-derived (C,S,C) pincer has not been explored to catalyze the Heck coupling of PhBr, but it gives moderate to good yields of the coupled product for deactivated aryl bromides at 1 mol% catalyst loading.62 The Heck reaction of 2,6-dibromopyridine is also successful, giving quantitatively the doubly-coupled product. Thus, it may be appropriate to conclude that Pd-complexes 23 and 24 of the (Se,N,Se) pincer ligand are very good catalysts for the coupling of ArBr among the complexes of pincer ligands that are not palladacycles. 26 may also be considered comparable to some extent to 23 and 24; of course the time required for comparable conversion at similar catalyst loading is much more.
The complexes of Pd(II) with Schiff base ligands are much less investigated for Heck coupling than Pd(II) pincer ligand complexes and/or palladacycles. Among Pd-complexes having Schiff base ligands, high efficiency of the diimine palladacycle system87 can be easily gauged from the two observations: (i) complex 34 shows catalytic activity for reactions involving both electronically activated and deactivated aryl chlorides at catalyst loadings of 3–5 mol% and (ii) 96–99% conversions in the reactions employing the electronically deactivated aryl bromides occur at 1.0 mol% catalyst loading and 135 °C, within a moderate reaction time of 4 to 12 h. The palladium complex of salicylaldehyde N(4)-ethylthiosemicarbazone (33) is also promising for the Heck reaction (TON 18000) of aryl bromides with styrene.86 In DMF, the reaction of bromobenzene (catalyst 33: substrate ratio of 1:1000) for 24 h at 150 °C using AcONa as a base and in the absence of any promoting additive gives a yield of 54%. It is interesting to note that catalysis with 33 can be performed in the presence of air.
The complexes 75–77,29,30 palladacycles of chalcogenoethers and complexes of type [PdCl2L2] (53116 and 7330) appear to be highly active Pd(II) species for catalyzing the Heck reaction of a variety of aryl bromides including activated, deactivated, and heterocyclic ones with n-butyl acrylate. All of them are remarkably air and moisture stable. The [PdCl2(SeAr2)2], 73, acts as an excellent catalyst for the Heck reaction (TON up to 223000 for PhBr).30 It is more reactive than the PdCl2(SEt2)2 (53),116 which gives a TON value of the order 450–760, only even in the presence of n-Bu4NBr as an additive. The complex 73 is also better than palladacycle 75 for PhBr (TON ∼ 120000).30 However, 75 gave a TON of the order 1.7 × 106 for 4-bromobenzaldehyde and also outperformed its sulfur analogue2976 (TON up to 23500 and 33000 for PhBr and 4-bromobenzaldehyde, respectively, in the presence of n-Bu4NBr). Furthermore, 75 is superior to its phosphine analogues, including Herrmann's P-palladacycle150 and Gibson's P-palladacycle151 for similar Heck reactions. The high activity of palladacycle 75 was also observed for the Heck reaction of styrene with various aryl bromides.3073 and 75, found to be better catalysts for aryl bromides than 53 and 76, are inactive for aryl chlorides, whereas the later ones show low to moderate activity for activated aryl chlorides. For example, the reaction of 4-nitrochlorobenzene with n-butyl acrylate mediated by 76 gave trans-n-butyl cinnamate in 49% yield. The reaction of chlorobenzene with n-butyl acrylate using 76 as a catalyst is very slow, giving less than 1.5% of trans-n-butyl cinnamate after 21 h (TON = 750) at 170 °C. On the other hand, higher concentrations of 53 (≥0.1 mol%) are required to couple electron poor aryl chlorides in good yields within reasonable reaction times.116 For example, in the reaction [catalyzed by 53 (0.1 mol%)] of 4-chloroacetophenone with styrene in DMA and in the presence of NaOAc as a base and additive n-Bu4NBr, 87% conversion was observed in the presence of air. However, complex 53 fails to catalyze the Heck reactions of electron-neutral or electron-rich aryl chlorides such as chlorobenzene or chloroanisole.116
The palladacycles discussed for Heck coupling so far have shown generally high catalytic activity, but surprisingly there are examples of such derivatives which show very low activity. For example, 69, 70 and 71 are inactive or poorly active (TON < 1000) for Heck coupling of even aryl bromides.100 The reaction temperature of 120 °C, the addition of sodium formate152,153 to promote reduction to Pd(0), Na2CO3 base,5,6,144n-Bu4NCl as a phase transfer reagent143,144 and stabilizer of nanoparticles, a high [Cl−]/[Pd] ratio154,155 and the use of anhydrous N,N-dimethylacetamide (DMA) as a polar solvent156–163 significantly improve the performance of 69–71. Similarly, low activity of dimetallic palladacycles 51 and 52 for the arylation of styrene with bromoacetophenone in N,N-dimethylacetamide (DMA) was observed under various experimental conditions.96 No conversion of aryl chlorides was observed, even after a prolonged reaction time. For the coupling of aryl bromides employing 51 and 52 as catalysts, a TON more than 480 (conversion ∼95%) could not be achieved even when the temperature was high (140 °C) and the reaction was carried out for 6 h.
54 and 55, which are not palladacycles but complexes of Pd with (N, S/Se) ligands,94 have been found to show good catalytic activity for aryl iodides (TON up to 83000) but only moderate for aryl bromides (TON up to 37000). The complexes 56 and 57, which are the Pd(II) complexes of selenated and tellurated benzotriazoles, respectively,95 are more catalytically active than 54 and 55 for activated aryl bromides. TON values are up to 91000 when n-butyl amine is the base used. Palladium complexes 58 and 59,99 similar to 54–57 in structure, show comparable catalytic activity for the Heck coupling of 4-bromoacetophenone with n-butylacrylate. It is proposed99 that the catalytic activities of isolated complexes are higher than those of in situ generated ones. The reason ascribed to it is that during in situ catalyst generation unreacted PdCl2 is left, resulting in a lower yield. However it does not seem to be very convincing. Further ligands L46–L52 have been tested in the presence of PdCl2 for the Heck coupling of various aryl bromides under aerobic conditions. Among them, the (Se,N) bidentate ligands L49–L52 were found to be more efficient in comparison to (Se,O) ligands L46–L48.99
Among non-palladacycle Pd(II) complexes (60–66) of substituted 2-organochalcogenomethylpyridines, the catalytic activity of 60 for Heck coupling of aryl bromides and chlorides is very good.100 The TON value is 43000 for 4-MeC6H4Br and 360000 for PhBr. The complexes 63 and 65 exhibit moderate activities with yields of 65–85% at 0.1 mol% catalyst loading. Surprisingly, complexes 61, 64 and 66, which contain an (N,Se) donor set have been reported to be almost inactive for 4-MeC6H4Br.100 4-Chloroacetophenone gave a more than >99% yield at a 1 mol% concentration of 60 under non-aqueous ionic liquid conditions,23,164,165 in which n-Bu4NCl was used and DMA, Na2CO3 and HCOONa were omitted.165 These results for 60–66 suggest that for small differences in complexes, a large variation in catalytic Heck coupling may be manifested. 67, which is a trimeric Pd(II) complex, and 68 (a dimeric complex) are either inactive or give very low conversions100 when used. Thus, a higher number of palladium atoms in a molecule does not always ensure a proportionately better catalytic performance. Such an observation has been made for other systems also, e.g. bis-pincer of (Se,C,Se) type.
The catalytic performance of complexes that have P as a donor along with S has also been evaluated. The efficient exclusion of air and the use of a pure and dry solvent and Heck reactants were necessary with 72,118 but not in case of 74117 as it has high thermal and air stability in both the solid state and in solution. Complex 72 can be applied successfully to catalyze118 the Heck reaction of styrene with several 4-substituted aryl bromides (from electron-rich to electron-poor) in DMF at 130 °C. The reaction time of 24 h and the use of AcONa as a base gives good conversion. Any promoting additive such as n-Bu4NBr is also not required. However, 72 has failed in the case of 4-bromophenol.118 The phosphino–thioether palladium complex 74 catalyzed Heck reaction of styrene with iodo and bromobenzenes not only produced exclusively trans-stilbenes in quantitative yields, but turnover numbers of the order 106 were observed surprisingly for bromobenzenes.117
The anionic palladium(II) complex (79) of heterodifunctional ligands containing O and S donors along with P has been found to be air stable and moderate for the Heck coupling of 4-bromoacetophenone with tert-butylacrylate. The 0.05 mol% loading of the catalyst is optimal, which gives a TON value of 1540.119 The anionic complex generated with such ligand systems having flexible coordination modes is expected to be more efficient than what these observations reveal as the rate of oxidative addition, a vital step in catalysis, increases.
