Gold nanoparticles as computerized tomography (CT) contrast agents
Da
Xi
a,
Sheng
Dong
*a,
Xiaoxi
Meng
a,
Qinghua
Lu
b,
Lingjie
Meng
*b and
Jin
Ye
c
aThe Third Affiliated Hospital, Second Military Medical University, Shanghai 200438, P.R. China. E-mail: dongsheng2828@hotmail.com
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, P.R. China. E-mail: menglingjie@sjtu.edu.cn
cJBridge LLC, 9009 Great Hills Trl, Austin, TX 78759, USA
First published on 20th September 2012
Abstract
Gold nanoparticles (GNPs) are emerging as one of the most promising contrast agents for computerized tomography (CT) due to their remarkable properties including high X-ray absorption coefficient, low cytotoxicity, tailored surface chemistry, excellent biocompatibility, and unique surface plasmon resonance. Moreover, GNPs with tumor cell targeting molecules can specifically show the tumor tissues, leading to an intriguing feature of molecular imaging. In this article we provide a historical perspective on the GNPs used as CT contrast agents. We summarize the CT imaging principles of GNPs, review the current status of available methodologies for their preparation and surface modification, and highlight the recent achievements in their contrast effect and toxicity. Finally, the future opportunities and challenges of GNPs used as CT contrast agents are addressed for our understanding.
1 Introduction
As a convenient and efficient image diagnostic technique, X-ray computerized tomography (CT) utilizes the difference in the absorption effect from different human tissues to produce images for body structures and tissues.1 With the widespread development and application of multi-layer spiral CT, the spatial resolution of CT has improved significantly so that CT has already become as good as the body anatomization technique and become an important part of modern medicine. However, due to the absence of natural contrast from some structures or tissues with their surroundings, it is necessary to introduce a substance (contrast agent) with higher or lower density compared to the target structure or organ into the organ or its surrounding gaps. This is known as contrast imaging.2 An ideal contrast agent should have high contrast density and low toxicity, and be ready to use and reasonably cheap.Currently the most commonly used CT contrast agents are organic molecules containing iodine.3 The normal water-soluble iodinated contrast agent can be eliminated by the liver and kidney so rapidly that there is not enough time to develop the image. Nano-grade iodine oil may extend the imaging time. For example, it is found that the conventional iodinated contrast agent can produce better image quality in the blood vessels of breast cancers after being coated and modified by liposomes.4,5 However, these improved iodinated contrast agents still have the downsides of causing allergic reactions and being toxic to the kidneys.
In contrast to organic molecules, inorganic nanomaterials have unique optical, electric and magnetic properties which are different from the bulk materials.6 Compared to normal body tissues, malignant tumor tissues are rich in blood vessels with wider gaps between the walls. They are also lacking in structural integrity and lymphatic circulation. So they are selective for nanoparticles with enhanced permeability and retention.8 This makes nanoparticles function as passive targets for tumor tissues. Among all the inorganic nanoparticles, nowadays gold nanoparticles (GNPs, with diameters less than 100 nm) have attracted considerable research interest for CT contrast agents. GNPs have superior physical and biological properties: high X-ray absorption coefficient (at 100 keV the coefficients of gold and iodine are 5.16 and 1.94 respectively), unique surface plasmon resonance, ease of surface chemical modification, good tissue compatibility, passive tumor targeting function, and a decent level of anti-oxidation and pro-apoptosis.9–12 For example, at a concentration of 0.5077 M, the GNPs contrast enhancement is up to 88% greater than iodine at low energies and up to 115% greater at high energies (Fig. 1). With more evidence of safety at high concentrations, GNPs, especially those having a proactive tumor targeting function, have already emerged as a hot research topic.7,9,12,13
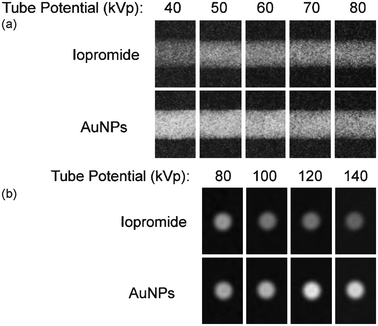 | ||
| Fig. 1 (a) and (b): Images of contrast phantom containing GNPs and iopromide. Exposures recorded at different tube potentials and laid side-by-side. Contrast media samples are imaged at equimolar concentration by a radiopaque element (0.5077 M). Low energy images in (a) are recorded in projection geometry (40–80 kVp) displaying a lateral view of each contrast media in a cylindrical volume. High-energy computed tomography images appear in (b) (80–140 kVp) as cross-sectional slices.7 | ||
Despite a large number of reviews on the synthesis and applications of GNPs with the majority focusing on catalysis,14,15 sensors,16 biocompatible markers,17 drug delivery, photothermal therapy and radiofrequency ablation,18–20 coverage in these reviews of CT has been elusive in the literature.
2 Imaging principle of GNPs
Under normal circumstance, the attenuation equation for an X-ray beam passing through any medium is the well-known Lambert–Beer law, I = I0e−kχ, where I and I0 denote the intensities of emergent and incident X-ray beams respectively. χ is the effective length for X-ray beam to traverse through the medium while k is the absorption coefficient of the medium and is inversely proportional to the medium density. Therefore, in order to obtain sufficient X-ray attenuation, one can choose a medium with relatively high density and concentration, or increase the medium thickness to increase χ. Given the fact that gold is much denser than molecular iodine, GNPs cause a much higher attenuation of an X-ray beam than iodine based contrast agents at the same thickness and concentration.7 Moreover, when the X-ray beam excites the surface electrons in the medium to ionize, it causes resonance absorption from atoms in the medium. This can also increase the absorption coefficient of the medium.7 As far as gold is concerned, the minimum energy required to ionize its outermost layer K electrons is 80.7 keV.21 Thus choosing an X-ray source with an output energy greater than 80.7 keV would allow GNPs to demonstrate an even better attenuation effect.Yusa et al. investigated the impact of the X-ray beam energy and the influence of GNP concentration on the imaging effect.10 Single color X-ray beams at 44 keV, 66 keV and 88 keV were used to illuminate the tumor with different mass fractions of GNPs, i.e. 0.1% and 1%. The X-ray attenuation results were also compared to those using a conventional X-ray beam. It was found that no desirable effects could be observed from using a single color X-ray beam to illuminate the GNP solutions with mass percentages of less than 0.1%. This indicates that using GNPs as contrast agents requires them to reach a threshold concentration. As for the tumor with a mass percentage of 1.0%, the image from using an 88 keV single color X-ray beam appears as bright as bone at the same thickness. However, there is no noticeable improvement if the energy of the X-rays was 44 keV or 66 keV. It well agrees with the previous report that the X-ray energy has to be above the K-absorption edge of GNPs, 80.7 keV, to show superior imaging capability.
To investigate the correlation between functionality and the dimensions of the GNPs, some research groups studied the relationship between the diameter of GNPs and the CT imaging capability.22,23 It was found that at the same concentration, the smaller the diameter of the GNPs, the greater attenuation the GNPs will have on the X-ray beam. When the size decreases to below 40 nm, the GNPs can exhibit better image quality than the commercialized CT contrast agent, Omnipaque, at the same concentration.
