Evidence of high rate visible light photochemical decolourisation of Rhodamine B with BiFeO3 nanoparticles associated with BiFeO3 photocorrosion†‡
Chang
Hengky
ab,
Xavier
Moya
c,
Neil D.
Mathur
c and
Steve
Dunn
*d
a180 Ang Mo Kio Ave 8, BioMEMS and Nanotechnology Group, School of Engineering (Manufacturing), Nanyang Polytechnic, Singapore
bSchool of Applied Science, Cranfield University, Bedfordshire, MK43 0AL, United Kingdom
cDepartment of Materials Science, University of Cambridge, Pembroke Street, CB2 3QZ, Cambridge, United Kingdom
dCentre for Materials Research, School and Engineering and Materials, Queen Mary University of London, E1 4NS, United Kingdom. E-mail: s.c.dunn@qmul.ac.uk
First published on 9th October 2012
Abstract
BiFeO3 nanopowders with a size distribution around 20 nm and optical absorption onset at 2.1 eV have been synthesized using self-combustion. These particles were used to photodecolourise Rhodamine B (RhB) dye under a solar simulator with AM1.5 irradiation. XRD analysis after illumination showed no change while XPS analysis on the particles showed significant changes to the chemical state of the Fe cations in BiFeO3 after irradiation. After 10 min of visible (AM 1.5) illumination, at pH 2, RhB showed >95% decolourisation, a rate of decolourisation that rivals other semiconductor systems. We conclude that BiFeO3 nanopowder is a highly active visible light reagent that rivals many of the doped titania systems in terms of activity, but it is not photostable due to photocorrosion of the material during illumination.
Introduction
The use of renewable or low energy sources for the degradation of organic pollutants has generated broad interest. Research on semiconductor photocatalysis came sharply into focus after the discovery that TiO2 photochemical electrodes could split water using ultraviolet light1 Following that, the photocatalytic oxidation of organic contaminants using TiO2-based semiconductors as a photocatalyst has been extensively investigated due to their excellent photochemical stability, high-efficiency, low cost, and non-toxicity. However, the photo-efficiency of TiO2 is limited by its restricted absorption in the visible-light region. Only 4% of terrestrial radiation is suitable for the photoexcitation of TiO22 rendering the process impractical due to the inefficiency of photocatalysis or photochemical reaction.A number of alternative systems have been investigated to generate systems that are photoactive under visible light stimulation. These include over 50 different semi-conductor systems, so called ‘z-system’ couples, doping and modification of wide band gap materials such as the doping of TiO23 and recently the use of plasmonic nanostructures grown on the surface of the catalyst. These represent a large and significant global effort to generate photocatalysts that are active in the visible region and so enhance the need to find materials that have a band gap in the visible region, are effective catalysts and stable. The use of BiFeO3 a material that exhibits low band gap (ca. 2.1 eV)16 could make a significant contribution to the visibly active photochemical reaction or photocatalysts.
A significant benefit of TiO2 is that it is photostable. For this the band positions of the semiconductor must ‘pinch’ the REDOX couples of available reactants and products. If the photocatalyst, or reagent, does not do this then it may be liable to photocorrosion.4 This is a well-known problem for many narrow band gap semiconductors such as CdTe, and has been a significant limiting factor in the development of visibly active systems. Recently it has been shown that perovskite ferroelectric materials can photocorrode5 when the REDOX couples are unfavourable during a photocatalytic process.
In recent years there has been growing interest in the use of ‘functional’ ferro or piezoelectric photocatalysts such as those from the perovskite ABO3 family such as BaTiO3 or analogous materials such as LiNbO3.6,7 The focus of work on these materials now also includes using the functional – pyroelectric,8 or piezoelectric9 – aspects of the materials to drive catalysis and produce active nanostructures.10 These materials demonstrate some unique photochemical properties, that can under appropriate circumstances be photocatalytic, that arise from the non-centrosymmetric nature of the crystal structure that can be harnessed to enhance the photochemistry performance of the catalyst.