4. Macromolecule–tethered Pd(II)-complexes and Heck coupling
Immobilization of Pd(II) complexes expected to be active for the Heck coupling reaction on a variety of macromolecular supports has been carried out and the resulting materials are explored as catalysts for such a coupling. It has been done in the hope that this may improve the recyclability of the catalyst, purification of products and commercialization of the catalyst. However, such efforts would be less fruitful if it happens at the cost of catalytic activity of the Pd centre, which may be marred by the attachment of its complex to a macromolecular matrix. It is essential for the success of a macromolecule tethered system that the complex is very robust. Undoubtedly, its properties are bound to vary to some extent upon immobilization on a support.The nature of the support is important as it may influence the properties of the immobilized complex. So far, Pd complexes immobilized on solid matrices for Heck coupling are those of (S,C,S) pincer ligands. To our knowledge, no Se and Te ligand anchored matrix has been explored for the Heck reaction. The macromolecular supports used for anchoring the (S,C,S) pincer are as follows:
Polyethylene glycol (PEG)
Polyethylene oligomers (PE)
Silica and its variants
Poly(ethylene oxide)
Poly(N-isopropylacrylamide)
Polyfluoroacrylates
Polystyrene
Polyisobutylenes
Polynorbornene
In Chart 8, various species having Pd complexes differing in the (S,C,S) pincer ligand tethered to a variety of supports are summarized. Such supports should contain unreactive bonds (except for the anchoring complex) and should be usable for anchoring a wide range of molecular catalysts. Their temperature dependent solubility is a special property, which may be useful in separating/recycling of the catalytic system. Furthermore, due to solubility, these materials can be analyzed by 1H NMR spectroscopy and thus monitoring of the course of a reaction becomes simplified.166 The Pd(II)–(S,C,S) pincer complex anchored PEG species 82 has been prepared by reacting [Pd(PhCN)2Cl2] with ligand anchored PEG. The first step in this synthesis involved the coupling of the ligand scaffold to a PEG support.
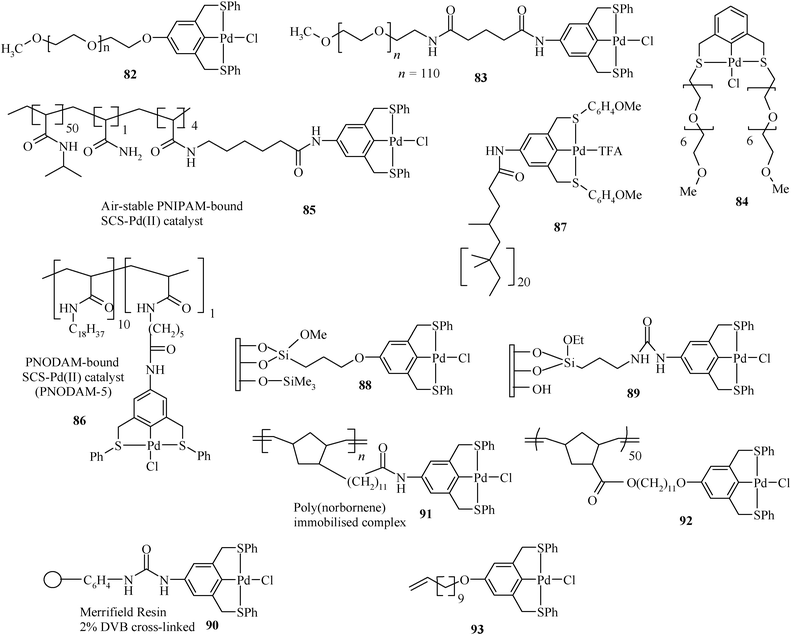 | ||
| Chart 8 Macromolecule tethered palladium complexes. | ||
Diethyl 5-hydroxyisophthalate was treated with potassium tert-butoxide and the phenolate so formed was allowed to react with mesylate-terminated 5000 molecular weight (Mn) poly(ethyleneglycol) monomethyl ether [CH3O(–CH2CH2O–)nCH2CH2–OMs]. The product of this reaction, a PEG-bound isophthalate diester, was then subjected to reduction with LAH in THF to obtain a diol. Subsequent treatment of the diol with MsCl in the presence of Et3N, followed by reaction with excess LiBr, resulted in the dibromo compound. The treatment of this dibromo derivative with PhSK in DMF yielded the desired ligand anchored PEG.54 The linkage generated between the support and the complex is ether in case of 82. The Pd(II) complex of L10, synthesized by the reaction of [Pd(PhCN)2Cl2] with the ligand, has been reacted with PEG having the NH2 end group [CH3O(–CH2CH2O–)nCH2CH2–NH2] to prepare 83, which has an amide linkage between the complex and the support. Purification at each step of the preparation was achieved by recrystallization of the polymeric species with 2-propanol.54,167 Oligo(ethyleneglycol) bound (S,C,S) pincer palladium complex 84, prepared from 1,3-bis(chloromethyl)benzene by its treatment with CH3O[CH2CH2O]6–CH2CH2–S–C(O)CH3, followed by cyclometallation with [Pd(PhCN)2Cl2], shows better solubility in water or the polar phase than PEG (its high molecular weight analogue). Its thermomorphic behaviour in mixed solvents is suitable for recycling.168 It has been used both in aqueous (for water soluble aryl halides) and in mixed phase systems (for water insoluble aryl halides). Recycling of the catalyst was feasible thermomorphically in a 1:2 v/v mixture of 10% aqueous DMA and heptane. It is claimed that the tethered complex does not exhibit any visual sign of decomposition on heating up to 200 °C in air or organic solvents/water. For reusing the catalyst, a fresh substrate solution only has to be added.
The (S,C,S) pincer palladium complex anchored on poly(N-isopropylacrylamide) (PNIPAM) (85) is air-stable and can be prepared through the route given in Scheme 14.169 It is used to catalyze Heck C–C coupling reactions in a liquid–liquid biphasic system made of aqueous DMA (90%) and heptane that exhibits an increase in phase miscibility at elevated temperature, together with solubility of 85, which otherwise has a strong preference for the polar phase at ambient temperature. This type of catalysis is known as thermomorphic catalysis and the solvent system used for it is known as thermomorphic catalyst recovery systems.170 A particular advantage of 85 is its insensitivity to oxygen.169 No catalyst degradation was observed, even after five cycles. Thus, time-consuming solvent purification and degassing protocols may be avoided. However, the use of 85 is limited when many reaction products or salt byproducts are polar and thus accumulate in the polar, catalyst-containing phase. Such products and byproducts are not separable from a polar polymer-bound catalyst. For such cases, a non-polar polymer support has been designed to stay quantitatively in the non-polar phase of a biphasic mixture (an inverse thermomorphic separation). This will facilitate the easy separation of polar products from the catalyst and its recovery. 86 (Scheme 15) is such a catalyst, having a (S,C,S) pincer–Pd(II) complex loaded on poly(N-octadecyl-acrylamide-co-N-acryloxy-succinimide) (PNODAM), which is soluble in the nonpolar phase of a mixture of polar and non-polar solvents (DMA and heptane) and used for Heck coupling. In 86, PNIPAM's N-alkyl group is a more lipophilic octadecyl group, which inverts the polymer solubility so that the polymer would selectively dissolve in the non-polar phase of a biphasic mixture but stay in solution at elevated temperature.171 Thus, use of 85 and 86 achieves easy separation of the catalysts from products, by tuning the preferential solubility of the catalyst in polar or nonpolar solvent systems, respectively.
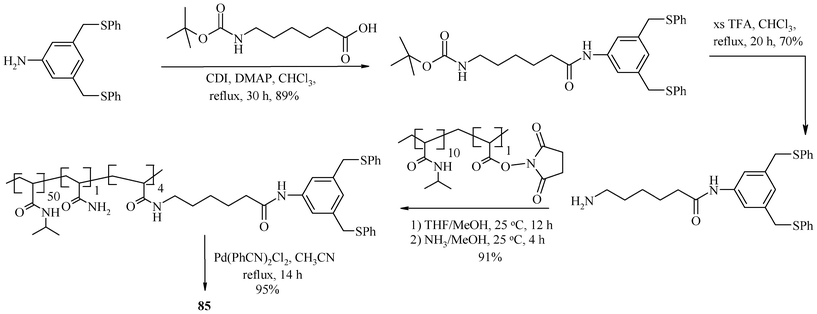 | ||
| Scheme 14 Synthesis of PNIPAM tethered (S,C,S) pincer-palladium complex. | ||
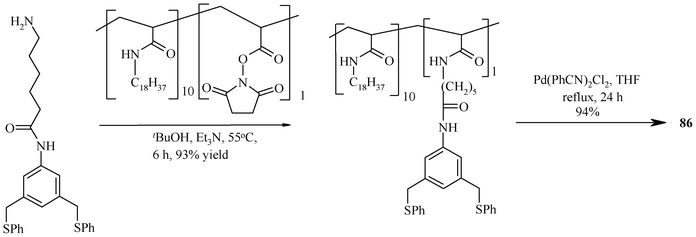 | ||
| Scheme 15 Methodology for tethering of a pincer complex on PNODAM. | ||
Polyisobutylene (PIB) oligomer based catalyst 87 was synthesized by attaching 3,5-bis{(p-methoxyphenyl)thiomethyl}aniline, a (S,C,S) pincer ligand, with PIB oligomer –(CH2–CMe2–)20–CH2–CH(CH3)–CH2CH2–COOH, via the terminal COOH group followed by cyclopalladation with Pd(TFA)2. 87 is selectively soluble in the non-polar phase and the oligomer matrix is an alternative to a terminally functionalized PEG oligomer support. The activity of the pincer complex attached to an oligomer is analogous to that of other soluble polymers or low molecular weight catalysts.16687 is readily recoverable in the non-polar phase by a liquid–liquid separation either after cooling of a thermomorphic solvent mixture or by perturbing a latently biphasic solvent mixture.