3 GNP preparation and surface modification
Though naming materials ‘nano’ started in the 1980s, glaziers in the Roman Empire unintentionally mixed gold and silver nanoparticles into glass to make the Lycurgus Cup as early as the fourth century A.D.24 The vessel appears yellowish green in reflected light, and turned ruby in transmitted light. Since Faraday first used phosphorus to reduce a solution of gold chloride in 1857,25 people began to deliberately prepare gold nanomaterials by all kinds of methods.26 Then Mie rationalized the visible absorption of GNPs using Maxwell's electromagnetic equations in 1908.27 So far, there are two major methods to prepare GNPs: the physical method and the chemical method. As a top-down approach, the physical method utilizes dispersion techniques to directly shatter large gold particles into GNPs. Meanwhile, the chemical method is a bottom-up method using various reducing agents to obtain GNPs from gold compound solutions. GNPs prepared by chemical methods have some advantages such as superior uniformity and adjustability of particle diameter, and readiness to control the shape, and have seen more applications. However, no matter which method is applied, the bare GNPs are unstable and prone to aggregation or Ostwald Ripening. For CT imaging applications, it is important to add stabilizing agents onto the GNP surface so that the electrostatic interaction and steric hindrance effect will prevent GNPs from getting closer to each other so as to ensure their stability in the solution.283.1 Physical preparation method
Gaseous metal normally stays in a single atom state, and the metal nanoparticles can be obtained when gaseous metal goes through a controllable condenser. But this method is very energy consuming and the diameter and shape of the resulting nanoparticles are difficult to control. To address these drawbacks and improve the stability of the nanoparticles, some stabilizers are often added to the gaseous metal for the purpose of dispersion. In recent years laser coblation technology based on the plasmon resonance principle has been applied against the downsides of the gaseous gold condensing method and achieved desired results.29,30 For example, using a laser with a wavelength of 532 nm to illuminate gold plate in sodium dodecyl sulfate solution can produce GNPs with diameters ranging from 1.7 nm to 3.4 nm.313.2 Chemical preparation method
GNP chemical preparation methods are generally based on a liquid chemical method. That is, with stabilizer, reducing gold precursors (mostly HAuCl4, or NaAuCl4) from aqueous solution using reducing agents. There are so many choices of stabilizers and reducers that the chemical preparation method is full of varieties.32–36 The reducers can be either strong reducers such as sodium borohydride, aldehydes, and amines, or some mild reducers including alcohol, trisodium citrate and ascorbic acid.32 There are even more types of stabilizers, and they can be any macromolecule or small molecule with functional groups such as nitrogen, phosphorus or sulfur which is capable of coordinate bonding with gold.37–48In early days, the most common chemical method was trisodium citrate reduction.32,37–39 In a typical experiment, trisodium citrate is added into boiling HAuCl4 solution to reduce gold atoms to form nuclei. The constantly produced gold atoms pile on gold nuclei to generate GNPs. Meanwhile, the citrate acid ions with negative charges attached to the surface of GNPs cause the particles to distribute evenly due to electrostatic repulsion. Adjusting the mass ratio of HAuCl4 to trisodium citrate can control the development of GNPs with different diameters from several nanometers to tens of nanometers.40 The downside of this approach is the low stability of the trisodium citrate. The dilution from continuously adding the solution may break the stable colloidal state of the gold nanoparticles. With the emergence of new types of stabilizers, this issue has been resolved. For example, phosphines and thiols can react with the gold atoms on the surface of the GNPs to produce stable Au–S and Au–P bonds.41–44 For instance, the diameters of GNPs do not vary obviously after using deionized water to dilute the 15–20 nm gold solution which is stabilized by P(C6H4SO3Na)3.45
The optical properties of gold and silver nanoparticles change drastically with nanoparticle shape. The photograph (Fig. 2) shows aqueous solutions of 4 nm gold nanospheres (vial 0) and progressively higher aspect ratio gold nanorods (1–5). The optical spectra and transmission electron micrographs for the particles in vials 1–5 are also shown. Scale bars in the micrographs are all 100 nm.37
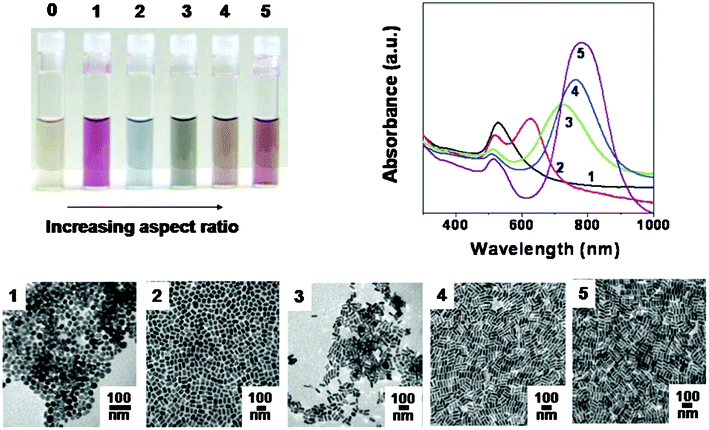 | ||
| Fig. 2 The optical properties of gold and silver nanoparticles change drastically with nanoparticle shape.37 | ||
In the past decade, preparation methods have also implemented the adjustability of GNPs' shape and diameter.46 For instance, using sodium borohydride as the reducer and organic thiol as the stabilizer can produce particles with diameters from 1 to 3 nm. Moreover the diameter of the particles can be randomly tuned in the range from 2 to 5 nm by adjusting the concentration of thiol.47 The seed mediated growth method has emerged as one important preparation method. For example, with ascorbic acid as the reducer and 3.5 nm GNPs as an initial gold core, changing the ratio of the initial gold core and the metal precursor can control the preparation of GNPs with 10 to 30 nm diameters.48 In addition, the optical properties of GNPs change drastically with nanoparticle shape and size (Fig. 2).
Most recently, ionic liquids, especially those with functional groups have also been used as solvents and stabilizers for the preparation of gold nanopatciles.49 With two charged parts—the cation and anion—in one molecule, the structure and functionality of the ionic liquids can be easily tuned.50 For example, thiol groups can be either incorporated into the cations51,52 or the anions53 and both can be used to control the topology of GNPs.
It is to note that there are manifold techniques that have been developed for the design and synthesis of GNPs. Numerous GNPs with some existing properties have been reported. However, not all these GNPs are investigated as CT contrast agents.
3.3 Surface modification of GNPs
The surface properties of GNPs are a key factor in determining their fate and behaviours when used as a biological reagent. Rational surface modification is very important to CT imaging applications. There are three major reasons to modify GNPs: 1) to enhance stability and water dispersion so as to prevent GNPs from agglomerating and depositing during storage and usage; 2) to improve blood circulation characteristics to reduce the elimination by the reticuloendothelial organs such as the liver and kidney so that the GNPs can effectively reach the imaging spot; 3) to enhance targeting characteristics by attaching the targeting molecules such as antibodies and folic acids onto the surface of GNPs to increase their accumulation at the lesion location, and thus improve imaging effects and reduce toxicity.The dendrimers usually have good water solubility and stereo-hindrance effects. It is a current research hot spot to use them to modify GNPs.48,54–57 For example, Wang et al. prepared a stable dendrimer–GNPs (DEN–GNPs) solution by using NaBH4 as a reducer to reduce chloroauric acid in the aqueous solution of amine-terminated generation 5 poly(amidoamine) dendrimers.57 And the stability may be improved much more if the positive ions on the surface of poly(amidoamine) dendrimers are neutralized through acetylation.