Perovskite-type BiFeO3 (BFO) materials have attracted much interest due to their multiferroic properties at room temperature.11,12 In particular, BFO thin films have been investigated intensively as novel materials for nonvolatile memory,13 magnetoelectric switching,14 and photovoltaic devices.15 In addition, there is increasing interest in these materials for photocatalytic processes under visible-light illumination.16 It is being established that ferroelectric materials can be highly photoactive with some exceptional properties due to the internal dipole of the material.17,18 Although the photocatalytic activity of BFO powders under visible light is ascribed mainly to small band-gap energy, attempts to elucidate other factors, such as their weak ferromagnetic properties, are still being made19 and further enhance interest in such functional materials as photocatalysts.
Previous work focusing on the photochemical degradation of dye compounds using BFO has focused on Methyl Orange (MO),16 Congo Red (CR)20 and Rhodamine B (RhB).21 In this previous work the ferroelectric or, in the case of BFO, multiferroic nature of the semiconductor has not been recognised or considered as important. However, there is a growing body of work that demonstrates materials with an electric field and spontaneous polarization behave in anomalous ways when photoexcited. This anomalous behaviour has recently been demonstrated on BFO surfaces where spatially selective REDOX chemistry was shown.22 It is, therefore, important to consider that BFO is not only a semiconductor but also a multiferroic material that demonstrates electron and hole separation due to the internal fields within the crystal lattice. In essence the internal electric field of BFO exhibits a spontaneous polarisation that acts like an internal p-n junction. This band structure can be considered a ‘self-junction’ as carriers are influenced by the field within, and inherent to the material. This, largely, determines the band bending at the interface and the surface where the mobile carriers can accumulate.
The effect of Gd doping on the photocatalytic response of BFO shows that the catalytic efficiency of the system increased when the band gap of the system increases from around 2 eV for BFO to 2.2 eV for Bi0.9Gd0.1FeO3. The maximum performance of photocatalytic activity for the BFO samples came from material with a band gap that exceeds 2 eV.23 The paper does not indicate whether there is a substantial change in the surface chemi- or physisorption interaction with the dye or other molecules in solution is the root cause for the enhanced performance.
It is typical for a semiconductor to experience movement of free carriers due to differences in local chemical potential that result in band bending when in contact with an ionic solution. In ferroelectric materials the depolarising fields screen the surface charge by drawing electrons to the positive C+ face and holes to the negative C− face.24 These regions of carrier accumulation induce bending at the surface causing downward bending at the C+ face and upward bending at the C− face. It has been shown that this band bending is sufficiently dominant that it determines the band bending irrespective of the chemical environment around the catalyst such as the solution or the dissolved species. Resulting in spatially distinct REDOX chemistry.25,26 Reduction occurs at the C+ face due to electron accumulation and oxidation at the C− face.27 The separation of carriers by the depolarisation fields also suppresses recombination rates thereby increasing carrier lifetimes. Evidence for this is provided by the long photoluminescence of up to 9 μs in LiNbO3.28
These intriguing properties of ferroelectrics have led to growing interest in their photochemistry. Selective deposition of metal nano particles has been extensively investigated27,29–35 and a wide range of different materials including lead zirconate titanate, barium titanate and lithium niobate have been used as a catalyst. Ferroelectrics have also been used to drive photocatalytic reactions such as artificial photo synthesis38,36 and water splitting.29,32 An investigation of different compositions of barium titanate to split water31,37 showed then reactivity of the materials to increase with strength of polarization.
Here we show that the multiferroic properties influence the band structure of the BFO and this interaction results in a material that demonstrates rapid dye decolourisation under artificial natural sunlight delivered by a solar simulator illumination (AM 1.5) to give 95% decolourisation in under 10 min. This competes favourably with the most rapid decolourisation for RhB over other semi-conductor systems and represents a significant step forward in visible light photochemistry design. However, the same multiferroic properties produce a band structure that results in photocorrosion of the BFO particles.