88 172 is a heterogeneous catalyst, but during catalysis all the Heck activity comes from leached soluble palladium species. However, it is not unambiguously clear whether the leached species result from pincer ligand–Pd bond rupture or via Si–O–Si bond rupture. It has been synthesized (Scheme 16) by covalently immobilizing the (S,C,S) pincer palladium(II) complex functionalized with an Si(OMe)3 end group onto SBA-15 (mesoporous silica support), utilizing a reaction between the –Si(OMe)3 group with surface silanols to form Si–O–Si bonds with the concomitant release of methanol (Scheme 16). The remaining accessible surface silanols were capped by reacting at room temperature with excess hexamethyldisilazane. A typical loading in 88 is approximately 0.12 mmol Pd per gram of SBA-15.
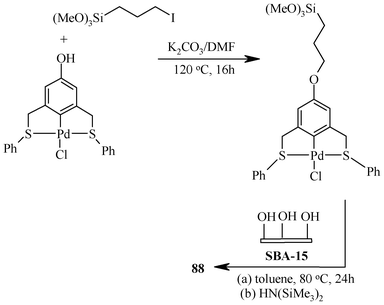 | ||
| Scheme 16 Immobilization of pincer complex on SBA-15. | ||
The (S,C,S) pincer–Pd(II) complex loaded porous silica 89173 was prepared by generating an amide or urea linkage. Reaction between a 3,5-bis(phenylthiomethyl)aniline with 3-isocyano-propyltriethoxysilane [(EtO)3Si–(CH2)3–NCO] followed by palladation with Pd(PhCN)2Cl2 gave an (S,C,S) pincer complex with a Si(OEt)3 end group in quantitative yield. This complex was covalently immobilized onto SBA-15 (mesoporous silica support), utilizing the reaction between the –Si(OEt)3 group and silanol groups present on the surface, resulting in Si–O–Si bonds with the concomitant release of ethanol. A typical loading for the SBA-15 supported complex 89 is approximately 0.10 mmol of Pd per gram support. Extensive washing with DMF and CH2Cl2 and filtration tests172 indicated that essentially all the (S,C,S) pincer–Pd(II) complexes in 88 and 89 are covalently bonded to the silica surface and there are no physisorbed species left behind.
Similarly, 90,173 which is an insoluble Merrifield resin-supported pincer–Pd complex, has been synthesized by treating 3,5-bis(phenylthiomethyl)aniline with isocyanate modified Merrifield resin, followed by metallation with [Pd(PhCN)2Cl2] in refluxing MeCN.
The poly(norbornene) based 91173 is a close replicate of PEG supported material such as 83,54 as both polymer supports have an amide linkage (in contrast to the urea linkage in 89 and 90). It was synthesized in five steps starting from 3,5-bis(phenylthiomethyl)aniline and cyclobutadiene (Scheme 17). In catalytic reactions, 91 goes into solution at elevated temperature (120 °C). Another polynorbornene-based system is 92, which contains a flexible C11 alkyl chain between the backbone and the terminal palladated (S,C,S) pincer ligand.174 It is reported that this chain was introduced to decouple the side-chain from the polymer backbone and to increase solubility. During its synthesis, first of all a monomer is synthesized by the reaction of CH2CHCOCl and cyclopentadiene. This monomer is condensed with 11-bromo-1-undecanol to obtain a bromoester, which is further reacted with 3,5-bis(phenylthiomethyl)phenol, a hydroxy functionalized (S,C,S) pincer ligand, in the presence of K2CO3. Palladation of so formed species with [Pd(CH3CN)4][BF4]2149 gives a monomeric cyclopalladated pincer complex. Its polymerization in the presence of a ruthenium catalyst gave 92.
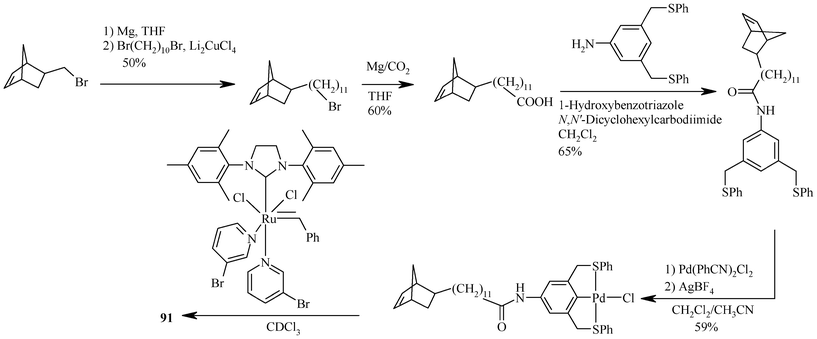 | ||
| Scheme 17 Synthesis of polynorbornene based precatalyst 91. | ||
All these tethered complexes have been applied to catalyze reactions (Table 2 and 3) of mainly aryl iodides, but TON values are generally ≤1000. The main advantage claimed for using them is that they are recovered quantitatively and can be reused in multiple reaction cycles. However, before reuse it is not fully ensured whether they are in original form or not. 88, 89 and 90 are insoluble in all common solvents and probably function as pre-catalysts only. The Pd(0) released from them has been described as a true catalyst. It is very likely that after the catalytic cycle, Pd(0) is re-deposited on the surface and catalyzes the Heck reaction in the next reuse. The evaluation of (S,C,S) pincer–Pd(II) complexes covalently bound to polymer supports and soluble in water/polar solvents (like DMF, DMA) or in non-polar solvents (like heptane) for Heck coupling reveals that tethering on different supports does not significantly change the % yield and TON values. Similarly, no effect of support on catalytic activity is observed if the catalyst system having loaded the Pd-complex is of an insoluble nature.
| 82 | 83 | 84 | 85 | 86 | |
|---|---|---|---|---|---|
| a First cycle. b Second cycle. c Third cycle. d Fourth cycle. | |||||
| Ar–X | Ph–I | Ph–I | Ph–I | Ph–I | Ph–I |
| Alkene | Methyl acrylate | Methyl acrylate | Methyl acrylate | t-Butyl acrylate | Acrylic acid |
| Mol% | 0.1 | 0.1 | 0.1 | 0.2 | 0.2 |
| Base | Et3N | Et3N | Et3N | Et3N | Et3N |
| Solvent | DMF | DMF | DMA | 90% aqueous DMA/heptane | Heptane/DMA |
| T/°C | 110 | 115 | 150 | 95 | 100 |
| Time | 4–6 h | 6.5 h | 10 min | 10 h | 24 h |
| Yield (%) | 83 | 85a | 74 | 80a | 96a |
| 87b | 98b | 99b | |||
| 95c | >99c | 99c | |||
| >99d | |||||
| TON | 1000 | — | — | — | |
| 87 | 88 | 89 | 90 | 91 | 92 | |
|---|---|---|---|---|---|---|
| a First Cycle. b Second Cycle. c Third Cycle. | ||||||
| Ar–X | Ph–I | Ph–I | Ph–I | Ph–I | Ph–I | Ph–I |
| Alkene | Methyl acrylate | n-Butyl acrylate | n-Butyl acrylate | n-Butyl acrylate | n-Butyl acrylate | t-Butyl acrylate |
| Mol (%) | 1 | 0.13 | — | — | — | — |
| Base | Et3N | Et3N | Et3N | Et3N | Et3N | Et3N |
| Solvent | DMA/heptane | DMF | DMF | DMF | DMF | DMF |
| T/°C | 100 | 120 | 120 | 120 | 120 | 120 |
| Time | 24 h | 2 h | 25 min | 1 h | 3.5 h | |
| Yield (%) | 63a | 99a | 93a | 92 | 99 | >99 |
| 98b | 97b | 92b | ||||
| 99c | 94c | |||||
| TON | — | 718–764 | — | — | — | 1000 |
5. Propagation of active palladium species from molecular pre-catalysts in Heck coupling
To better appreciate Heck coupling reactions catalyzed with palladium(II)-organochalcogen ligand complexes, it is important to briefly review the functioning of other catalysts known for the Heck reaction, a subject of intense investigation in the past one and a half decades, in which many catalysts have been developed, including palladium–phosphine/carbene complexes. Those showing promise prominently among them are palladacycles, pincer ligand complexes, heterogeneous palladium catalysts, palladium colloids and ligand-free palladium, e.g. Pd(OAc)2.11 The typical working temperature for many such complexes is between 120 and 160 °C and it has been shown that none of these catalysts is stable at these high temperatures. They all have a tendency to form palladium clusters, colloids or nanoparticles,11 which have the Pd(0) responsible for carrying out the Heck reaction. The attack of the arylating agent on the palladium atoms in the outer rim of these species takes place, setting up the catalytic cycle (Scheme 18). Also, in case of ligand-free palladium catalysts such as Pd(OAc)2, Pd(0) species have been found important for the Heck catalytic process.10a,11,175 In the case of heterogeneous catalysts having anchored Pd, the metal is dissolved from the surface under reaction conditions by a chemical reaction (complex formation and/or oxidative addition of the aryl halide), forming extremely active coordinatively unsaturated Pd species. Pd is partially or completely redeposited onto the support at the end of the reaction when the aryl halide is used up. The Pd dissolution–redeposition processes correlate with the reaction rate and are strongly influenced by the reaction conditions.176 By monitoring the Heck reactions with FT-NIR spectroscopy and quick scanning EXAFS, the formation and involvement of Pd(0) containing species in the Heck process has been supported.12,13 Thus, the role of Pd(0) containing nano-particles, clusters or other forms appears to be unequivocally established in the case of high temperature Heck catalytic processes reported using palladium complexes of non-chalcogen ligands.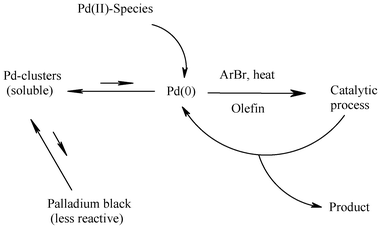 | ||
| Scheme 18 Transformations of palladium in Heck reaction of aryl bromides. | ||