Polyethylene glycol (PEG) and its derivatives present excellent water solubility and good biological compatibility, and therefore are often used to modify other materials for biological applications.58–63 For example, upon adding PEG with thiol groups into the GNP solution, the PEG can bond with the surface of the GNPs through Au–S covalent bonds and produce PEG–GNPs.48,61,63 PEG functionalized gold nanoparticles show excellent stability and biological compatibility. Also importantly, PEG can lower the nonspecific phagocytosis of GNPs by the body cells so as to prolong the GNPs circulation time in the blood (as in a mouse model).48
As a type of natural biological molecule, Gum Arabic (GA) shows good biological compatibility and was also used to modify and stabilize GNPs.64–67 For instance, stable and non-toxic GA–GNPs can be produced by simple mixing of NaAuCl4 with an aqueous solution of amino acid reducing agent in the presence of GA.65,68 The GA matrix can absorb onto the surface of the GNPs and provide in vitro and in vivo stability.
Other biological molecules such as high density lipoproteins (HDL) and DNA can also be used to stabilize GNPs.69–75 Cormode et al. used HDL to modify GNPs and found the HDL–GNP conjugates displayed fine results for CT, MRI and fluorescence imaging. Furthermore, they compared imaging results from using HDL–GNPs and PEG–GNPs with Omnipaque and discovered that HDL could greatly increase the Hounsfield unit (HU, which is an arbitrary unit of X-ray attenuation) value of gold.69
Besides using biocompatible molecules to modify and stabilize GNPs, some researchers tried other types of functional materials to modify GNP surfaces to achieve multi-functionality and high performance.76–78 For example, Alri and his co-workers used gadolinium-chelate to coat GNPs and fabricated one kind of nanohybrid. As a commonly used magnetic resonance imaging (MRI) contrast agent, Gd and its complex compounds can enhance MRI signals. Thus these nanoparticles present a new class of dual-mode imaging (CT/MRI) contrast agent.78
In cancer diagnosis and therapy, targeted delivery in a localized way is one of the key challenges. While GNPs show passive targeting functionality of tumors to some extent,79 it is still necessary to attach specific targeting molecules to the surface of GNPs in order to improve further the enrichment effect of GNPs on lesion locations and reduce the impacts on normal tissues. Among different kinds of targeting molecules, the antibody has been applied extensively in the targeted delivery of nanomaterials due to its high affinity and specificity.80,81 Currently there are many reports about successfully using different kinds of antibodies such as the epidermal growth factor receptor (EGFR), infliximab and certolizumab to modify GNPs.59,82–84 These antibodies can be attached to the surface of GNPs by disulfide bonds and ether bonds. Generally, these antibody functionalized GNPs show much higher tumor targeting efficiency and specificity than passive targeting.
4 Imaging effect of GNPs
After being modified by biological and targeting molecules, GNPs show solid biological compatibility and can be dispersed in water forming a stable physiological solution. As discussed above in part 2, in principle, the imaging effect is related to the GNP concentration, particle diameter and X-ray frequency. In fact, the surface modification and the state of aggregation of GNPs will also determine the imaging effect. Understanding the structure and properties of the surface modifiers is key in using GNPs for CT imaging. Therefore, we will further discuss the imaging effect of GNPs according to different surface modification.Dendrimers are a class of highly branched, mono-dispersed, synthetic macromolecules with well-defined architecture and unique physicochemical properties,85,86 and therefore can be used as templates or stabilizers to generate dendrimer coated GNPs (DEN–GNPs). Wang and his co-workers found that the DEN–GNPs can be detected through the attenuation of X-rays and the xenograft tumor model can be imaged using CT after both intratumoral and intraperitoneal administration of the DEN–GNPs. The DEN–GNPs can be taken up predominantly by the lysosomes of the cells and are non-cytotoxic at the concentration range of 0–3000 nM.57 Guo et al. combined gold and iodine within one single dendrimer-based nanodevice for potential CT imaging applications.54 The DEN–GNPs nanocomplexes formed possessed a good stability in aqueous solution and display a much higher attenuation effect than that of both the commercial iodine-based contrast agent at the same iodine concentration and pure GNPs at the same gold concentration. In addition, other recent studies also proved that DEN–GNPs have good X-ray attenuation and a substantial circulation time, and have the potential to be an excellent contrast agent for CT.55,56
PEG and its derivatives are often used to modify other nanomaterials for biological applications because PEG can help the nanomaterials to escape elimination from the reticuloendothelial organs and extend the circulation period in the blood. Kim et al. recently functionalized uniform GNPs (approximately 30 nm in diameter) with PEG to extend their lifetime in the bloodstream. They found that the attenuation of PEG–GNPs is 5.7 times higher than that of the current iodine-based CT contrast agent, Ultravist. Furthermore, when injected intravenously into rats, the PEG–GNPs had a much longer blood circulation time (>4 h) than Ultravist (<10 min).58 Cai and his co-workers injected PEG–GNPs of 38 nm into six adult mice via subcutaneous injection with a concentration of 2.5 mmol g−1.87 Compared to the precontrast CT image (before PEG–GNP injection), the CT can produce clear images of the vascularity even 24 h after injection (see Fig. 3), indicating that the PEG–GNPs have great potential as a blood-pool agent for CT imaging. Kim's group used the PEG–GNPs for CT imaging via transdermal drug delivery to penetrate the stratum corneum.88 The PEG–GNPs could enter the blood and reach the lesion location, and improved the diagnostic sensitivity for early stage cancer (modeled on hamster oral tissues) by 20–33%.
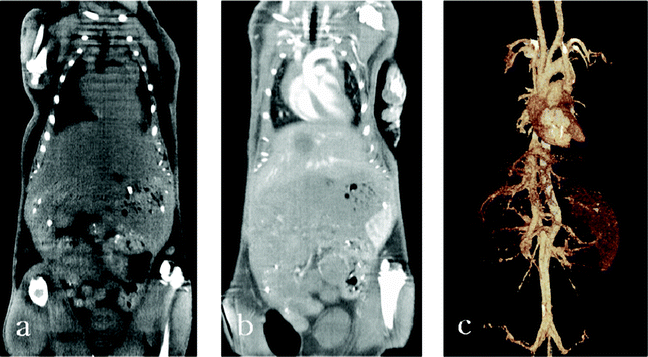 | ||
| Fig. 3 Micro CT images: precontrast (a), contrast enhanced at 6 h after AuNP–PEG injection (b), and 3D-volume rendered images (c). The major vessels are well visualized throughout the mouse and the smaller branches can also be traced.87 | ||
Gum Arabic is a type of natural polysaccharide and was used to functionalize GNPs. Boote et al. injected swine with GA stabilized Au nanoparticles (GA-GNPs), up to 85 mg per kg body weight.68 They found the concentrations of Au in tissues increased the CT numbers in the liver by approximately 22 HU per mg Au concentration at 80 kVp and 27 HU per mg Au concentration at 140 kVp. Kattumuri's group has also made similar conclusions.65 Interestingly, Chanda's team found the GA-GNPs have a high tumor affinity in severely compromised immunodeficient (SCID) mice bearing human prostate tumor xenografts.89
Theoretically, GNPs modified by other biological molecules can also be used as CT contrast agents. For instance, Cormode's group used apolipoprotein E knockout (apo E-KO) mice as a model for atherosclerosis and compared CT images generated from high-density lipoprotein coated GNPs (HDL-GNPs), with Omnipaque and calcium phosphate in mice. They discovered that HDL significantly increased the HU value of gold.69
The design and preparation of multifunctional nanoparticales is a current hotspot as they may be used for multi-mode or high performance diagnostics. Kim et al. prepared a kind of amphiphilic polymer coated hybrid nanoparticle, composed of iron oxide and gold nanoparticles.77 The hybrid nanoparticles have high CT attenuation due to the presence of GNPs, meanwhile they display a good MRI signal, attributable to the incorporated iron oxide nanoparticles. These results suggest that the hybrid nanoparticles may be useful as CT/MRI dual contrast agents for in vivo hepatoma imaging. Park et al. synthesised GNPs coated with Gd-chelate (GdL–GNPs) with an average diameter of 14 nm, where L is a conjugate of DTPA and cysteine.78 These particles exhibit very high spin–lattice relaxation rate (R1) relaxivity, X-ray attenuation and macrophage-specific properties, rendering themselves as a new class of T1 multimodal CT/MRI contrast agent, where T1 is spin–lattice relaxation time which relates to the recovery of the magnetization along the z direction after a radio frequency pulse. They also used DTPA and 4-aminothiophenol to combine Gd-chelate and GNPs to obtain GdL–GNPs with a diameter of 12 nm.90 Each particle contains 1.4 × 103 gadolinium atoms and exhibits similar CT/MRI image quality.