Experimental procedure
Commercially available reagent grade bismuth nitrate pentahydrate (Bi(NO3)3·5H2O) and iron nitrate nonahydrate (Fe(NO3)3·9H2O) (purity >99%, International Laboratory) were used as raw materials. 70% nitric acid (HNO3) or de-ionised (DI) water (18.3 MΩ) was used to dissolve the pre-cursors in a proportion of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (molar ratio) to create a 0.2 M solution. Bismuth nitrate pentahydrate salts were dissolved in nitric acid. Iron nitrate nonahydrate was dissolved in DI water. Citric acid >98% purity reagent grade (Sigma Aldrich) was used as the fuel for this autocombustion technique. The Bi–Fe with citric acid solution was then heated to 300 °C until dry. This was followed by auto ignition at 300 °C with a dwell for 30 min. The resultant flakes were ground and annealed in a furnace for 3 h at 650 °C.
:1 (molar ratio) to create a 0.2 M solution. Bismuth nitrate pentahydrate salts were dissolved in nitric acid. Iron nitrate nonahydrate was dissolved in DI water. Citric acid >98% purity reagent grade (Sigma Aldrich) was used as the fuel for this autocombustion technique. The Bi–Fe with citric acid solution was then heated to 300 °C until dry. This was followed by auto ignition at 300 °C with a dwell for 30 min. The resultant flakes were ground and annealed in a furnace for 3 h at 650 °C.
The BFO powders were characterized using FESEM (JEOL-JSM7500F) and TEM (JEOL-2010F). EDS spectra were taken using JEOL-JSM7500F under 5500 times magnification and 15 KV acceleration. XRD patterns were obtained using PANAlytical X′Pert Pro MPD advanced powder X-ray diffractometer (using Cu-Kα = 1.54056 A° radiation) with a scanning range of 20° to 70° (2θ) and step size of 0.001.
Visible absorption was determined using a Perkin Elmer model Lambda 950 UV-Vis-NIR spectrophotometer with integrating sphere. Magnetic measurements were performed using a Princeton Measurements Corporation Vibrating Sample Magnetometer (VSM).
Photo decolourisation of the RhB dye was performed by mixing 4.8 mg of RhB dye powder in 1 litre of DI water. 300 mg of BFO was loaded into 100 mL of RhB dye solution. This was transferred into quartz petri dish and exposed under solar simulator (SAN-EI solar simulator – XES Series) with AM1.5. 3 mL of solution was taken for sampling every interval followed by centrifugation at 12000 RPM for 10 min before measurement of the absorption spectra.
The zeta potential of BFO powder dispersed in DI water was measured using a Malvern Zeta Sizer Nano ZS Series at pH values of 6.7 (normal water pH), 4.8, 3.2 and 2.2. X-ray photo-electron spectrometry (XPS) was performed using a Thermo Fisher Scientific Theta Probe. The X-ray source was monochromatic Al Kα (1486.5 eV) at a voltage of 15 kV and 100 W. XPS spectra were gathered using a hemispherical energy analyzer operated at pass energy of 200 eV for survey scans and 40.0 eV for high resolution elemental scans.
Results and discussions
Synthesis of the BiFeO3 was achieved using an autocombustion technique with a citric acid/metal molar ratio of 1:1 and annealing at 650 °C for 3 h. The XRD data, shown in Fig. 1, obtained for the BiFeO3 (BFO) sample after the autocombustion shows that the material contains a very minor trace of secondary phases with the majority phase fitting JCPDS card No. 71-2494. The XRD pattern of the BFO sample produced using this technique matches very closely with the accepted XRD pattern published in previous work for BFO and shows the peak splitting associated with BFO ceramics.16Fig. 2 shows the optical band gap of the BFO nanopowders calculated using Tauc's relationship formula of αhν = A(hν − Eg)n using n = ½. The onset of absorption taken from Fig. 2 is 2.10 eV and is consistent with the previous reports by Gao et al.16 where the BiFeO3 nanoparticle size is in a similar range of 20–50 nm. While the optical bandgap value is smaller than for BiFeO3 nanowires with a band gap of 2.5 eV19 and a thin film BiFeO3 with a bandgap of 2.54 eV38 we ascribe this surface or interfacial phenomena of the BFO and not due to the quantisation as the De Broglie wavelength of BFO is likely to be significantly smaller than the 20–50 nm of the BFO particles produced.