The ligand free palladium species, such as Pd(OAc)2, based cross coupling processes, said to be industrially viable, also need to be mentioned here, as the role of Pd-halide anionic species has also been invoked in their case. For ligand-free catalysis some advantages reported are: (i) possibility of faster catalysis, (ii) easy work-up and (iii) low cost. De Vries and co-workers carried out studies on using ligand free systems such as PdCl2 and Pd(OAc)2.10a,11,175 They have studied the C–C bond formation reaction of butyl acrylate with benzoic anhydride (used as the arylating agent) using PdCl2 as the catalyst in the presence of sodium halide.177 At the onset of the reaction, rapid formation of Pd(0) was observed with the help of EXAFS. The presence of a number of anionic monomeric (such as PhPdCl2−) and dimeric palladium species was also noticed during ES-MS analysis.11 Thus a very large amount of the palladium is not only present in the form of soluble nanoparticles, but there is also a role of monomeric or possibly dimeric anionic species in the actual catalysis. The halide salts added have in fact two fold utility. They stabilise the colloids, preventing their further growth to palladium black and provide halide ligands that coordinate with palladium, resulting in anionic intermediates that also drive the actual catalytic cycle.11 Similarly in the course of catalysis of the Heck coupling reaction between iodobenzene and n-butyl acrylate using palladium acetate as the catalyst, the presence of PhPdI2− and PdI3− was detected using ES-MS by de Vries and coworkers.178 Further investigations by Evans et al. using EXAFS showed the presence of large amounts of the dimeric species Pd2I6− in this reaction.179 During the course of such coupling reactions of aryl iodides, hardly any palladium colloid was observed (of course for a lower substrate to catalyst ratio it may appear) because oxidative addition is fast with ArI. However, at the end of the reaction palladium nano-particle formation takes place rapidly, eventually leading to the formation of palladium black.11 In the case of aryl bromides the picture changes dramatically. The oxidative addition of aryl bromides is slower than that of aryl iodides and this now becomes the rate-determining step. As a consequence, most of the palladium will be in the form of Pd(0). It is quite likely that under these circumstances colloid formation will be the faster reaction. If the nanoparticles grow beyond a certain size, palladium black formation will occur and the Heck reaction stops (Scheme 18).
Dupont and co-workers have shown that in the presence of an ionic liquid the sizes of the nanoparticles at the beginning and the end of the Heck reaction are significantly different.180 To explain this, de Vries11 has suggested that oxidative addition of bromoarene takes place continuously at the outer rim of the nanoparticles and is followed by detachment of the anionic complex from the colloid. Of course it is difficult to pinpoint the stage of detachment from the colloid. It is possible that ArPdBr2− or its dimer is the leaving species. The detachment may also be assisted by reaction with the olefin.
It also seems very unlikely that the entire catalytic cycle takes place on the surface of the colloids, as not enough coordination sites would be available to complete the cycle. ES-MS study has revealed the presence of minor amounts of PdBr3− in solution. Thus most of the palladium remains locked up in the colloidal form.11
6. Palladium complexes of organochalcogen ligands: real or pre-catalysts
The mechanistic aspects of Heck reactions catalyzed by palladium complexes of non chalcogen ligands of diverse types have been studied in detail and the role of Pd(0) colloids or nanoparticles is strongly advocated and supported experimentally, as mentioned above. Furthermore, the debate on the actual role of a complex in Heck C–C coupling reactions may be concluded in a large number of cases in the following manner. The real catalysts are the Pd(0) species and the complexes are their dispensers. The ligands appear to play a role in controlling the rate of formation of Pd(0) species. Collaterally to these studies, mechanistic aspects of the Heck reaction catalyzed by Pd(II)-complexes of organochalcogen pincer ligands have been studied, largely in the case of organosulphur ligands. On the basis of several investigations,19,58,59,134,172,173,181 a common conclusion for palladium complexes of organochalcogen ligands is that such pincer complexes decompose under the applied (often harsh) conditions, and the catalytic activity arises due to heterogeneous Pd(0), formed probably from homogeneous Pd(0) generated from the complex. Furthermore, it is also believed that involvement of Pd NPs in the Heck reaction may not require such a high temperature as in the Suzuki reaction. Their role is shown at low temperatures also.10b Thus, for Heck coupling reactions, the efficiently performing pincer complexes are not direct catalysts but dispensers of palladium(0) containing species of high activity. The control of ligand architecture on rate of formation and nature of these small sized species (as mentioned in the previous section) appears to be a reason for the variation in catalytic efficiencies with the ligand. The formation of palladium nano-particles or Pd(0) catalysts can be detected using various methods, which have also been employed several times in the case of organosulphur ligands. However, precipitation of palladium-black (amorphous metallic Pd) may be taken as an indication for the decomposition of the applied pincer complex catalyst. Some observations that throw light on the mechanistic aspects of Pd-complexes of organosulphur ligands are as follows: Rocaboy and Gladysz122 have reported that palladacycle 81 acts as a catalyst in the coupling of phenyl iodide with methyl acrylate in the presence of Et3N in DMF at 140 °C via a Pd(0) species (non recyclable) which bleeds out of the complex, which is found to be recyclable but with diminishing activity. The recycled palladacycle, upon use gives fewer active catalytic species, resulting in lower activity. Gladysz and co-workers,58 in the cross-coupling reaction of phenyl iodide and methyl acrylate in the presence of 1, have also supported formation of palladium(0) nanoparticles on the basis of the reddish colour of the reaction mixture and TEM investigations. Curran and co-workers57 found that in the reaction of 4-bromoacetophenone and methylacrylate catalyzed by 3 in presence of n-Bu3N in DMA at 140 °C, traces of palladium (0.45% of the original amount of palladium added as complex 3) were present in the cross coupled product. In the case of 53, the washed reaction vessel and the magnetic stirring bar were found to contain the ultra trace amounts of residual palladium, which has been detected by ICP-MS analysis,116 and therefore this vessel was also found to show some activity for the Heck reaction after the first run. Thus, the organochalcogen ligand assisted Heck reaction may have the role of Pd(0) nanoparticles, and the classical mechanism shown in Scheme 19 may be an operative pathway. Unfortunately, the issue of the real catalyst has not been studied or commented on in the context of a large number of palladium(II)–organochalcogen ligand complexes explored for Heck coupling in solution. They include Schiff base complexes, as well as those designed by using other chalcogenoethers. In the case of Pd(II) complexes of (S,C,S) pincer ligands tethered on thermomorphic polymers, the observations that may lead to a decision about active species for catalysis are of diverse types. Some polymer-supported complexes have been surprisingly reported to be stable at high temperatures and in the presence of oxygen. For example, it has been reported that 85, having a Pd-complex of (S,C,S) pincer ligand linked via an amide group,169 does not show any sign of decomposition.9 It could be recovered (nearly quantitatively) and recycled multiple times. No decrease in catalysis rate was observed during its reuse. This is probably due to the large amount of anchored Pd. On the other hand 82, based on a polymer anchored with (S,C,S) ligand54via an ether linkage between the aromatic ring and the polymer, was found to form palladium black9 in the course of the reaction. This formation is a tell-tale sign of catalyst decomposition and shows that catalysis probably occurs by leached Pd(0).9 Bergbreiter and co-workers54,61 and Jones and Weck172,173 have also shown independently that active soluble palladium leaches out to catalyze the Heck reaction when some immobilized (S,C,S) pincer–Pd(II) complexes (82, 86, 88, 89, 90, 91, 92 and 93) were used as the catalyst. The mercury test supported the formation and the direct involvement of colloidal Pd(0) species (and not pincer complexes) in many coupling reactions.58,61,172,173,181 Further poly(vinylpyridine) (PVPy) test for the several coupling reactions58,172,173,181 also excludes catalysis by palladium complexes tethered on supports. Furthermore, the possibility of a Pd(II)–Pd(IV) catalytic cycle in the case of the Pd(II) complex tethered on supports appears to be unlikely.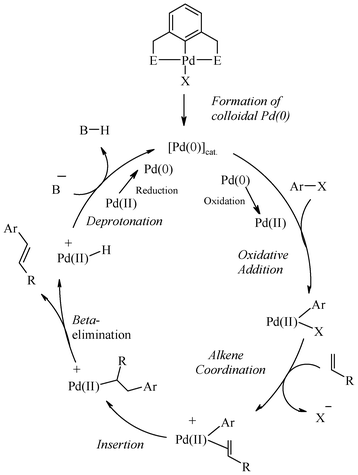 | ||
| Scheme 19 Pd(0)–Pd(II) catalytic cycle for Heck coupling. | ||
Weck, Jones and co-workers134,172,173,181 have proposed the role of Et3N in the formation of the Pd(0) species. The high strain of complexes of palladium(II) with pincer ligands, resulting from their distorted-square-planar configuration makes them prone to cleavage Scheme 20.134 Theoretical calculations have revealed that the one-arm-off configuration is only 7.0 kcal mol−1 higher in energy than that of the (S,C,S) pincer–Pd(II) complex in the presence of an amine base. Furthermore, removal of the second arm is calculated to be downhill, relative to the one-arm-off configuration by 0.6 kcal mol−1.134 These calculations suggest that the possibility shown in Scheme 20 is feasible. It appears to be favourable in the case of (S,C,S) pincers in comparison to (P,C,P) ones, as palladium black formation occurs by adding only 1 equiv triethylamine to a poly(norbornene)-supported (S,C,S) pincer palladium complex, whereas 7 equiv triethylamine is required for visible decomposition in the case of the (P,C,P) pincer.47,54 Generally the Pd–P bond is considered stronger than the Pd–S bond, which is consistent with this observation. Most probably the pincer ligand has practically no effect on the Heck reaction, except on releasing the catalytic Pd(0) species.