Modification with targeting molecules can deliver more GNPs to specific tumor cells to increase the HU value at the tumor spot. It is anticipated that the targeted delivery of GNPs might significantly reduce the dosage of GNPs with better specificity, enhanced efficacy and lower toxicities. Recently researchers demonstrated that targeted GNPs are capable of producing contrast enhancement (increased X-ray attenuation) so that CT can detect cancer based on molecular markers rather than on anatomical structures. The strong selective X-ray attenuation by GNPs is distinct from the attenuation of other cell types and tissues, and therefore transforms the targeted cancer into highly distinct features that are easy to diagnose in CT imaging.91 Popovtzer et al. reported targeted GNPs that enable cancer detection at the cellular and molecular level, with standard clinical CT.84 The targeted GNPs selectively and sensitively target tumor selective antigens while inducing distinct contrast in CT imaging. For head and neck cancer, the results showed that the attenuation coefficient for the molecularly targeted cells is over 5 times higher than for identical but untargeted cancer cells or for normal cells. Chanda's team used bombesin (BBN) to synthesize BBN–GNPs targeting prostate cancer, breast cancer and small cell lung cancer.89 The obtained BBN–GNPs produced excellent imaging results for these cancers. Wang et al. summarized the modification of GNPs using antibodies including transferrin and HER-2 and so on.92 Besides, GNPs modified by antibodies targeting the urokinase-type plasminogen activator (uPA) also produced high efficiency CT imaging diagnosis.93
For the tumors without internal blood vessels, the tumor targeting transport efficiency and specificity of GNPs dependent on the enhanced permeability and retention (EPR) effect will be a challenge. Fortunately, Kennedy et al. found that the efficiency of the use of T cells as a vehicle to deliver 45 nm GNPs into tumors is four times that of freely injected PEG–GNPs .94
These early results are encouraging, and future research will still focus on the in-depth study of targeting mechanics, and the further design and preparation of CT contrast agents with higher targeting efficiency. It is also crucial to systemically study the relationship of the size, shape and surface modification of GNPs with their imaging effect and pharmacokinetic properties.
5 Toxicity of GNPs
The safety of any materials is of utmost importance when considering their biomedical applications. Though GNPs are usually safe and nontoxic to human bodies from the long history of using GNPs, a high dose of any substance would cause potential harm to human bodies. It is essential to conduct research on toxicity issues of GNPs with the level of concentration being used for imaging. Moreover, during the preparation and stabilization of GNPs, it is possible that surfactants or other chemical reagents are added. Thus the safety issues of surface modifiers should also be considered.5.1 In vitro studies
For research on the safety issues of any material, it is normal to conduct in vitro cytotoxicity studies in order to understand the toxicity mechanics, sensitivity and intensity to provide references for in vivo tests afterwards. Due to ‘nano’ effects, GNPs can easily penetrate cell membranes, enter cells and deposit inside organelles such as the endoplasmic reticulum and Golgi apparatus.9The toxicity of GNPs seems to be closely related to the surface properties.95 For example, Goodman et al. investigated the toxicity of anionic and cationic GNPs with diameters of 2 nm to Cos-1 cells and to erythrocytes.96 The cationic particles functionalized with quaternary amines proved to be sevenfold more toxic (lethal concentration LC50, 1 mM) than anionic particles, prepared by substituting carboxylic groups for amine groups (LC50, 7.37 mM). Similarly, it was reported that cetyltrimethyl ammonium bromide functionalized GNPs (CTAB-GNPs) showed cytotoxicity at low concentrations but further study indicated that the surfactant CTAB was the actual toxicity source and using biological molecules such as Lecithin could significantly reduce the cytotoxicity.32 GNPs modified by other biological molecules including dendrimers, cytosine, DNA, citrate and glucose also did not show noticeable cytotoxicity.48,97–101 Based on these results, it is rationalised that the low toxicity of GNPs being found in some research is probably from the adverse reaction of modifiers and the GPNs modified by biological molecules should be safe for CT imaging.
Recent investigations also demonstrate that the size and shape of the GNPs also determine its toxicity. For instance, Simon and his co-workers investigated a series of phosphine-functionalized GNPs with diameters ranging from 0.8 to 15 nm. They found that particles with a 1.4 nm diameter were toxic, whereas particles with a 15 nm diameter were nontoxic, even at up to 100-fold higher concentrations.102,103 In the case of the particles with a 1.4 nm diameter, evidence is presented that toxicity results from necrosis. Interestingly, none of the particles with a 1.2 or 1.8 nm diameter display this same effect.103 Chan et al. found that particles of 40 and 50 nm in size have the greatest effect on cell signaling functions by investigating the cell response to herceptin-coated GNPs within the 2–100 nm size range.104 Schaeublin et al. have recently demonstrated that the shape of GNPs is a major factor in establishing the biological response. They found that gold nanorods (length: 43.8 nm, diameter: 16.7 nm) had substantial detrimental effects on the human keratinocyte cell line (HaCaT) from viability, mitochondrial stress, gene, and protein studies, while gold nanospheres (diameter: 20 nm) are nontoxic.105
In addition, some reports also claimed that the toxicity of GNPs was related to the selected cell lines.9,32,60 Based on the above discussion, the toxicity of GNPs might be related to their particle sizes and shapes, functionalization methods, cell types, and particle doses, and so on. Therefore, it is rational that the reported data on GNP toxicity are sometimes contradictory. The toxicity of GNPs still needs to be further explored by using a series of GNPs with different sizes and shapes and exact chemical surface functionality.
5.2 In vivo studies
Given that in vitro studies can only inspect the toxicity of GNPs on a number of cell lines, in vivo studies of toxicity in animals will provide significant guidance for assessing the biological safety of GNPs.Cho et al. first investigated the biodistribution of 13 nm PEG–GNPs in vivo.106 GNPs were intravenously administered to mice at doses of 0.17, 0.85, and 4.26 mg kg−1. For up to 7 days after injection, the nanoparticles were found to accumulate in the liver and spleen and have prolonged blood circulation times. In addition, TEM analysis revealed PEG–GNPs in numerous cytoplasmic vesicles and lysosomes of liver Kupffer cells and spleen macrophages. They also found that the PEG–GNPs caused acute inflammation and apoptosis in the liver.