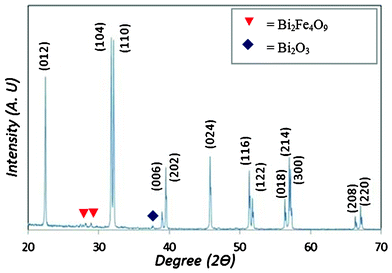 | ||
| Fig. 1 XRD pattern for as produced BiFeO3 annealed at 650 °C for 3 h. | ||
 | ||
| Fig. 2 UV-vis analysis of particles with Tauc relationship showing an absorption onset at 2.1 eV. | ||
The magnetization of the optimized nanopowder was measured out to an applied field of μ0H = 0.2 T and is shown in Fig. 3. Bulk BFO is antiferromagnetic,39 but our nanoparticles show magnetic hysteresis and the magnetization does not saturate, as expected for sol–gel nanoparticles.40 The magnetization at our maximum measurement field corresponds well to the value for thin-film BFO,41 but is an order of magnitude smaller than the magnetization of sol–gel BFO nanoparticles.42 This discrepancy is attributed to the differences in synthesis technique and the concomitant differences in particle-size distribution.
 | ||
| Fig. 3 Room temperature magnetization versus field for BiFeO3 nanoparticles annealed at 650 °C for 3 h. | ||
SEM micrographs show that the BFO exhibits a largely spherical morphology that consists of large loosely bound agglomerates (supporting information Fig. S1†). TEM analysis of the powders post light ultrasonic treatment, to break up the agglomerates, shows that the material is formed of dispersed nanoparticles with a particle size distribution ranging from 20 to 100 nm, Fig. 4. A histogram of the particle size distribution derived from the TEM micrograph indicates the particle size ranges from 10 nm–100 nm (supporting information Fig. S2†).
 | ||
| Fig. 4 TEM micrograph of BiFeO3 particles, insert shows hexagonal morphology of individual particles. | ||
Zeta potential measurements indicated that at a pH of 6.7, the neutral pH of the RhB solution in water the BFO particles exhibited a strongly negative surface charge of −16 mV. Changing the pH to strongly acidic (pH 2.2) by titration with diluted nitric acid (HNO3) reduces the zeta potential to −1.62 mV, and the isoelectric point was extrapolated to be pH 2 (supporting information Fig. S3†).
The photo decolourisation of the RhB dye was performed using the as produced BFO nanopowder as a photochemical agent at a pH of 6.7, 4.0 and 2. In all cases there was negligible decolourisation of the dye without illumination in the presence of the BFO and no measurable decolourisation in the timeframe of the experiments under illumination in the absence of BFO nanopowder .
A representative example of dye decolourisation over the BFO nanopowder at pH 2, 4 and 6.7 is shown in Fig. 5. Two trends are visible; the first shows that there is a clear association between the initial rate of decolourisation and the pH of the dye solution. The second shows that there are two reaction rates, with the second reaction rate being largely independent of pH. In all cases equilibrium absorption of RhB on the BFO was achieved before the system was illuminated.
 | ||
| Fig. 5 Decolourisation profiles for RhB with BiFeO3 photoreagent under simulated visible light (AM1.5) illumination. Two distinct reactions rates are observable, one a function of pH the other largely independent of pH with a lower rate of dye decolourisation. | ||
A change in solution pH influences the Stern and double layer formation around the BFO particles as indicated by the Zeta potential measurements. There is a further complication in that the double layer at the surface of a ferroelectric is heavily influenced by the surface charge associated with the spontaneous dipole inherent within the ferroelectric. A combination of the variation in pH and the influence on the surface charge through the multiferroic nature of the BFO will influence the binding and proximity of the dye molecules to the BFO particles. In order for the dye to be decolourised, photoexcited carriers must be able to migrate from catalyst to a suitable species that can subsequently react with the dye or a reactive species must interact directly with the dye, as indicated in eqn (1)–(7).