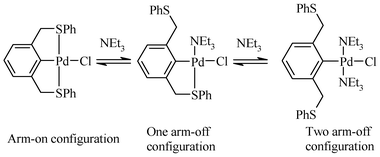 | ||
| Scheme 20 Suggested pathway for dissociation of the palladium(II) pincer complex. | ||
Van Koten, Klein Gebbink and co-workers59 have shown that pincer–porphyrin complexes 5–8 have different catalytic activities depending on the metal atom (M) coordinated to the porphyrin ligand. Based on kinetic and spectroscopic studies, it has been concluded that the reaction is catalyzed by Pd(0) species arising from decomposition of 5–8. However, the decomposition rate, and thus the amount of catalytically active Pd(0) species, were probably controlled by the electronic effects of the metal atom present in the porphyrin ligand. The catalytic activity of 5–8 was found to increase in the order M = MnCl < 2H < Ni < Mg, which coincides with increasing electron density in the porphyrin ring.
The anionic complex 79 containing O, N, or S donor atoms along with P does not seem to decompose during reduction of palladium(II) to Pd(0). This complex is reported to have a high rate of oxidative addition, a vital step in homogeneous catalysis. A mechanism (Scheme 21) involving the Pd(0)/Pd(II) cycle is postulated in this case, in which the Pd–P bond is preserved. During catalysis the anionic complex 79 exchanges the cation with the base (i.e. K2CO3) to form [Pd-chloro complex]−[K]+ (94), causing faster oxidative addition due to the presence of K+.119 The metal cation interacts with the halide anions ligated to palladium by ion pairing, to afford the more reactive Pd(0) complex 96. In this complex, coordination with sulphur provides a temporary platform for the active catalytic species during an oxidative addition step and also extra stability to the catalyst precursor. Addition of a drop of mercury to the reaction mixtures involving 79 did not affect the activity of the catalyst, thus showing them to be truly homogeneous systems.
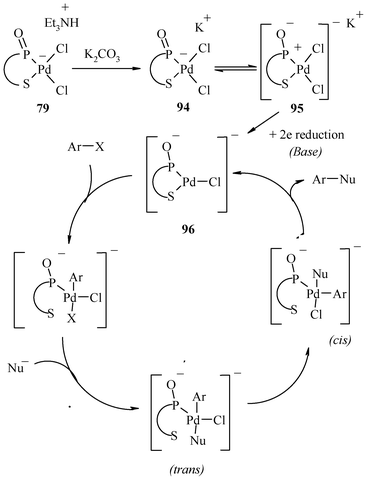 | ||
| Scheme 21 Plausible catalytic cycle for the cross-coupling reactions. | ||
It is claimed that the ditopic pincer palladium complex 25 remains active for at least 10 additional cycles, with no evidence of the metallic palladium deposit. Even the influence of elemental mercury, known to poison heterogeneous Pd(0) catalysts, was also not observed on yields and product distributions. On this basis, the author's assertion that colloidal palladium was not involved in catalysis by 25 needs further verification.65 Phosphino–thioether palladium complex (74) has also been claimed not to decompose during catalysis on the basis of 31P analysis of the reaction mixtures. However, this conclusion will become invalid if only a small fraction of the complex is converted to Pd(0) species; the signal in 31P NMR may remain intact. However, the halide of 74 undergoes exchange during the course of catalysis.117 In addition to the Heck reaction, the generation of Pd(0) containing species and their active role has also been noticed in Sonogashira coupling.182
7. Conclusion and outlook
The present survey, undertaken to understand Heck reactions assisted by palladium(II) complexes of organochalcogen ligands, suggests the following:(i) The catalytic efficiency shown by some of the complexes is as good as that of Pd(II)-complexes of phosphines or carbenes. Sometimes it is also better. For example, some molecular species of high promise are 22, 23, 24, 60, 73 and 75.
(ii) Their easy synthesis and better stability can be additional advantages.
(iii) In aerobic conditions, the Heck coupling may be catalyzed with them.
(iv) Generally, the pathway of Heck's catalytic process for Pd(II)-organochalcogen ligands also appears to be through Pd(0) containing species.
(v) These complexes have been found in most cases, not the direct catalysts of the coupling reaction but dispensers of catalytically active palladium(0) containing species, evidenced directly as well as indirectly. Of course, on the basis of just visual observation of precipitation of palladium-black, such inferences sometimes appear premature. For example, in the case of fluorous complexes (1–3 and 81) and [PdCl2(SEt)2],116 occurrence of such precipitation has been interpreted in this manner. More investigations to strengthen such conclusions and understand the function of true catalytic species in depth are required.
(vi) In several instances, complexes themselves have been claimed to be catalysts. For example, in the case of 14, 25, 72 and 79, catalysis of coupling reactions is ascribed directly to complexes. Similarly, in the cases of 4 and 76–78,29 though n-Bu4NBr was used as a promoter, there was no noticeable formation of metallic palladium and the final reaction mixture was pale yellow or almost colourless. However, these cases need further investigation, as such observations may be misleading.
The pathway of Heck coupling, as an alternative to the one based on Pd(0), may involve oxidation of palladium(II) to Pd(IV), but it is believed to be thermodynamically disfavoured in the absence of strong oxidants. However, in the absence of palladium particle formation, the presumption of the Pd(II)–Pd(IV) pathway in some reports65,117 is not very convincing. It has also been argued that in some cases none of these two conventional possibilities regarding coupling mechanisms is followed.183–186 Furthermore, if the ligand is separated completely from palladium, when Pd(0) is formed the activity of the palladium catalyst should not be affected by the ligand at all. However, this is not true because the coexisting ligand's architecture mostly affects the performance of its palladium complex as the catalyst. This may be due to its influence on the size and release of Pd(0) containing species.187 Thus, designing of new efficient catalytic systems through ligand variation continues to be rewarding. At least the catalysis of Heck coupling by palladium(II) complexes of organochalcogen ligands is worth exploring further.
For organochalcogen ligand-assisted Heck coupling at least the following are worth exploring:
(i) Tethered complexes with Se donor atoms, as several molecular species having such donor sites have shown promise.
(ii) Palladium(II) complexes of tellurium ligands, as they are little explored at the moment.
(iii) A generalized route for synthesis of unsymmetrical as well as symmetrical pincer ligands, possibly by a click reaction.
(iv) Mechanistic aspects in detail to understand real catalytic species.