Zhang et al. found that the administration routes affect the toxicity of GNPs remarkably.107 The oral and intraperitoneal routes showed a high toxicity, but the tail vein injection showed a very low toxicity. The toxicity of GNPs was also studied in Sprague Dawley rats by inhalation.108 Seven-week-old rats, weighing approximately 200 g (males) and 145 g (females), were exposed to GNPs (average diameter 4–5 nm) for 6 h per day, 5 days per week, for 90 days in a whole-body inhalation chamber. The tissue distribution of GNPs showed a dose-dependent accumulation of GNPs in only lungs and kidneys with a gender-related difference in GNP content. Therefore, the targeted GNPs administered by tail vein injection may be more suitable for the enhancement of radiotherapy, photothermal therapy, and related medical diagnostic procedures.
It is remarkable that the biodistribution and toxicity of GNPs depend largely on the surface functionalities and diameter. For instance, GNPs modified by biocompatible amphiphilic molecules could reduce the uptake by reticuloendothelial cells and prolong the duration in the blood.107,110 Modifying the surface of the GNPs by incorporating immunogenic peptides ameliorated their toxicity. This reduction in toxicity is associated with an increase in the ability to induce an antibody response. In mice to which GNPs with different diameters (15, 50, 100 and 200 nm) were intravenously administered, all the GNPs were found in the liver, lung and spleen but the distribution among different organs depended on the diameter.111 The GNPs of 15 nm to 50 nm in size were not only found in organs such as the blood, liver, lung, spleen and stomach but also penetrated the blood brain barrier. Very few GNPs of 200 nm in size entered and were deposited in the blood, brain, stomach and pancreas. Similar results were observed in another experiment in which mice were intravenously injected with GNPs of 10, 50, 100 and 250 nm diameters.32 These results indicated that smaller GNPs of 10 nm in size appeared in many organs while GNPs with larger diameters were detected only in the blood, liver and spleen. On the other hand, Chen and his co-workers used BALB/c mice as a model and injected them with naked GNPs ranging from 3 to 100 nm at a dose of 8 mg kg−1 per week.112 GNPs with diameters of 3, 5, 50, and 100 nm did not show harmful effects; however, GNPs with diameters ranging from 8 to 37 nm induced severe sickness in the mice. Mice injected with GNPs in this range showed fatigue, loss of appetite, change of fur colour, and weight loss. Starting from day 14, mice in this group exhibited a camel-like back and crooked spine. The majority of mice in these groups died within 21 days. Injection of GNPs with diameters of 5 nm and 3 nm, however, did not induce sickness or death in mice.
To evaluate the gene expression profile and mechanism at the molecular level of PEG–GNPs and the effect of particle size, Cho et al. applied an expression profiling approach to test the toxicity of GNPs with different diameters.113 PEG–GNPs with sizes of 4 and 100 nm were intravenously administered to BALB/c mice (4.26 mg per kg body weight). Histology of the liver tissues did not indicate any pathological changes in all treatment groups after injection of PEG–GNPs for 30 min. Only 0.38% (170 genes) and 0.50% (224 genes) of the total genes (45,000 genes) were significantly induced by the treatment of 4 and 100 nm PEG–GNPs, respectively. In addition, 114 genes out of the two groups of changed genes (67.1% and 50.9%) are identical.
In order to study the relationship of toxicity with GNP dosage and post-injection timing, Lasagna-Reeves et al. intravenously administered GNPs to mice with dosages of 40, 200, and 400 μg kg−1 per day.109 They found that the gold levels in the blood did not increase with the dose administered, whereas all the organs examined can effectively take up the GNPs (Fig. 4). Although the brain was the organ containing the lowest quantity of injected GNPs, GNPs are able to cross the blood-brain barrier and accumulate in the neural tissue. Importantly, no evidence of toxicity was observed in any of the diverse studies performed, including survival, behaviour, animal weight, organ morphology, blood biochemistry and tissue histology. Zhang et al. increased the dosage to 137.5–2200 μg kg−1 per day, and fed the mice for 14 to 28 days.107 The result showed that low concentrations of GNPs (<550 μg kg−1) did not cause an obvious decrease in body weight or appreciable toxicity, even after their breakdown in vivo,. while high concentrations of gold nanoparticles (>1100 μg kg−1) induced decreases in body weight, red blood cells, and hematocrit. Rocha et al. investigated the interactions between GNPs and the nervous system of the discoid or false dead-head roach.114 The negatively charged GNPs affected the insect's locomotion, but had no major impacts on the life expectancy of the cockroach after two months of observation. This indicated that GNPs had no long term toxicity in organisms.
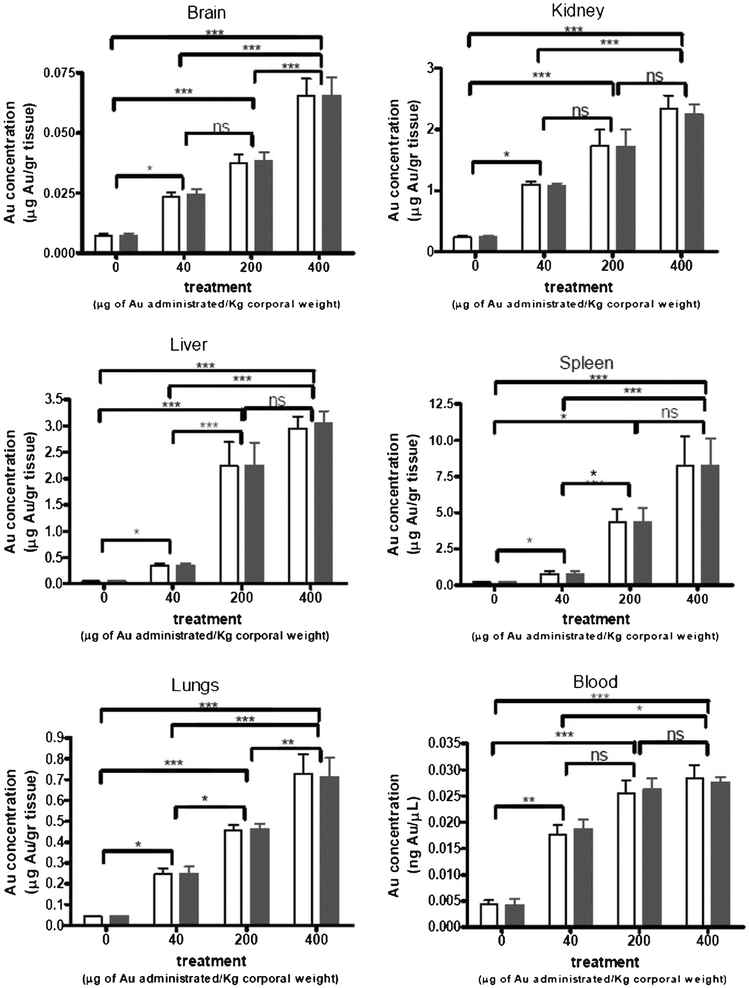 | ||
| Fig. 4 Gold accumulation in body tissues. Animals were injected intraperitoneally with various concentrations of GNPs (40, 200, and 400 μg kg−1) for 8 consecutive days. After 1 day of withdrawal following the last injection, animals were sacrificed and the blood and organs were extracted and analyzed by graphite furnace atomic absorption spectrophotometry (white bar) and inductively coupled plasma-mass spectrometry (black bar) to measure the accumulated gold. The values correspond to the gold concentration relative to the weight of the dry tissue. Bars represent mean ± standard error; data were analyzed by ANOVA with Bonferroni's post test; ns: no significant difference. *p < 0.05, **p < 0.01, and ***p < 0.001.109 | ||
6 Conclusions and outlook
GNPs modified with suitable biomolecules and targeting molecules show excellent water dispersion and stability, and no obvious toxicity within the concentration range being used for CT imaging. With incorporation of high X-ray absorption coefficients and passive tumor targeting functionality, GNPs have been proven to show excellent CT imaging results on animal models. In addition, the unique fluorescence and photothermal effects from the surface plasmon resonance of GNPs will also provide further possibilities for future therapy. Owing to the possibility of tailored surface chemistry of GNPs, biomolecule stabilizers, targeting molecules, fluorescent molecules and other functional molecules can be easily attached simultaneously onto GNP surfaces, making it a platform for multimode diagnosis and therapy on tumors.Despite many exciting advances in the field, GNP based CT image agents are still in their infancy, and some challenges must be addressed in order to provide an effective clinical product. (1): Currently materials scientists and chemists have been able to produce GNPs with different sizes and shapes such as sphere, rod, sheet, polyhedron and cage. These parameters may affect directly GNP biodistribution, toxicity and specificity. Moreover, the different crystal plane characteristics of the surface could affect the absorption properties for X-rays. To optimize the CT imaging functionality of GNPs, it is essential to conduct systematically research on the effects of these factors on the CT imaging, blood circulation characteristics and biological toxicity. (2): More in-depth study of the targeting mechanics is still required. One key issue is the design and preparation of targeting image agents with higher efficiency so that it is possible to enhance the imaging quality while lowering the quantity of GNPs entering the human body and reducing the side effects. (3): The modification of GNP surfaces with other functional materials to implement multi-functionality and high performance should be further explored in order to realize early diagnosis and synchronized therapy of cancer to meet the needs of clinical practice and patients.