| PC + hv → e−cb + h+vb | (1) |
| h+vb + OH− → OH | (2) |
| e−cb + O2ads → O·−2 | (3) |
| Dye + OH· → Degradation products | (4) |
| Dye + e−cb → Reduction products | (5) |
| Dye + h+vb → Oxidation products | (6) |
| Dye+· + O2−· → Degredation products | (7) |
For BFO particles in DI water at pH 6.7, the surfaces of the nanoparticles are negatively charged. The surfaces will have a tight and overcrowded co-ions screening layer on the BFO particle surface (supporting information Fig. S4†). The development of the charge co-ions screening on the BFO particle surface is due to OH− ions present in the suspension as the dye was dissolved in DI water. This observation was consistent with the work by Dunn et al.43 where the development of a screening layer on a ferroelectric surface was fully explained. Such a screening layer of co-ions will prevent the dye molecules being adsorbed directly onto the BFO particle surface. The result of this is that the dye molecules will not undergo photo REDOX reaction by the photo excited carriers from the BFO. This would effectively reduce the overall reaction rate as an intermediate step is required for decolourisation to occur. At lower pH values, the negative charge screening layer on BFO surface is reduced and becomes less negative towards the iso-electric point at pH 2. This allows the dye molecules to be adsorbed at the BFO surface and undergo direct photo-reduction reactions under exposure of the simulated solar spectrum. At pH2, the isoelectric point of the particles there we show the highest level of dye absorption on the BFO surface (supporting information Fig. S5†).
Indicative band positions of BFO relative to the accepted positions of NHE, water and RhB are shown in Fig. 6. It is important to note that the band bending of the ferroelectric is controlled not by the exchange of carriers across the interface but by the internal field driven by the spontaneous depolarisation field. Earlier work has shown that BFO exhibits spatially selective photochemistry and so the accepted band diagram for a non-polarisable semiconductor is not valid in this case. The band structure also indicates why there may be some instability in a photoexcited BFO aqueous solution of RhB. In the band diagram the valence band of the BFO lies above the lower energy levels of both the dye and water. As the valence and conduction band of BFO do not ‘pinch’ the reactive species at the interface there is instability in the system. This instability is characteristic of semiconductor systems that are liable to photocorrosion. While it is not possible for the BFO to inject holes into the RhB dye due to the alignment of bands, RhB dye molecules are able to inject holes into the BFO. This leads to oxidation of the BFO and initiates photocorrosion. A possible and plausible route for this is the photoexcitation of the RhB to produce a photoexcited species.
 | ||
| Fig. 6 Indicative band alignment illustrations for BiFeO3 in a RhB aqueous environment. The band bending of the BiFeO3 has been drawn as flat in (a) but drawn according to ferroelectric dipole interactions in (b) and (c) indicating the transfer of excited carriers between proximal species. | ||
There are two significant differences from the previous work that has focused on BFO from the work that is presented here. The first is that the band gap of the BFO in previous reports has been measured as larger than that reported here. This small shift in band gap could push the valence band below the oxidation potentials of the species in solution thereby reducing the photocorrosion of the BFO. An examination of the band structures presented indicates that the valence band edge of BFO is on a cusp of stability/instability in aqueous environments. Our results may also indicate why in earlier work material with a larger band gap performed as a better catalyst – the larger band gap allowed for a complete REDOX couple. The other difference is in the size of the catalyst material. The BFO powder we have produced is significantly smaller than previous BFO powders examined, but we do not believe that the size of the BFO is approaching a quantum confined system and instead propose that the higher surface area – volume ratio of the samples tested in this series of experiments may explain the differences found as the smaller particle size enables more rapid photochemistry and so enhances the rate of photodegradation. Increasing the surface area of a material is well known to enhance the reactivity.
Samples of BFO before and after illumination with the dye were measured using X-ray diffraction and XPS. The X-ray diffraction data indicated no change before and after illumination for 6 h (see supporting information Fig. S6†), while the XPS data, shown in Fig. 7, shows considerable changes. We attribute the lack of change in the crystallography on the nature of the technique. X-ray diffraction produces data averaged over the full penetration of the X-rays used and is not surface sensitive.
 | ||
| Fig. 7 XPS spectra for BiFeO3 before and after exposure to irradiation in RhB pH 6.7 solution. (a) indicates the O1S peak with some minor changes to the peak shape and position, (b) the Bi1F peak with a slight shift in peak position, and (c) the Fe2p peak showing a significant change in the relative ratios of Fe3+ peaks at at 711.5 and 725.8 eV after illumination. | ||
XPS is a highly surface sensitive technique and returns information about the chemical environment at the surface. The XPS data, Fig. 7, shows typical spectra for an as prepared sample of BFO and in agreement with other report by Popa et al.44 The position of Fe 2p is expected to be at 711 eV for Fe3+ and 709.5 eV for Fe2+.45 The 3/2 and 1/2 spin orbit doublet components of the Fe 2p photoemission located at 711.5 and 725.8 eV respectively, were identified as Fe3+. Hence, no Fe2+ and Fe were found. The XPS results show that BFO nanoparticle surface has a single phase with Fe present in the 3+ valence state.