Acknowledgements
The work was supported by Council of Scientific and Industrial Research (CSIR) India through the award of RA/SRF to A. K. and G. K. R., respectively. The financial support from DST (project no. SR/S1/IC–40/2010) is gratefully acknowledged by the authors.References
- (a) R. F. Heck, Acc. Chem. Res., 1979, 12, 146 CrossRef CAS; (b) S. Bräse and A. de Meijere, Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich, P. J. Stang, Wiley-VCH, Weinheim, 1998, p. 99 Search PubMed; (c) A. de Meijere and S. Bräse, J. Organomet. Chem., 1999, 576, 88 CrossRef CAS; (d) W. A. Herrmann, Applied Homogeneous Catalysis with Organometallic Compounds, Wiley-VCH, Weinheim, 2000, p. 712 Search PubMed; (e) I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef CAS.
- N. J. Whitcombe, K. K. Hii and S. E. Gibson, Tetrahedron, 2001, 57, 7449 CrossRef CAS.
- A. M. Trzeciak and J. J. Ziolkowski, Coord. Chem. Rev., 2005, 249, 2308 CrossRef CAS.
- (a) T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CrossRef CAS; (b) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS.
- A. de Meijere and F. E. Meyer, Angew. Chem., Int. Ed. Engl., 1994, 33, 2379 CrossRef.
- A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS.
- J. G. de Vries, Can. J. Chem., 2001, 79, 1086 CrossRef CAS.
- R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283 CrossRef CAS.
- N. T. S. Phan, M. Van der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS.
- (a) M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559 RSC; (b) P. J. Ellis, I. J. S. Fairlamb, S. F. J. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820 CrossRef CAS.
- J. G. de Vries, Dalton Trans., 2006, 421 RSC.
- G. Rothenberg, S. C. Cruz, G. P. F. van Strijdonck and H. C. J. Hoefsloot, Adv. Synth. Catal., 2004, 346, 467 CrossRef CAS.
- S. Reimann, J. Stötzel, R. Frahm, W. Kleist, J.-D. Grunwaldt and A. Baiker, J. Am. Chem. Soc., 2011, 133, 3921 CrossRef CAS.
- E. Burello, D. Farrusseng and G. Rothenberg, Adv. Synth. Catal., 2004, 346, 1844 CrossRef CAS.
- E. Burello and G. Rothenberg, Adv. Synth. Catal., 2003, 345, 1334 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbai and C. B. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS.
- J. Dupont, M. Pfeffer and J. Spencer, Eur. J. Inorg. Chem., 2001, 1917 CrossRef CAS.
- J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, J. Organomet. Chem., 2004, 689, 4055 CrossRef CAS.
- For a review of the reactivity of chloroarenes, see: A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS.
- D. Morales-Morales, R. Redon, C. Yung and C. M. Jensen, Chem. Commun., 2000, 1619 RSC.
- V. P. W. Böhm and W. A. Herrmann, Chem.–Eur. J., 2000, 6, 1017 CrossRef.
- D. A. Alonso, C. Najera and M. C. Pacheco, Adv. Synth. Catal., 2002, 344, 172 CrossRef CAS.
- A. Schnyder, A. F. Indolese, M. Studer and H. U. Blaser, Angew. Chem., Int. Ed., 2002, 41, 3668 CrossRef CAS.
- S. Iyer and A. Jayanthi, Synlett, 2003, 1125 Search PubMed.
- C. S. Consorti, M. L. Zanini, S. Leal, G. Ebeling and J. Dupont, Org. Lett., 2003, 5, 983 CrossRef CAS.
- E. Diez-Barra, J. Guerra, V. Hornillos, S. Merino and J. Tejeda, Organometallics, 2003, 22, 4610 CrossRef CAS.
- A. S. Gruber, D. Zim, G. Ebeling, A. L. Monteiro and J. Dupont, Org. Lett., 2000, 2, 1287 CrossRef CAS.
- Q. Yao, E. P. Kinney and C. Zheng, Org. Lett., 2004, 6, 2997 CrossRef CAS.
- A. Panda, Coord. Chem. Rev., 2009, 253, 1056 CrossRef CAS.
- A. K. Singh and S. Sharma, Coord. Chem. Rev., 2000, 209, 49 CrossRef CAS.
- T. Chivers and R. W. Hilts, Coord. Chem. Rev., 1994, 137, 201 CrossRef CAS.
- A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2010, 110, 4357 CrossRef CAS.
- L. C. Roof and J. W. Kolis, Chem. Rev., 1993, 93, 1037 CrossRef CAS.
- M. Mazzeo, M. Lamberti, A. Massa, A. Scettri, C. Pellechia and J. C. Peters, Organometallics, 2008, 27, 5741 CrossRef CAS.
- J. Takaya and N. Iwasawa, Organometallics, 2009, 28, 6636 CrossRef CAS.
- J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2008, 130, 15254 CrossRef CAS.
- R. A. Begum, D. Powell and K. Bowman-James, Inorg. Chem., 2006, 45, 964 CrossRef CAS.
- (a) J. Liu, H. Wang, H. Zhang, X. Wu, H. Zhang, Y. Deng, Z. Yang and A. Lei, Chem.–Eur. J., 2009, 15, 4437 CrossRef CAS; (b) A. Kumar, G. K. Rao, F. Saleem and A. K. Singh, Dalton Trans., 2012, 41, 11949 RSC.
- H. Wang, J. Liu, Y. Deng, T. Min, G. Yu, X. Wu, Z. Yang and A. Lei, Chem.–Eur. J., 2009, 15, 1499 CrossRef CAS.
- B. Inés, R. SanMartin, M. J. Moure and E. Domínguez, Adv. Synth. Catal., 2009, 351, 2124 CrossRef.
- C. S. Consorti, G. Ebeling, F. R. Flores, F. Rominger and J. Dupont, Adv. Synth. Catal., 2004, 346, 617 CrossRef CAS.
- E. M. Schuster, M. Botoshansky and M. Gandelman, Angew. Chem., Int. Ed., 2008, 47, 4555 CrossRef CAS.
- A. Khalid and A. K. Singh, Polyhedron, 1997, 16, 33 CrossRef CAS.
- M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750 CrossRef CAS.
- M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS.
- R. B. Bedford, Chem. Commun., 2003, 1787 RSC.
- I. Moreno, R. SanMartin, B. Inès, M. T. Herrero and E. Domínguez, Curr. Org. Chem., 2009, 13, 878 CrossRef CAS.
- D. Morales-Morales and C. M. Jensen, The Chemistry of Pincer Compounds, Elsevier, Amsterdam, 2007 Search PubMed.
- D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201 CrossRef CAS.
- D. Morales-Morales, Mini-Rev. Org. Chem., 2008, 5, 141 CrossRef CAS.
- J. M. Serrano-Becerra and D. Morales-Morales, Curr. Org. Synth., 2009, 6, 169 CrossRef CAS.
- D. E. Bergbreiter, P. L. Osburn and Y.-S. Liu, J. Am. Chem. Soc., 1999, 121, 9531 CrossRef CAS.
- J. Errington, W. S. McDonald and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1980, 2309 RSC.
- J. Errington, W. S. McDonald and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1980, 2312 RSC.
- D. P. Curran, K. Fischer and G. M. Letts, Synlett, 2004, 1379 CrossRef CAS.
- R. C. da Costa, M. Jurisch and J. A. Gladysz, Inorg. Chim. Acta, 2008, 361, 3205 CrossRef.
- B. M. J. M. Suijkerbuijk, S. D. H. Martinez, G. van Koten and R. J. M. K. Gebbink, Organometallics, 2008, 27, 534 CrossRef CAS.
- B. M. J. M. Suijkerbuijk, M. Lutz, A. L. Spek, G. van Koten and R. J. M. K. Gebbink, Org. Lett., 2004, 6, 3023 CrossRef CAS.
- D. E. Bergbreiter, P. L. Osburn and J. D. Frels, Adv. Synth. Catal., 2005, 347, 172 CrossRef CAS.
- H. V. Huynh, D. Yuan and Y. Han, Dalton Trans., 2009, 7262 RSC.
- D. Das, G. K. Rao and A. K. Singh, Organometallics, 2009, 28, 6054 CrossRef CAS.
- D. Das, P. Singh, M. Singh and A. K. Singh, Dalton Trans., 2010, 39, 10876 RSC.
- M. A. Hossain, S. Lucarini, D. Powell and K. Bowman-James, Inorg. Chem., 2004, 43, 7275 CrossRef CAS.
- M. Gandelman, A. Vigalok, L. J. W. Shimon and D. Milstein, Organometallics, 1997, 16, 3981 CrossRef CAS.
- J.-F. Gong, Y.-H. Zhang, M.-P. Song and C. Xu, Organometallics, 2007, 26, 6487 CrossRef CAS.
- J.-E. Backvall, Y. I. M. Nilsson and R. G. P. Gatti, Organometallics, 1995, 14, 4242 CrossRef.
- Synthesis of Acetylenes, Allenes and Cumulenes: A Laboratory Manual, Elsevier, Amsterdam, 1981 Search PubMed.
- R. Huisgen, Proc. Chem. Soc., 1961, 357 Search PubMed.
- V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., 2002, 114, 2708 CrossRef Angew. Chem., Int. Ed., 2002, 41, 2596.
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420 CrossRef CAS.