Acknowledgements
This work was funded by the National Science Foundation of China (20904030, 21174087), the High Technology Research and Development Program of China (863 Porject: 2009AA03Z329), the Key Fundamental Research Project of Science and Technology Commission of Shanghai Municipal Government (08JC1412300), the Shanghai Leading Academic Discipline Project (No.B202) and the Shanghai Municipal Natural Science Foundation (11ZR1448200).References
- C. A. Carlsson, Phys. Med. Biol., 1999, 44, R23–R56 CrossRef CAS.
- J. Li, A. Chaudhary, S. J. Chmura, C. Pelizzari, T. Rajh, C. Wietholt, M. Kurtoglu and B. Aydogan, Phys. Med. Biol., 2010, 55, 4389–4397 CrossRef.
- F. Hallouard, N. Anton, P. Choquet, A. Constantinesco and T. Vandamme, Biomaterials, 2010, 31, 6249–6268 CrossRef CAS.
- S. J. Lim, J. S. Lim, J. Choi, J. Y. Choi, W. J. Hyung, H. S. Kim, J. Suh and K. W. Kim, Acad. Radiol., 2010, 17, 985–991 CrossRef.
- E. Samei, R. S. Saunders, C. T. Badea, K. B. Ghaghada, L. W. Hedlund, Y. Qi, H. Yuan, R. C. Bentley and S. Mukundan, Int. J. Nanomed., 2009, 4, 277–282 CrossRef CAS.
- C. C. Chien, C. H. Wang, C. L. Wang, E. R. Li, K. H. Lee, Y. Hwu, C. Y. Lin, S. J. Chang, C. S. Yang, C. Petibois and G. Margaritondo, Anal. Bioanal. Chem., 2010, 397, 2109–2116 CrossRef CAS.
- P. A. Jackson, W. N. W. Abd Rahman, C. J. Wong, T. Ackerly and M. Geso, Eur. J. Radiol., 2010, 75, 104–109 CrossRef.
- K. Greish, in Cancer Nanotechnology: Methods and Protocols, ed. S. R. M. B. M. Grobmyer, 2010, pp. 25–37 Search PubMed.
- M. Y. Chang, A. L. Shiau, Y. H. Chen, C. J. Chang, H. H. W. Chen and C. L. Wu, Cancer Sci., 2008, 99, 1479–1484 CrossRef CAS.
- N. Yusa, M. Jiang, K. Mizuno and M. Uesaka, Radiol. Phys. Technol., 2009, 2, 33–39 CrossRef.
- W.-C. Law, K.-T. Yong, A. Baev and P. N. Prasad, ACS Nano, 2011, 5, 4858–4864 CrossRef CAS.
- S. BarathManiKanth, K. Kalishwaralal, M. Sriram, S. R. K. Pandian, H.-S. Youn, S. Eom and S. Gurunathan, J. Nanobiotechnol., 2010, 8, 1–16 CrossRef.
- Z. Zhang, R. D. Ross and R. K. Roeder, Nanoscale, 2010, 2, 582–586 RSC.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS.
- H. H. Wang, C. H. Su, Y. J. Wu, C. A. J. Lin, C. H. Lee, J. L. Shen, W. H. Chan, W. H. Chang and H. I. Yeh, Int. J. Gerontol., 2012, 6, 1–4 CrossRef.
- E. C. Dreaden, A. M. Alkilany, X. H. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- S. Jain, D. G. Hirst and J. M. O'Sullivan, Br. J. Radiol., 2012, 85, 101–113 CrossRef CAS.
- S. Rana, A. Bajaj, R. Mout and V. M. Rotello, Adv. Drug Delivery Rev., 2012, 64, 200–216 CrossRef CAS.
- M. Geso, Br. J. Radiol., 2007, 80, 64–65 CrossRef CAS.
- C. Xu, G. A. Tung and S. Sun, Chem. Mater., 2008, 20, 4167–4169 CrossRef CAS.
- Z. Wang, L. Wu and W. Cai, Chem.–Eur. J., 2010, 16, 1459–1463 CrossRef CAS.
- D. J. Barber and I. C. Freestone, Archaeometry, 1990, 32, 33–45 CrossRef.
- M. H. Magnusson, K. Deppert, J.-O. Malm, J.-O. Bovin and L. Samuelson, J. Nanopart. Res., 1999, 1, 243–251 CrossRef CAS.
- B. Zheng, L. Qian, H. Yuan, D. Xiao, X. Yang, M. C. Paau and M. M. F. Choi, Talanta, 2010, 82, 177–183 CrossRef CAS.
- H. Horvath, J. Quant. Spectrosc. Radiat. Transfer, 2009, 110, 787–799 CrossRef CAS.
- N. Bogliotti, B. Oberleitner, A. Di-Cicco, F. Schmidt, J. C. Florent and V. Semetey, J. Colloid Interface Sci., 2011, 357, 75–81 CrossRef CAS.
- J. E. Giles, N. K. Worley and N. Telusca, Int. J. Pediatr. Otorhinolaryngol., 2009, 73, 1274–1277 CrossRef.
- F. Mafune, J. Y. Kohno, Y. Takeda and T. Kondow, J. Phys. Chem. B, 2002, 106, 7575–7577 CrossRef CAS.
- J. E. Millstone, S. J. Hurst, G. S. Metraux, J. I. Cutler and C. A. Mirkin, Small, 2009, 5, 646–664 CrossRef CAS.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- L. A. Dykman and V. A. Bogatyrev, Russ. Chem. Rev., 2007, 76, 199–213 CrossRef.
- Y.-C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- K.-T. Yong, M. T. Swihart, H. Ding and P. N. Prasad, Plasmonics, 2009, 4, 79–93 CrossRef CAS.
- Z. Y. Zhong, K. B. Male and J. H. T. Luong, Anal. Lett., 2003, 36, 3097–3118 CrossRef CAS.