A comparison of the spectra obtained for the samples illuminated at pH 6.7 indicates that there is significant change in the chemical environment of the Fe3+ cations and no visible changes in the environment for Bi and O. The changes to the XPS spectra for Fe are consistent with dissolution of the Fe within the BFO lattice in the surrounding solution. BFO has been considered as a solid solution of the oxides of Fe and Bi and in this case the Fe–O bonds are the least stable and so most able to corrode. It seems likely that the pathway for photocorrosion is the dissolution and breaking of the Fe–O bonds to effectively etch Fe from the surface of the BFO lattice. While EDS spectra cannot be considered as quantitative results, EDS obtained during SEM analysis does show a drop in Fe atomic% of 3.5% across an average of 5 samples after illumination. The atomic% of Bi and O increased to compensate for the drop in Fe content.
The removal of Fe from the lattice would leave a surface that is highly defected at a crystallographic level and also induce a number of trap states into the band structure. The surface of the material would also no longer exhibit ferroelectric properties, while it could be presumed that the bulk region would still do so. This would change the interaction between the BFO and the dye during exposure. Such changes would have an impact on the surface photochemistry as is well known for the various polymorphs of TiO2.
The reduction in reaction rate for the decolourisation of the dye is therefore associated with the photocorrosion of the BFO. During reaction chemisorbed dye, or secondary species, molecules inject holes into the valance band of the BFO. This disrupts the Fe–O bond structure and causes the BFO to photocorrode. The surface of the BFO then becomes less suitable for the production of reactive species; hence the rate of decolourisation slows.
Nanostructured BFO had shown promise as a photochemical reagent under visible light, an experimental method was developed to mitigate the photocorrosion problem of BFO and determine the extent to which BFO could be used in a photochemical reaction to decolourise a typical dye compound. A series of dye decolourisation experiments were conducted with replacement of the BFO nanopowder at regular intervals, every minute, into the dye solution. The results, shown in Fig. 8, indicate that decolourisation of RhB is at more than 95% after 10 min of illumination under simulated solar irradiation with BFO at pH 2. This compares to 30 min for 90% decolourisation of RhB with TiO2. The decolourisation performance for BFO at pH 6.7 shows that the pH 6.7 solution has a performance closely matched to TiO2. The differences in the performance for BFO with pH have been explained earlier and stem from the interaction of reactive species with the BFO.
 | ||
| Fig. 8 Photodecolourisation of RhB over BiFeO3 at pH 2 and 6.7 under AM1.5 illumination. Decolourisation at pH 2 shows greater than 95% decolourisation after 10 min, the inset shows the decolourisation of RhB over nanostructured (Degussa P25) TiO2 for comparison. | ||
Conclusions
In this work we have demonstrated that BFO nanoparticles can be synthesized using an autocombustion method and that these particles have a measurable magnetization typical of thin films and an optical band gap around 2.1 eV. We explain the photochemical behaviour of BFO in relation with the ferroelectric nature of the material, and also explain the non-photostablility of BFO under illumination which is attributed to photocorrosion. BFO produced using this technique is not photostable and photocorrodes through the dissolution of Fe from Fe–O bond in the solid solution; the Bi2O3 component remaining largely unchanged as shown by XPS analysis. This dissolution is also evidenced by EDS studies on BFO powders before and after the photodecolourisation experiment. We explain the photocorrosion effect of the BFO in aqueous RhB dye in terms of the band positions of our BFO with respect to the band position of the RhB dye, where holes can be injected from RhB dye HOMO level to BFO valence band. It was shown that BFO is able to photodecolourise RhB dye under visible light simulated by solar simulator with AM = 1.5, and work best under pH 2 environment where negative charge co-ions screening on the BFO particle surface is minimum (iso-electric point). It was also observed that there is a trend of photodecolourisation rate slowdown in all pH values. The slowdown of decolourisation rate of the RhB dye was caused by the photocorrosion of the BFO and is the limiting feature for the decolourisation of the dye. We have proven with an experiment by replacing BFO at regular intervals then decolourisation of the dye proceeds at a rate exceeding titania, and it is indeed a promising material for visible-light driven photochemistry.References
- A. Fujishima and K. Honda, Nature, 1972, 37, 238 Search PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr., Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- S. George, S. Pokhrel and A. E. Nel, J. Am. Chem. Soc., 2011, 133, 11270–11278 CrossRef CAS.