- A. Martinez, C. Hemmert, C. Loup, G. Barré and B. Meunier, J. Org. Chem., 2006, 71, 1449 CrossRef CAS.
- A. Arbaoui, C. Redshaw and D. L. Hughes, Chem. Commun., 2008, 4717 RSC.
- L. F. Lindoy, Prog. Macrocycle Chem. ed. R. M. Izatt and J. J. Christensen, John Wiley and Sons, Inc., New York, 1987, vol. 3, ch. 2 Search PubMed.
- N. E. Borisova, M. D. Reshetova and Y. A. Ustynyuk, Chem. Rev., 2007, 107, 46 CrossRef CAS.
- N. E. Borisova, M. D. Reshetova and Y. A. Ustynyuk, Russ. Chem. Rev., 2007, 76, 785 CrossRef CAS.
- D. Pou, C. López, S. Pérez, X. Solans, M. Font-Bardía, P. W. N. M. van Leeuwen and G. P. F. van Strijdonck, Eur. J. Inorg. Chem., 2010, 1642 CrossRef CAS.
- S. Pérez, C. López, A. Caubet, X. Solans and M. Font-Bardia, J. Organomet. Chem., 2004, 689, 3184 CrossRef.
- G. R. Owen, H. A. Burkill, R. Vilar, A. J. P. White and D. J. Williams, J. Organomet. Chem., 2005, 690, 5113 CrossRef CAS.
- J. W. Faller and J. Parr, Organometallics, 2000, 19, 1829 CrossRef CAS.
- A. Kumar, M. Agarwal and A. K. Singh, Polyhedron, 2008, 27, 485 CrossRef CAS.
- A. Kumar, M. Agarwal and A. K. Singh, J. Organomet. Chem., 2008, 693, 3533 CrossRef CAS.
- A. Kumar, M. Agarwal and A. K. Singh, Inorg. Chim. Acta, 2009, 362, 3208 CrossRef CAS.
- D. K. -Demertzi, P. N. Yadav, M. A. Demertzis, J. P. Jasiski, F. J. Andreadaki and I. D. Kostas, Tetrahedron Lett., 2004, 45, 2923 CrossRef.
- M.-T. Chen, C.-A. Huang and C.-T. Chen, Eur. J. Inorg. Chem., 2006, 4642 CrossRef CAS.
- C.-T. Chen, Y. -S. Chan, Y. -R. Tzeng and M. -T. Chen, Dalton Trans., 2004, 2691 RSC.
- A. R. Katritzky, Y. J. Xu, H. Y. He and S. Mehta, J. Org. Chem., 2001, 66, 5590 CrossRef CAS.
- A. Habtemariam, B. Watchman, B. S. Potter, R. Palmer, S. Parsons, A. Parkin and P. J. Sadler, J. Chem. Soc., Dalton Trans., 2001, 1306 RSC.
- A. K. Singh and V. Srivastava, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 47, 471 CrossRef CAS.
- A. Khanna, A. Bala and B. L. Khandelwal, J. Organomet. Chem., 1995, 494, 199 CrossRef CAS.
- V. V. Singh, G. K. Rao, A. Kumar and A. K. Singh, Dalton Trans., 2012, 41, 1142 RSC.
- P. Singh, M. Singh and A. K. Singh, J. Organomet. Chem., 2009, 694, 3872 CrossRef CAS.
- D. Das, P. Singh and A. K. Singh, J. Organomet. Chem., 2010, 695, 955 CrossRef CAS.
- A. Poulain, A. Neels and M. Albrecht, Eur. J. Inorg. Chem., 2009, 1871 CrossRef CAS.
- G. Mugesh, A. Panda, H. B. Singh, N. S. Punekar and R. J. Butcher, J. Am. Chem. Soc., 2001, 123, 839 CrossRef CAS.
- G. Mugesh, A. Panda, H. B. Singh and R. J. Butcher, Chem.–Eur. J., 1999, 5, 1411 CrossRef CAS.
- T. Chakraborty, K. Srivastava, H. B. Singh and R. J. Butcher, J. Organomet. Chem., 2011, 696, 2559 CrossRef CAS.
- R. C. Jones, A. J. Canty, M. G. Gardiner, B. W. Skelton, V. -A. Tolhurst and A. H. White, Inorg. Chim. Acta, 2010, 363, 77 CrossRef CAS.
- R. C. Jones, B. W. Skelton, V.-A. Tolhurst, A. H. White, A. J. Wilson and A. J. Canty, Polyhedron, 2007, 26, 708 CrossRef CAS.
- L. Canovese, F. Visentin, P. Uguagliati, G. Chessa, V. Lucchini and G. Bandoli, Inorg. Chim. Acta, 1998, 275–276, 385 CrossRef CAS.
- L. Canovese, F. Visentin, P. Uguagliati, G. Chessa and A. Pesce, J. Organomet. Chem., 1998, 566, 61 CrossRef CAS.
- L. Canovese, F. Visentin, G. Chessa, P. Uguagliati and A. Dolmella, J. Organomet. Chem., 2000, 601, 1 CrossRef CAS.
- L. Canovese, F. Visentin, C. Santo, G. Chessa and P. Uguagliati, Polyhedron, 2001, 20, 3171 CrossRef CAS.
- L. Canovese, G. Chessa, C. Santo, F. Visentin and P. Uguagliati, Inorg. Chim. Acta, 2003, 346, 158 CrossRef CAS.
- R. C. Jones, A. J. Canty, J. A. Deverell, M. G. Gardiner, R. M. Guijt, J. A. Smith, T. Rodemann and V.-A. Tolhurst, Tetrahedron, 2009, 65, 7474 CrossRef CAS.
- L. Bauer and L. A. Gardella, J. Org. Chem., 1963, 28, 1320 CrossRef CAS.
- F. Haviv, R. W. de Net, R. J. Michaels, J. D. Ratajczyk, G. W. Carter and P. R. Young, J. Med. Chem., 1983, 26, 218 CrossRef CAS.
- B. M. Trost and R. Braslau, J. Org. Chem., 1988, 53, 532 CrossRef CAS.
- E. Reisner, T. C. Abikoff and S. J. Lippard, Inorg. Chem., 2007, 46, 10229 CrossRef CAS.
- H. L. Holland, C. D. Turner, P. R. Andreana and D. Nguyen, Can. J. Chem., 1999, 77, 463 CAS.
- K. K. Bhasin, J. Singh and K. N. Singh, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 597 CrossRef CAS.
- E. Ghera, Y. Ben-David and H. Rapoport, J. Org. Chem., 1983, 48, 774 CrossRef CAS.
- E. M. -Wieczorkowska and J. Michalski, Roczniki Chem., 1957, 31, 543 Search PubMed.
- A. S. Gruber, D. Pozebon, A. L. Monteiro and J. Dupont, Tetrahedron Lett., 2001, 42, 7345 CrossRef CAS.
- D. M. -Morales, R. Redón, Y. Zheng and J. R. Dilworth, Inorg. Chim. Acta, 2002, 328, 39 CrossRef.
- I. D. Kostas, B. R. Steele, A. Terzis and S. V. Amosova, Tetrahedron, 2003, 59, 3467 CrossRef CAS.
- B. Punji, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2007, 46, 11316 CrossRef CAS.
- R. M. L. Mercado, A. Chandrasekaran, R. O. Day and R. R. Holmes, Organometallics, 1999, 18, 906 CrossRef CAS.
- B. Punji, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2006, 45, 9454 CrossRef CAS.
- C. Rocaboy and J. A. Gladysz, New J. Chem., 2003, 27, 39 RSC.
- C. Rocaboy, D. Rutherford, B. L. Bennett and J. A. Gladysz, J. Phys. Org. Chem., 2000, 13, 596 CrossRef CAS.
- C. Rocaboy, F. Hampel and J. A. Gladysz, J. Org. Chem., 2002, 67, 6863 CrossRef CAS.
- T. Soós, B. L. Bennett, D. Rutherford, L. P. B. Rosa and J. A. Gladysz, Organometallics, 2001, 20, 3079 CrossRef.
- J.-M. Vincent, A. Rabion, V. K. Yachandra and R. H. Fish, Angew. Chem., Int. Ed. Engl., 1997, 36, 2346 CrossRef CAS.
- Y. Guindon, R. Frenette, R. Fortin and J. Rokach, J. Org. Chem., 1983, 48, 1357 CrossRef CAS.
- C. Naud, P. Calas, H. Blancou and A. Commeyras, J. Fluorine Chem., 2000, 104, 173 CrossRef CAS.
- N. Mureau, F. Guittard and S. Geribaldi, Tetrahedron Lett., 2000, 41, 2885 CrossRef CAS.
- F. Szonyi and A. Cambon, J. Fluorine Chem., 1989, 42, 59 CrossRef CAS.
- H. Rimml and L. M. Venanzi, J. Organomet. Chem., 1983, 259, C6 CrossRef CAS.