- D. T. Nguyen, D.-J. Kim, M. G. So and K.-S. Kim, Adv. Powder Technol., 2010, 21, 111–118 CrossRef CAS.
- I. Ojea-Jimenez and V. Puntes, J. Am. Chem. Soc., 2009, 131, 13320–13327 CrossRef CAS.
- J. Polte, T. T. Ahner, F. Delissen, S. Sokolov, F. Emmerling, A. F. Thuenemann and R. Kraehnert, J. Am. Chem. Soc., 2010, 132, 1296–1301 CrossRef CAS.
- S. K. K, A. R, P. Arumugam and S. Berchmans, ACS Appl. Mater. Interfaces, 2011, 3, 1418–1425 Search PubMed.
- C. K. Yee, R. Jordan, A. Ulman, H. White, A. King, M. Rafailovich and J. Sokolov, Langmuir, 1999, 15, 3486–3491 CrossRef CAS.
- D. Zanchet, B. D. Hall and D. Ugarte, J. Phys. Chem. B, 2000, 104, 11013–11018 CrossRef CAS.
- J. Petroski, M. H. Chou and C. Creutz, Inorg. Chem., 2004, 43, 1597–1599 CrossRef CAS.
- W. W. Weare, S. M. Reed, M. G. Warner and J. E. Hutchison, J. Am. Chem. Soc., 2000, 122, 12890–12891 CrossRef CAS.
- V. Armendariz, J. G. Parsons, M. L. Lopez, J. R. Peralta-Videa, M. Jose-Yacaman and J. L. Gardea-Torresdey, Nanotechnology, 2009, 20, 1–8 CrossRef.
- C. J. Murphy, A. M. Gole, S. E. Hunyadi, J. W. Stone, P. N. Sisco, A. Alkilany, B. E. Kinard and P. Hankins, Chem. Commun., 2008, 544–557 RSC.
- P. Pengo, S. Polizzi, M. Battagliarin, L. Pasquato and P. Scrimin, J. Mater. Chem., 2003, 13, 2471–2478 RSC.
- C. Kojima, Y. Umeda, M. Ogawa, A. Harada, Y. Magata and K. Kono, Nanotechnology, 2010, 21, 1–6 CrossRef.
- C. Vollmer and C. Janiak, Coord. Chem. Rev., 2011, 255, 2039–2057 CrossRef CAS.
- S. Bulut, P. Klose, M. M. Huang, H. Weingartner, P. J. Dyson, G. Laurenczy, C. Friedrich, J. Menz, K. Kummerer and I. Krossing, Chem.–Eur. J., 2010, 16, 13139–13154 CrossRef CAS.
- C. M. Crudden, M. Sateesh and R. Lewis, J. Am. Chem. Soc., 2005, 127, 10045–10050 CrossRef CAS.
- C. J. Weiss and T. J. Marks, J. Am. Chem. Soc., 2010, 132, 10533–10546 CrossRef CAS.
- K. S. Kim, D. Demberelnyamba and H. Lee, Langmuir, 2004, 20, 556–560 CrossRef CAS.
- R. Guo, H. Wang, C. Peng, M. W. Shen, L. F. Zheng, G. X. Zhang and X. Y. Shi, J. Mater. Chem., 2011, 21, 5120–5127 RSC.
- C. Peng, L. F. Zheng, Q. Chen, M. W. Shen, R. Guo, H. Wang, X. Y. Cao, G. X. Zhang and X. Y. Shi, Biomaterials, 2012, 33, 1107–1119 CrossRef CAS.
- H. Wang, L. F. Zheng, R. Guo, C. Peng, M. W. Shen, X. Y. Shi and G. X. Zhang, Nanoscale Res. Lett., 2012, 7, 1–8 CrossRef.
- H. Wang, L. F. Zheng, C. Peng, R. Guo, M. W. Shen, X. Y. Shi and G. X. Zhang, Biomaterials, 2011, 32, 2979–2988 CrossRef CAS.
- D. Kim, S. Park, J. H. Lee, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2007, 129, 7661–7665 CrossRef CAS.
- M. P. Melancon, W. Lu, M. Zhong, M. Zhou, G. Liang, A. M. Elliott, J. D. Hazle, J. N. Myers, C. Li and R. J. Stafford, Biomaterials, 2011, 32, 7600–7608 CrossRef CAS.
- A. S. Thakor, R. Luong, R. Paulmurugan, F. I. Lin, P. Kempen, C. Zavaleta, P. Chu, T. F. Massoud, R. Sinclair and S. S. Gambhir, Sci. Transl. Med., 2011, 3, 1–11 CrossRef.
- F. K. Wang, N. Phonthammachai, K. Y. Mya, W. W. Tjiu and C. B. He, Chem. Commun., 2011, 47, 767–769 RSC.
- S. Zhang, P. Xiong, X. Yang and X. Wang, Nanoscale, 2011, 3, 2169–2174 RSC.
- S. Harakeh, R. M. Abdel-Massih, P. Rivera Gil, R. A. Sperling, A. Meinhardt, A. Niedwiecki, M. Rath, W. J. Parak and E. Baydoun, Nanotoxicology, 2010, 4, 177–185 CrossRef CAS.
- C. C. Wu and D. H. Chen, Gold Bull., 2010, 43, 234–240 CrossRef CAS.
- V. Kattumuri, K. Katti, S. Bhaskaran, E. J. Boote, S. W. Casteel, G. M. Fent, D. J. Robertson, M. Chandrasekhar, R. Kannan and K. V. Katti, Small, 2007, 3, 333–341 CrossRef CAS.
- P. Kan, H. P. Engelbrecht, L. D. Watkinson, T. L. Carmack, J. R. Lever, C. J. Smith, R. Kannan, K. V. Kattesh, S. S. Jurisson and C. S. Cutler, J. Labelled Comp. Rad., 2009, 52, S521–S521 Search PubMed.
- G. M. Fent, S. W. Casteel, D. Y. Kim, R. Kannan, K. Katti and N. Chanda, Nanomed.: Nanotechnol., Biol. Med., 2009, 5, 128–135 CrossRef CAS.
- E. Boote, G. Fent, V. Kattumuri, S. Casteel, K. Katti, N. Chanda, R. Kannan and R. Churchill, Acad. Radiol., 2010, 17, 410–417 CrossRef.
- D. P. Cormode, E. Roessl, A. Thran, T. Skajaa, R. E. Gordon, J. P. Schlomka, V. Fuster, E. A. Fisher, W. J. M. Mulder, R. Proksa and Z. A. Fayad, Radiology, 2010, 256, 774–782 CrossRef.
- C. S. Thaxton, W. L. Daniel, D. A. Giljohann, A. D. Thomas and C. A. Mirkin, J. Am. Chem. Soc., 2009, 131, 1384–1385 CrossRef CAS.
- D. P. Cormode, T. Skajaa, M. M. van Schooneveld, R. Koole, P. Jarzyna, M. E. Lobatto, C. Calcagno, A. Barazza, R. E. Gordon, P. Zanzonico, E. A. Fisher, Z. A. Fayad and W. J. M. Mulder, Nano Lett., 2008, 8, 3715–3723 CrossRef CAS.
- T. Solomun, H. Sturm, R. Wellhausen and H. Seitz, Chem. Phys. Lett., 2012, 533, 92–94 CrossRef CAS.
- J. A. Diaz, D. M. Grewer and J. M. Gibbs-Davis, Small, 2012, 8, 873–883 CrossRef CAS.