- V. Pleskov, Semiconductor Photochemistry, Springer , 1986, ISBN: 978-0306109836 Search PubMed.
- P. Jones, D. E. Gallardo and S. Dunn, Chem. Mater., 2008, 20, 5901–5906 CrossRef CAS.
- K. Saito, K. Koga and A. Kudo, Dalton Trans., 2011, 40, 3909–3913 RSC.
- M. Stock and S. Dunn, J. Phys. Chem. C, 2012, 116, 20854–20859 CAS.
- E. Gutmann, A. Benke, K. Gerth, H. Böttcher, E. Mehner, C. Klein, U. Krause-Buchholz, U. Bergmann, W. Pompe and D. C. Meyer, J. Phys. Chem. C, 2012, 116, 5383–5393 CAS.
- M. B. Starr, J. Shi and X. Wang, Angew. Chem., 2012, 124(6), 6064–6068 Search PubMed.
- N. C. Carville, M. Manzo, S. Damm, M. Castiella, L. Collins, D. Denning, S. A. L. Weber, K. Gallo, J. H. Rice and B. J. Rodriguez, ACS Nano, 2012, 6, 7373–7380 CrossRef CAS.
- J. R. Teague, R. Gerson and W. J. James, Solid State Communications, 1970, 8(13), 1073–4 CrossRef CAS.
- I. Sosnowskat, T. Peterlin-Neumaier and E. Steichele, J. Phys. C: Solid State Phys., 1982, 15(23), 4835–46 CrossRef.
- D. Lebeugle, D. Colson, A. Forget and M. Viret, Appl. Phys. Lett., 2007, 91(2), 022907 CrossRef.
- S. M. Wu, S. A. Cybart, P. Yu, M. D. Rossell, J. X. Zhang, R. Ramesh and R. C. Dynes, Nat. Mater., 2010, 9(9), 756–761 CrossRef CAS.
- T. Choi, S. Lee, Y. J. Choi, V. Kiryukhin and S.-W. Cheong, Science, 2009, 5923, 324, 63–66 Search PubMed.
- F. Gao, X. Chen, K. Yin, S. Dong, Z. Ren, F. Yuan, T. Yu, Z. Zou and J.-M. Liu, Adv. Mater., 2007, 19(19), 2889–92 CrossRef CAS.
- S. Dunn, P. M. Jones and D. E. Gallardo, J. Am. Chem. Soc., 2007, 129(28), 8724–8728 CrossRef CAS.
- D. Tiwari and S. Dunn, J. Mater. Sci., 2009, 44(19), 5063–5079 CrossRef CAS.
- F. Gao, Y. Yuan, K. F. Wang, X. Y. Chen, F. Chen and J.-M. Liu, Appl. Phys. Lett., 2006, 89, 102506–8 CrossRef.
- Shun Li, Yuan-Hua Lin, Bo-Ping Zhang, Ce-Wen Nan and Yao Wang, J. Appl. Phys., 2009, 105, 056105 CrossRef.
- C. Madhu, B. Manjunath, Belakki and V. Manivannan, Indian J Eng. Mater. Sci., 2010, 17, 131–139 CAS.
- M. Schultz Andrew, Yiling Zhang, Paul A. Salvador and Gregory S. Rohrer, ACS Appl. Mater. Interfaces, 2011, 3, 1562–1567 Search PubMed.
- R. Guo, L. Fang, W. Dong, F. Zheng and M. Shen, J. Phys. Chem. C, 2010, 114, 21390–21396 CAS.
- J. F. Scott, Ed, Ferroelectric memories, Springer, 2000 Search PubMed.
- S. Dunn, D. Cullen, E. Abad-Garcia, C. Bertoni, R. Carter, D. Howorth and R. W. Whatmore, Appl. Phys. Lett., 2004, 85, 3537 CrossRef CAS.
- J. Garra, J. M. Vohs and D. A. Bonnell, Surf. Sci., 2009, 603, 1106 CrossRef CAS.
- J. L. Giocondi and G. S. Rohrer, J. Phys. Chem. B, 2001, 105, 8275 CrossRef CAS.
- A. Harhira, L. Guilbert, P. Bourson and H. Rinnert, Phys. Status Solidi C, 2007, 4, 926 CrossRef CAS.
- P. M. Jones and S. Dunn, J. Phys. D, 2009, 42 Search PubMed.
- S. Dunn and D. Tiwari, Appl. Phys. Lett., 2008, 93 Search PubMed.
- P. M. Jones, D. E. Gallardo and S. Dunn, Chem. Mater., 2008, 20, 5901 CrossRef CAS.
- S. Dunn, P. M. Jones and D. E. Gallardo, J. Am. Chem. Soc., 2007, 129, 8724 CrossRef CAS.
- S. Dunn, D. Tiwari, P. M. Jones and D. E. Gallardo, J. Mater. Chem., 2007, 17, 4460 RSC.
- P. M. Jones and S. Dunn, Nanotechnology, 2007, 18 Search PubMed.
- J. L. Giocondi and G. S. Rohrer, Photochemical reduction and oxidation reactions on barium titanate surfaces, presented at Materials Research Society Symposium – Proceedings, 2001 Search PubMed.
- J. L. Giocondi and G. S. Rohrer, Chem. Mater., 2001, 13, 241 CrossRef CAS.
- J. Xu, G. Zhang, F. Li, X. Zhang, Q. Sun, S. Liu, F. Song, Y. Kong, X. Chen, H. Qiao, J. Yao and Z. Lijuan, Opt. Lett., 2000, 25, 129 CrossRef CAS.
- K. Takahashi, N. Kida and M. Tonouchi, Phys. Rev. Lett., 2006, 96, 117402 CrossRef.
- G. Smolenskii and I. Chupis, Sov. Phys. Usp., 1982, 25, 475 CrossRef.
- T.-J. Park, et al. , Nano Lett., 2007, 7, 766 CrossRef CAS.
- W. Eerenstein, F. D. Morrison, J. Dho, M. G. Blamire, J. F. Scott and N. D. Mathur, Science, 2005, 307, 1203a CrossRef.
- T.-J. Park, et al. , Nano Lett., 2007, 7, 766 CrossRef CAS.
- S. Dunn, D. Cullen, E. Abad-Garcia, C. Bertoni, R. Carter and R. W. Whatmore, Appl. Phys. Lett., 2004, 85(16), 3537–3539 CrossRef CAS.
- M. Popa, S. Preda, V. Fruth, K. Sedlácková, C. Balázsi, D. Crespo and J. M. Calderón-Moreno, Thin Solid Films, 2009, 517, 2581–2585 CrossRef CAS.
- T. Schedel-Niedrig, W. Weiss and R. Schlogl, Phys. Rev., 1995, B 52, 17449 Search PubMed.
Footnotes |
| † Electronic Supplementary Information (ESI) available: Scanning Electron Micrograph (SEM) image of BiFeO3 nanopowders, histogram of the particle size distribution analysed from TEM image, Zeta potential measurement results of BiFeO3 powder at various pH values, schematic illustration of BFO ferroelectric surface and its interaction with RhB dye molecules, the results of RhB dye molecules adsorption on BFO particle surface at various pH values, and XRD patterns of BiFeO3 before and after illumination are presented. See DOI: 10.1039/c2ra22211f |
| ‡ X.M. acknowledges support from the Herchel Smith Fellowship fund. |
| This journal is © The Royal Society of Chemistry 2012 |