- R. A. Baber, R. B. Bedford, M. Betham, M. E. Blake, S. J. Coles, M. F. Haddow, M. B. Hurthouse, A. G. Orpen, L. T. Pilarski, P. G. Pringle and R. L. Wingad, Chem. Commun., 2006, 3880 RSC.
- R. B. Bedford, S. M. Draper, P. N. Scully and S. L. Welch, New J. Chem., 2000, 24, 745 RSC.
- W. J. Sommer, K. Yu, J. S. Sears, Y. Ji, X. Zheng, R. J. Davis, C. D. Sherrill, C. W. Jones and M. Weck, Organometallics, 2005, 24, 4351 CrossRef CAS.
- M. Gagliardo, N. Selander, N. C. Mehendale, G. van Koten, R. J. M. K. Gebbink and K. J. Szabo, Chem.–Eur. J., 2008, 14, 4800 CrossRef CAS.
- J.-C. Yuan and S.-J. Lu, Organometallics, 2001, 20, 2697 CrossRef CAS.
- A. Sen and T.-W. Lai, J. Am. Chem. Soc., 1981, 103, 4627 CrossRef CAS.
- M. Albrecht, P. Dani, M. Lutz, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 2000, 122, 11822 CrossRef CAS.
- V. Gómez-Benítez, S. Hernández-Ortega, R. A. Toscano and D. Morales-Morales, Inorg. Chim. Acta, 2007, 360, 2128 CrossRef.
- V. Gómez-Benítez, S. Hernández-Ortega and D. Morales-Morales, Inorg. Chim. Acta, 2003, 346, 256 CrossRef.
- E. Bloch, V. Eswarakrishnan, M. Gernon, G. Ofori-Okai, C. Saha, K. Tang and J. Zubieta, J. Am. Chem. Soc., 1989, 111, 658 CrossRef.
- J. Chatt and F. G. Mann, J. Chem. Soc., 1939, 1622 Search PubMed.
- T. Jeffery, J. Chem. Soc., Chem. Commun., 1984, 19, 1287 RSC.
- T. Jeffery, Tetrahedron, 1996, 52, 10113 CrossRef CAS.
- T. Jeffery and M. David, Tetrahedron Lett., 1998, 39, 5751 CrossRef CAS.
- T. Jeffery, Tetrahedron Lett., 1994, 35, 3051 CrossRef CAS.
- T. Jeffery, Advances in Metal–Organic Chemistry, ed. L. S. Liebeskind, Jai Press, Greenwich, CT, 1996, vol. 5 Search PubMed.
- M. Ohff, A. Ohff, M. E. van der Boom and D. Milstein, J. Am. Chem. Soc., 1997, 119, 11687 CrossRef CAS.
- W. T. S. Huck, F. C. J. M. van Veggel, B. L. Kropman, D. M. A. Blank, E. G. Keim, M. M. A. Smithers and D. M. Reinhoudt, J. Am. Chem. Soc., 1995, 117, 8293 CrossRef CAS.
- W. A. Herrmann, C. Brossmer, C.-P. Reisinger, T. H. Priermeier, K. Öfele and M. Beller, Chem.–Eur. J., 1997, 3, 1357 CrossRef CAS.
- S. Gibson, D. F. Foster, G. R. Eastam, R. P. Tooze and D. J. Cole-Hamilton, Chem. Commun., 2001, 779 RSC.
- W. A. Herrmann, V. P.W. Bohm and C.-P. Reisinger, J. Organomet. Chem., 1999, 576, 23 CrossRef CAS.
- A. F. Schmidt, A. Halaiqa, L. O. Nindakova and O. S. Skripina, React. Kinet. Catal. Lett., 1999, 67, 301 CrossRef CAS.
- Y. Ben-David, M. Portnoy, M. Gozin and D. Milstein, Organometallics, 1992, 11, 1995 CrossRef CAS.
- W. Cabri and I. Candiani, Acc. Chem. Res., 1995, 28, 2 CrossRef CAS.
- T. Schultz, N. Schnees and A. Pfaltz, Appl. Organomet. Chem., 2004, 18, 595 CrossRef CAS.
- A. Scrivanti, M. Bertoldini, U. Matteoli, V. Beghetto, S. Antonaroli, A. Marini and B. Crociani, J. Mol. Catal. A: Chem., 2005, 235, 12 CrossRef CAS.
- M. T. Reetz, E. Westermann, R. Lohmer and G. Lohmer, Tetrahedron Lett., 1998, 39, 8449 CrossRef CAS.
- X. Cui, Z. Li, C.-Z. Tao, Y. Xu, J. Li, L. Liu and Q.-X. Guo, Org. Lett., 2006, 8, 2467 CrossRef CAS.
- W. A. Herrmann, C. Broßmer, K. Öfele, M. Beller and H. Fischer, J. Organomet. Chem., 1995, 491, C1 CrossRef CAS.
- W. A. Herrmann, C. Broßmer, K. Ofele, M. Beller and H. Fischer, J. Mol. Catal. A: Chem., 1995, 103, 133 CrossRef CAS.
- V. Farina, Adv. Synth. Catal., 2004, 346, 1553 CrossRef CAS.
- J. P. Knowles and A. Whiting, Org. Biomol. Chem., 2007, 5, 31 CAS.
- D. E. Kaufmann, M. Nouroozian and H. Henze, Synlett, 1996, 1091 CrossRef.
- V. Calò, A. Nacci and A. Monopoli, J. Mol. Catal. A: Chem., 2004, 214, 45 CrossRef.
- D. E. Bergbreiter and J. Li, Chem. Commun., 2004, 42 RSC.
- A. Harada, J. Li and M. Kamachi, J. Am. Chem. Soc., 1994, 116, 3192 CrossRef CAS.
- D. E. Bergbreiter and S. Furyk, Green Chem., 2004, 6, 280 RSC.
- D. E. Bergbreiter, P. L. Osburn, A. Wilson and E. M. Sink, J. Am. Chem. Soc., 2000, 122, 9058 CrossRef CAS.
- D. E. Bergbreiter, Y.-S. Liu and P. L. Osburn, J. Am. Chem. Soc., 1998, 120, 4250 CrossRef CAS.
- D. E. Bergbreiter, P. L. Osburn and J. D. Frels, J. Am. Chem. Soc., 2001, 123, 11105 CrossRef CAS.
- K. Yu, W. Sommer, M. Weck and C. W. Jones, J. Catal., 2004, 226, 101 CrossRef CAS.
- K. Yu, W. Sommer, J. M. Richardson, M. Weck and C. W. Jones, Adv. Synth. Catal., 2005, 347, 161 CrossRef CAS.
- J. M. Pollino and M. Weck, Org. Lett., 2002, 4, 753 CrossRef CAS.
- A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285 CrossRef CAS.
- K. Kö1hler, W. Kleist and S. S. Prö1ckl, Inorg. Chem., 2007, 46, 1876 CrossRef.
- M. S. Stephan, A. J. J. M. Teunissen, G. K. M. Verzijl and J. G. de Vries, Angew. Chem., Int. Ed., 1998, 37, 662 CrossRef CAS.
- A. H. M. de Vries, F. J. Parlevliet, L. Schmieder-van de Vondervoort, J. H. M. Mommers, H. J. W. Henderickx, M. A. N. Walet and J. G. de Vries, Adv. Synth. Catal., 2002, 344, 996 CrossRef CAS.
- J. Evans, L. O'Neill, V. L. Kambhampati, G. Rayner, S. Turin, A. Genge, A. J. Dent and T. Neisius, J. Chem. Soc., Dalton Trans., 2002, 2207 RSC.
- C. C. Cassol, A. P. Umpierre, G. Machado, S. I. Wolke and J. Dupont, J. Am. Chem. Soc., 2005, 127, 3298 CrossRef CAS.
- M. Weck and C. W. Jones, Inorg. Chem., 2007, 46, 1865 CrossRef CAS.
- I. J. Fairlamb, A. R. Kapdi, A. F. Lee, G. Sánchez, G. López, J. L. Serrano, L. García, J. Pérez and E. Pérez, Dalton Trans., 2004, 3970 RSC.
- J. Kjellgren, J. Aydin, O. A. Wallner, I. V. Saltanova and K. J. Szabo, Chem.–Eur. J., 2005, 11, 5260 CrossRef CAS.
- S. Bonnet, M. Lutz, A. L. Spek, G. van Koten and R. J. M. K. Gebbink, Organometallics, 2010, 29, 1157 CrossRef CAS.
- S. Bonnet, J. H. van Lenthe, M. A. Siegler, A. L. Spek, G. van Koten and R. J. M. K. Gebbink, Organometallics, 2009, 28, 2325 CrossRef CAS.
- T. Takemoto, S. Iwasa, H. Hamada, K. Shibatomi, M. Kameyama, Y. Motoyama and H. Nishiyama, Tetrahedron Lett., 2007, 48, 3397 CrossRef CAS.
- G. K. Rao, A. Kumar, B. Kumar, D. Kumar and A. K. Singh, Dalton Trans., 2012, 41, 1931 RSC.
| This journal is © The Royal Society of Chemistry 2012 |