- M. Fujita, Y. Katafuchi, K. Ito, N. Kanayama, T. Takarada and M. Maeda, J. Colloid Interface Sci., 2012, 368, 629–635 CrossRef CAS.
- X. Zhang, M. R. Servos and J. W. Liu, Langmuir, 2012, 28, 3896–3902 CrossRef CAS.
- S. Narayanan, B. N. Sathy, U. Mony, M. Koyakutty, S. V. Nair and D. Menon, ACS Appl. Mater. Interfaces, 2012, 4, 251–260 CAS.
- D. Kim, M. K. Yu, T. S. Lee, J. J. Park, Y. Y. Jeong and S. Jon, Nanotechnology, 2011, 22, 1–7 Search PubMed.
- J. A. Park, H. K. Kim, J. H. Kim, S. W. Jeong, J. C. Jung, G. H. Lee, J. Lee, Y. Chang and T. J. Kim, Bioorg. Med. Chem. Lett., 2010, 20, 2287–2291 CrossRef CAS.
- P. Puvanakrishnan, J. Park, D. Chatterjee, S. Krishnan and J. W. Tunnell, Int. J. Nanomed., 2012, 7, 1251–1258 CrossRef CAS.
- F. E. Escorcia, M. R. McDevitt, C. H. Villa and D. A. Scheinberg, Nanomedicine, 2007, 2, 805–815 CrossRef CAS.
- S. Pietronave, M. Iafisco, D. Locarno, L. Rimondini and M. Prat, J. Appl. Biomater. Biom., 2009, 7, 77–89 CAS.
- M. Fournelle, W. Bost, I. H. Tarner, T. Lehmberg, E. Weiss, R. Lemor and R. Dinser, Nanomed.: Nanotechnol., Biol. Med., 2012, 8, 346–354 CrossRef CAS.
- P. Puvanakrishnan, P. Diagaradjane, S. M. S. Kazmi, A. K. Dunn, S. Krishnan and J. W. Tunnell, Lasers Surg. Med., 2012, 44, 310–317 CrossRef.
- R. Popovtzer, A. Agrawal, N. A. Kotov, A. Popovtzer, J. Balter, T. E. Carey and R. Kopelman, Nano Lett., 2008, 8, 4593–4596 CrossRef CAS.
- A. Hirao and H. S. Yoo, Polym. J., 2011, 43, 2–17 CrossRef CAS.
- D. Taton, X. S. Feng and Y. Gnanou, New J. Chem., 2007, 31, 1097–1110 RSC.
- Q. Y. Cai, S. H. Kim, K. S. Choi, S. Y. Kim, S. J. Byun, K. W. Kim, S. H. Park, S. K. Juhng and K. H. Yoon, Invest. Radiol., 2007, 42, 797–806 CrossRef CAS.
- C. S. Kim, Y. C. Ahn, P. Wilder-Smith, S. Oh, Z. P. Chen and Y. J. Kwon, Biomed. Opt. Express, 2010, 1, 106–113 CrossRef CAS.
- N. Chanda, P. Kan, L. D. Watkinson, R. Shukla, A. Zambre, T. L. Carmack, H. Engelbrecht, J. R. Lever, K. Katti, G. M. Fent, S. W. Casteel, C. J. Smith, W. H. Miller, S. Jurisson, E. Boote, J. D. Robertson, C. Cutler, M. Dobrovolskaia, R. Kannan and K. V. Katti, Nanomed.: Nanotechnol., Biol. Med., 2010, 6, 201–209 CrossRef CAS.
- N. S. Md, H.-K. Kim, J.-A. Park, Y. Chang and T.-J. Kim, Bull. Korean Chem. Soc., 2010, 31, 1177–1181 CrossRef CAS.
- T. Reuveni, M. Motiei, Z. Romman, A. Popovtzer and R. Popovtzer, Int. J. Nanomed., 2011, 6, 2859–2864 CAS.
- M. Wang and M. Thanou, Pharmacol. Res., 2010, 62, 90–99 CrossRef CAS.
- G. P. Goodrich, L. Bao, K. Gill-Sharp, K. L. Sang, J. Wang and J. D. Payne, J. Biomed. Opt., 2010, 15, 018001 CrossRef.
- L. C. Kennedy, A. S. Bear, J. K. Young, N. A. Lewinski, J. Kim, A. E. Foster and R. A. Drezek, Nanoscale Res. Lett., 2011, 6, 1–11 CrossRef.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647–1671 RSC.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS.
- N. Pernodet, X. H. Fang, Y. Sun, A. Bakhtina, A. Ramakrishnan, J. Sokolov, A. Ulman and M. Rafailovich, Small, 2006, 2, 766–773 CrossRef CAS.
- J. J. Li, L. Zou, D. Hartono, C. N. Ong, B. H. Bay and L. Y. L. Yung, Adv. Mater., 2008, 20, 138–142 CrossRef CAS.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, P. C. Patel, J. E. Millstone, N. L. Rosi and C. A. Mirkin, Nano Lett., 2007, 7, 3818–3821 CrossRef CAS.
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941–1949 CrossRef CAS.
- Y. Pan, A. Leifert, D. Ruau, S. Neuss, J. Bornemann, G. Schmid, W. Brandau, U. Simon and W. Jahnen-Dechent, Small, 2009, 5, 2067–2076 CrossRef CAS.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS.
- N. M. Schaeublin, L. K. Braydich-Stolle, E. I. Maurer, K. Park, R. I. MacCuspie, A. Afrooz, R. A. Vaia, N. B. Saleh and S. M. Hussain, Langmuir, 2012, 28, 3248–3258 CrossRef CAS.
- W. S. Cho, M. J. Cho, J. Jeong, M. Choi, H. Y. Cho, B. S. Han, S. H. Kim, H. O. Kim, Y. T. Lim and B. H. Chung, Toxicol. Appl. Pharmacol., 2009, 236, 16–24 CrossRef CAS.
- X. D. Zhang, H. Y. Wu, D. Wu, Y. Y. Wang, J. H. Chang, Z. B. Zhai, A. M. Meng, P. X. Liu, L. A. Zhang and F. Y. Fan, Int. J. Nanomed., 2010, 5, 771–781 CrossRef CAS.
- J. H. Sung, J. H. Ji, J. D. Park, M. Y. Song, K. S. Song, H. R. Ryu, J. U. Yoon, K. S. Jeon, J. Jeong, B. S. Han, Y. H. Chung, H. K. Chang, J. H. Lee, D. W. Kim, B. J. Kelman and I. J. Yu, Part. Fibre Toxicol., 2011, 8, 1–33 CrossRef.
- C. Lasagna-Reeves, D. Gonzalez-Romero, M. A. Barria, I. Olmedo, A. Clos, V. M. S. Ramanujam, A. Urayama, L. Vergara, M. J. Kogan and C. Soto, Biochem. Biophys. Res. Commun., 2010, 393, 649–655 CrossRef CAS.
- G. D. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS.
- K. Kandasamy, C. S. Choi and S. Kim, Nanotechnology, 2010, 21, 1–11 CrossRef.
- Y.-S. Chen, Y.-C. Hung, I. Liau and G. S. Huang, Nanoscale Res. Lett., 2009, 4, 858–864 CrossRef CAS.
- W.-S. Cho, S. Kim, B. S. Han, W. C. Son and J. Jeong, Toxicol. Lett., 2009, 191, 96–102 CrossRef CAS.
- A. Rocha, Y. Zhou, S. Kundu, J. M. Gonzalez, S. BradleighVinson and H. Liang, J. Nanobiotechnol., 2011, 9, 1–9 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |