Relative and inherent reactivities of imidazolium-based ionic liquids: the implications for lignocellulose processing applications†
Alistair W. T.
King
*a,
Arno
Parviainen
a,
Pirkko
Karhunen
a,
Jorma
Matikainen
a,
Lauri K. J.
Hauru
b,
Herbert
Sixta
b and
Ilkka
Kilpeläinen
*a
aDepartment of Chemistry, University of Helsinki, FI-00014, Helsinki, PO Box 55 (A. I. Virtasen Aukio 1), Finland. E-mail: alistair.king@helsinki.fi; ilkka.kilpelainen@helsinki.fi; Tel: +358 (0)50527 9446
bDepartment of Forest Products Technology, Aalto University, FI-00076, Espoo, PO Box 16300 (Vuorimiehentie 1 A), Finland. Tel: +358 (0)94702 4201E-mail: herbert.sixta@aalto.fi
First published on 27th June 2012
Abstract
Novel methods for the fractionation of wood, as a major renewable chemical and material feedstock, are in demand. Ionic liquids, such as 1-ethyl-3-methylimidazolium acetate ([emim][OAc]), are promoted as potential media for these processes. However, the chemical stabilities of such ionic liquids are in question as they may have an effect on process sustainability or efficiency. With anion nucleophilicity and basicity being implicated more in ionic liquid reactivity, a rough scale of the relative reactivities for [emim]-based ionic liquids is demonstrated, based upon their TGA decomposition temperatures. These values are compared to the proton affinities for the anions of those ionic liquids, as a crude measure of nucleophilicity or basicity. The implications for the temperature-dependent chemical stability of imidazolium-based ionic liquids are discussed, in regard to their interactions with wood biopolymers. It is observed that for ionic liquids with less diffuse anions (more nucleophilic or basic), such as [emim][OAc], they unfortunately become more unstable. This is exhibited by a decrease in the thermal stability and an increase in the degree of interaction with the biomass, to the point of better solvation and even covalent interactions with dissolved components. The ab initio proton affinities, dipole moments, van der Waals surface area, and volumes, are presented for an extended series of anions, commonly used in ionic liquids.
Introduction
There are now several important recent literature reports detailing the nucleophilicities of species, in ionic liquid media.1–4 They all describe, via kinetic studies, the relative reactivity of nucleophiles in common ionic liquids, such as the non-basic 1-ethyl-3-methylimidazolium bistriflimide ([emim][NTf2]) or 1-ethyl-3-methylimidazolium triflate ([emim][OTf]). An article by Ranieri et al.2 successfully employs Kamlet–Taft parameters, for molecular solvents and ionic liquids, in the prediction of the nucleophilicity of primary, secondary and tertiary amines, using a LSER (linear solvation energy relationship) approach. An article by Landini and Maia4 describes the relative nucleophilicity of additional anions in a short series of ionic liquids and molecular solvents. An article by Bini et al.3 identified counter-intuitive reactivity, when considering the relative SN reactivity of Br−vs. [NTf2]− toward an aryldiazonium salt. In this case, [NTf2]− was found to be more reactive than Br−. Hallett et al.1 even describes an ‘ionic effect’, in comparison to molecular solvents, when studying the reaction of Cl− with a dialkylarylsulfonium salt. Both of these reported phenomena were deemed to be as a result of strong vs. weak ion pairing in solution. These types of reports are fundamental to the understanding of ionic liquid reactivity, with a range of solutes. The ‘effects’ described by individual reports are however isolated to the particular kinds of reactivity at play, whether it be SN1, SN2 or other types of chemical reactivity. They may also be dependent on the differing degrees of the solubility of the solutes in the different classes of ionic liquid. From our own research application perspective, which concerns the processing of lignocellulosics with ionic liquids, the reactivity of different kinds of species in ionic liquid media most certainly has implications for the sustainability and efficiency of prospective processes. Ionic liquids are now well known solvents for cellulose5–8 and are now also known to be capable of solvating wood to a high degree.9–11 However, four reports have surfaced which appear to describe the selective reactivity of ionic liquids, such as 1-ethyl-3-methylimidazolium acetate ([emim][OAc]), with lignocellulosic solutes. Publications by Liebert and Heinze 12 and Ebner et al.13 describe the potential for the reaction of the imidazolium C2 with the C1 reducing end groups in cellulose. This is likely to be dependent on the basicity of the anion in solution, catalysing the reaction via an initial deprotonation step at C2–H. A publication by Çetinkol et al.14 describes the deacetylation of xylan residues and the simultaneous acetylation of lignin residues when Wiley milled Eucalyptus globulus wood was treated with [emim][OAc] at 120 °C for 3 h. Hauru et al.15 also described the deacetylation of residues in Birch after treatment with [emim][OAc] at 100 °C for 30 h. This selective reactivity may occur via a hydrolysis mechanism or it could also be considered to occur via a transacetylation mechanism, involving an acetic anhydride intermediate or acetylated carbene intermediate. The reactivity in question for both sets of publications suggests the potential involvement of the acetate anion in a nucleophilic or basic capacity (Scheme 1). Revealing the abilities of ionic liquids, in this regard, is therefore an important step for determining their applicability for the processing of complex materials, such as lignocellulosics. This reactivity may also be considered to be beneficial in the area of wood fractionation as some native or artificial lignin-carbohydrate complexes (LCCs) are considered to be labile linkages, such as esters or p-hydroxybenzyl ethers (or esters).16![The common mechanisms for the decomposition of [emim][OAc], reliant upon the basicity or nucleophilicity imparted by the acetate anion.](/image/article/2012/RA/c2ra21287k/c2ra21287k-s1.gif) | ||
| Scheme 1 The common mechanisms for the decomposition of [emim][OAc], reliant upon the basicity or nucleophilicity imparted by the acetate anion. | ||
In addition to the covalent interactions of ionic liquids with lignocellulosics, depending on the basicity of the anions, cellulose and wood themselves are commonly reported to be soluble in the more basic of the ionic liquids.5,6,11 This however is reported to be a non-covalent interaction, whereby the ionic liquid can simultaneously break the hydrogen-bonds between the cellulose chains, in the substrate, and stabilize the polymer in solution.
As a result of the above considerations, we suggest the comparison of thermogravimetric analysis (TGA) decomposition temperatures, for neat ionic liquids, as a relative scale of inherent reactivity for certain ionic liquids. Proton affinities, as a measure of diffusivity and a crude measure of basicity or nucleophilicity, can also be used to extend the stability scale. In this respect, it should be emphasised that ab initio calculations are gas-phase calculations and that by comparing gas-phase vs. solution-phase, only approximate correlations are achievable. Nucleophilicity is also not likely to be accurately predictable by a simple parameter, such as the proton affinity. In addition to this, certain ionic liquids will decompose via alternative mechanisms to an SN2 mechanism. This becomes even more complicated as you introduce additional solutes. However, the point remains that relative thermal decomposition temperatures, for neat ionic liquids, may offer an indication of their reactivity with alternative solutes, such as those derived from wood, whether it be via an SN2, base-catalysed or an alternative mechanism. This reactivity is correlated to the diffusivity of the anions, for similar cations.
Experimental
Materials and methods
1-Ethyl-3-methylimidazolium acetate ([emim][OAc], >95%), 1-ethyl-3-methylimidazolium nitrate ([emim][NO3], >98%), 1-ethyl-3-methylimidazolium bromide ([emim]Br, 99%), 1-ethyl-3-methylimidazolium iodide ([emim]I, >98%), 1-ethyl-3-methylimidazolium tosylate ([emim][OTs], 99%) and 1-ethyl-3-methylimidazolium trifluoroacetate ([emim][CF3CO2], >97%) were purchased from IoLiTec GmbH and used without further purification. 1-Ethyl-3-methylimidazolium ethylsulphate ([emim][EtSO4], 99%), 1-ethyl-3-methylimidazolium bis(trifluoromethylsulphonyl)imide ([emim][NTf2], 99%) was provided by Solvent Innovation GmbH and were used without further purification. 1-Ethyl-3-methylimidazolium chloride ([emim]Cl, ‘for synthesis’), 1-ethyl-3-methylimidazolium thiocyanate ([emim][NCS], ‘for synthesis’) and 1-ethyl-3-methylimidazolium tris(pentafluoroethyl)trifluorophosphate ([emim][FAP], ‘high purity’) were provided by Merck KGaA and used without further purification. Dimethylphosphite, methyl methanesulphonate, methyl trifluoromethanesulphonate, silver acetate and trimethylphosphate were purchased from Aldrich and used without further purification.[emim][OAc] was vacuum distilled using both a sublimation apparatus and a Büchi Kugelrohr attached to an oil pump and a turbo pump (see the ESI†).
TGAs were consistently recorded on 10–15 mg of sample with a 10 °C min−1 heating rate (see the ESI†).
Proton affinities were determined using GAMESS17 2009 compiled for 64-bit linux. Geometries were optimised and zero-point energies were calculated using MP2/6-311+G(d,p)//MP2/6-311+G(d,p) (see the ESI†).
Ionic liquid syntheses
1-Ethyl-3-methylimidazolium nitrite ([emim][NO2]), was synthesised to high purity by anion metathesis using [emim]Cl and silver nitrite. The water content was determined to be negligible by NMR (see the ESI†). The chloride and silver content was determined to be negligible by ion chromatography and argentometry (see the ESI†). 1-Ethyl-3-methylimidazolium methanesulphonate ([emim][OMs]), 1-ethyl-3-methylimidazolium trifluoromethanesulphonate ([emim][OTf]), 1-ethyl-3-methylimidazolium dimethylphosphate ([emim][Me2PO4]) and 1-ethyl-3-methylimidazolium methylhydrogenphosphonate ([emim][MeHPO3]) were synthesised to high purity by the quaternisation of 1-ethylimidazole with the respective electrophiles (see the ESI†). The water content was 1% less than by NMR.Results and discussion
Relative ionic liquid nucleophilicity/basicity/diffusivity
It is now well documented that the most basic dialkylimidazolium-based ionic liquids can thermally decompose via the SN2 attack of the ionic liquid anion at the alpha-positions on the imidazolium rings (similar to that depicted for [emim][OAc] in Scheme 1).18–20 This has been observed to occur for dialkylimidazolium bromides and chlorides, such as 1-allyl-3-methylimidazolium chloride ([amim]Cl); a common ionic liquid for lignocellulose processing, and now [emim][OAc] (see later in the text). Similarly, it has been experimentally shown that C2–H is also susceptible to deprotonation with basic ionic liquids.21,22 A publication by Hollóczki et al.21 has also demonstrated that at low enough pressures, in the molecular distillation regime, [emim][OAc] will vaporize to give the C2 deprotonated 1-ethyl-3-methylimidazol-2-ylidene (carbene) and acetic acid species. This is a direct result of the basicity of the acetate anion in the gas-phase. It is uncertain whether the initial deprotonation occurs in the neat ionic liquid, at the phase-boundary or in the gas-phase starting from a single ion pair. As mentioned previously however, carbenes also react with lignocellulosic solutes in the solution phase.12,13 This is emphatically exemplified in a publication by Rodríguez et al.,22 whereby elemental sulphur (electrophilic) reacts with neat [emim][OAc], even at 25 °C, to form 1-ethyl-3-methylimidazole-2-thione. In other cases, where the ionic liquids contain more diffuse anions, TGA suggests that decomposition is also occurring via alternative mechanisms.For the comparison of the thermal decomposition of different imidazolium-based ionic liquids, of different anions but the same cation, a series of [emim]-based ionic liquids were analysed using TGA. In most cases the samples were commercial and included [emim][OAc], 1-ethyl-3-methylimidazolium chloride ([emim]Cl), 1-ethyl-3-methylimidazolium thiocyanate ([emim][NCS]), 1-ethyl-3-methylimidazolium nitrate ([emim][NO3]), 1-ethyl-3-methylimidazolium bromide ([emim]Br), 1-ethyl-3-methylimidazolium iodide ([emim]I), 1-ethyl-3-methylimidazolium ethylsulphate ([emim][EtSO4]), 1-ethyl-3-methylimidazolium tosylate ([emim][OTs]), 1-ethyl-3-methylimidazolium trifluoroacetate ([emim][CF3CO2]), 1-ethyl-3-methylimidazolium tris(pentafluoroethyl)trifluorophosphate ([emim][FAP]) and [emim][NTf2]. 1-Ethyl-3-methylimidazolium nitrite ([emim][NO2]) was synthesised by anion metathesis from [emim]Cl (see the ESI†). 1-Ethyl-3-methylimidazolium dimethylphosphate ([emim][Me2PO4]), 1-ethyl-3-methylimidazolium methylhydrogenphosphonate ([emim][MeHPO3]), 1-ethyl-3-methylimidazolium mesylate ([emim][OMs]) and [emim][OTf] were synthesised by the quaternisation (Menschutkin reaction) of 1-ethylimidazole with the corresponding methylating reagent (dimethylphosphite, trimethylphosphate, methylmesylate or methyltriflate, see the ESI†). The TGA method used a relatively slow heating rate (10 °C min−1) to ensure more accurate decomposition temperatures were obtained. The results were separated into two classes: (1) those, which exhibit a relatively uniform decomposition curve, consistent with SN2 decomposition (although not necessarily indicative of, Fig. 1a), and (2) those, which exhibit a multi-step decomposition or obvious polymerization (Fig. 1b).
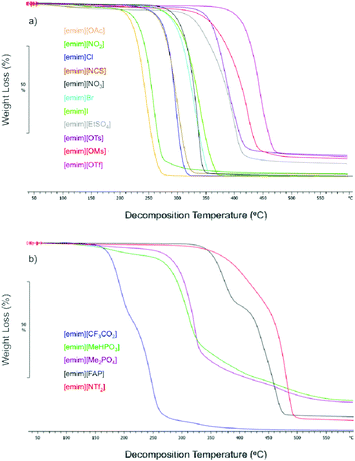 | ||
| Fig. 1 TGA traces for ionic liquids giving uniform decomposition rate (a) and multi-step decomposition or clear polymerization products (b). | ||
From the TGA traces in Fig. 1a, the sulphate or sulphonate-based ionic liquids show an obvious polymeric residue, in comparison to the other structures. It is likely that the sulphates and sulphonates are not decomposing via an SN2 mechanism, as the major mechanism, although further experimentation is necessary for each structure. For these structures, the release of smaller fragments such as SO2 or SO3 would be entropically favourable. The rest of the structures, decomposing at lower temperatures, in Fig. 1a do not have that mode of decomposition and are all known as good nucleophiles in polar aprotic molecular solvents. Based on existing literary evidence, it is unknown if [emim][OAc] will predominantly decompose via an SN2 mechanism at atmospheric pressure.
Predicting diffusivity and basicity
As a measure of anion basicity, proton affinities (ΔHPA, MP2/6-311+G(d,p)//MP2/6-311+G(d,p)) were calculated for all anions of the ionic liquids, in Fig. 1a, and the [NTf2]− anion. These values are tabulated along with the temperatures at which the maximum rate of decomposition is observed (Tk(max)) for each ionic liquid23 (Table 1). A reasonably strong correlation between the ΔHPA and Tk(max) values is evident (Fig. 2).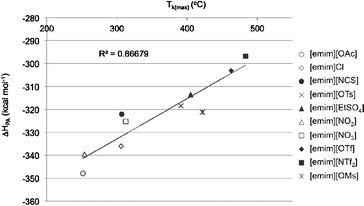 | ||
| Fig. 2 Correlation between the proton affinities (ΔHPA) and maximum rate of decomposition temperatures (Tk(max)) for a series of ionic liquids and corresponding anions. | ||
| Species Name | Common Species Abbreviation | ΔHPA (kcal mol−1) | Ionic Liquid Tk(max) (°C) | Anion Dipole Moment (D)a | VDV Surface Area/Volume (Å2 Å−3) a | Cellulose Solvation ([emim]+)b | ||
|---|---|---|---|---|---|---|---|---|
| a MP2/6-311+G(d,p)//MP2/6-311+G(d,p). b Dissolution scale: **** (highly effective) to * (slight dissolution) to — (not effective or not reported at all). c B3-LYP/6-31+G(d)//MP2/aug-cc-pVTZ.24 d Br and I are not supported for this basis set. e Value calculated for the benzenesulphonate anion (comparative to tosylate). f Protonated anions dissociate in the gas-phase, at this level.34 g Tris(perfluoromethyl)trifluorophosphate. | ||||||||
| 6311+G(d,p)a | 6311+G(d,p)a | aug-cc-pVTZc | ||||||
| [Absolute Values]a | [Relative to H2O] | [Relative to H2O] | ||||||
| Hydroxide | [OH]− | −391.28 | −225.67 | — | — | 1.55 | 28.24/14.77 | Not Synthesised |
| Methoxide | [MeO]− | −383.68 | −218.07 | −216.25 | — | 2.28 | 33.96/22.87 | Not Synthesised |
| Ethoxide | [EtO]− | −380.10 | −214.49 | — | — | 3.82 | 44.01/33.29 | Not Synthesised |
| Fluoride | F− | −371.39 | −205.78 | −205.53 | — | 0.00 | 22.90/10.31 | Not Synthesised |
| Methylacetamide | [AcMeN]− | −361.58 | −195.97 | — | — | 0.99 | 58.28/46.72 | Not Synthesised |
| Methylmesylamide | [MsMeN]− | −350.53 | −184.92 | — | — | 2.17 | 67.89/56.76 | Not Synthesised |
| Guaiacolate (Phenolate) | — | −348.44 | −182.83 | — | — | 3.45 | 82.10/71.34 | Not Synthesised |
| Acetate | [OAc]− | −347.96 | −182.35 | −181.93 | 251.5 | 3.45 | 52.39/39.48 | ****28 |
| Propionate | [CO2Et]− | −347.34 | −181.73 | — | — | 5.06 | 58.23/47.72 | ****29 |
| Butyrate | [CO2Pr]− | −346.69 | −181.08 | — | — | 7.28 | 63.17/54.45 | ****29 |
| Formate | [CO2H]− | −343.89 | −178.28 | — | — | 1.39 | 44.14/29.31 | ****29 |
| Nitrite | [NO2]− | −339.56 | −173.95 | — | 253.5 | 0.24 | 46.17/29.27 | — |
| Methylcarbonate | [MeCO3]− | −338.89 | −173.28 | — | — | 4.35 | 57.25/45.11 | — |
| Chloride | Cl− | −336.06 | −170.45 | −168.96 | 306.0 | 0.00 | 41.17/24.84 | ****30 |
| Phosphinate | [H2PO2]− | −333.74 | −168.13 | — | — | 2.29 | 53.34/37.89 | ****31 |
| Methyltriflimide | [TfMeN]− | −332.41 | −166.80 | — | — | 4.04 | 75.86/66.35 | Not Synthesised |
| Dimethylphosphate | [Me2PO4]− | −330.66 | −165.05 | — | — | 4.69 | 73.96/62.80 | ****6 |
| Methylphosphonate | [MeHPO3]− | −329.70 | −164.09 | — | — | 4.67 | 64.60/51.11 | ****6 |
| Nitrate | [NO3]− | −325.27 | −159.66 | −160.00 | 312.5 | 0.00 | 52.30/35.00 | — |
| Dimethylthiophosphate | [Me2PO3S]− | −324.04 | −158.43 | — | — | 5.01 | 78.12/69.04 | ****32 |
| Trifluoroacetate | [CF3CO2]− | −322.95 | −157.34 | −159.31 | — | 4.92 | 61.42/47.33 | — |
| Thiocyanate | [NCS]− | −322.03 | −156.42 | — | 306.5 | 1.47 | 60.63/43.32 | — |
| Mesylate | [OMs]− | −321.18 | −155.57 | −154.56 | 422.0 | 3.80 | 61.73/50.02 | — |
| Bromide | Br− | — d | — d | — | 338.0 | — d | — d | *33 |
| Iodide | I− | — d | — d | — | 344.5 | — d | — d | — |
| Tosylate | [OTs]− | −318.50 | −152.89 | −150.90e | 391.0 | 10.22 | 98.86/86.36 | *33 |
| Ethylsulfate | [EtSO4]− | −313.56 | −147.95 | — | 405.0 | 5.31 | 71.41/63.72 | — |
| Methylsulfate | [MeSO4]− | −313.22 | −147.61 | — | — | 3.72 | 65.45/52.60 | — |
| Dicyanamide | [N(CN)2]− | −307.68 | −142.07 | −141.02 | — | 0.89 | 69.32/51.08 | — |
| Tricyanomethanide | [C(CN)3]− | −304.68 | −139.07 | −136.64 | — | 0.01 | 88.65/69.28 | — |
| Triflate | [OTf]− | −303.12 | −137.51 | −135.81 | 462.5 | 4.22 | 69.44/55.80 | — |
| Bistriflimide | [NTf2]− | −296.82 | −131.21 | −129.62 | 483.0 | 0.25 | 104.57/98.44 | — |
| Tetrafluoroborate | [BF4]− | −294.79f | −129.18 | — | — | 0.02 | 57.05/41.54 | — |
| Hexafluorophosphate | [PF6]− | −282.08f | −116.47 | — | — | 0.01 | 52.66/41.21 | — |
| FMPg | [PF3(CF3)3]− | −279.98f | −114.37 | — | — | 1.07 | 81.20/79.83 | — |
| Bis(oxalato)borate | [(C2O4)2B]− | −271.94 | −106.33 | — | — | 0.01 | 107.92/94.69 | — |
| Tetrachloroaluminate | [AlCl4]− | −265.57f | −99.96 | — | — | 0.02 | 113.99/96.05 | — |
| Tetracyanoborate | [B(CN)4]− | −263.01 | −97.40 | 0.00 | 105.43/88.89 | |||
| [emim]-Carbene | [emim]: | −262.90 | −97.27 | — | — | 2.62 | 75.71/67.98 | — |
| Water | H2O | −165.61 | 0 | — | — | 2.19 | 25.30/13.79 | — |
A thorough study into the validation of the levels of theory, for the calculation of the anion proton affinities, has been completed by Izgorodina et al.24 In this report the authors use proton affinities as a measure of diffusivity, a more general concept describing the distribution of the electron density throughout the anion. The results indicate that high levels of theory, at least MP2 or CCSD(T), in combination with heavily diffuse basis sets (e.g. aug-cc-pVTZ) is important to achieve a high degree of accuracy. The use of diffuse basis sets at the MP2 level was found to be more important than using the computationally intensive CCSD(T). In regards to this publication, the values calculated here, using a lower quality basis set, were sufficient to observe a relatively healthy correlation between ΔHPA and Tk(max) temperatures. Mihichuk et al.25 have also used gas-phase proton affinities as a crude measure of basicity but note that these values are not equivalent to the basicities in the condensed phases. It is also important to note that the observed correlation between ΔHPA and Tk(max) is not proof of SN2 decomposition for all the ionic liquids in Table 1. However, at least we can say that for the ionic liquids containing anions of lower basicity (more diffuse), they are not nucleophilic enough to decompose via an SN2 mechanism, as opposed to the predominant mechanism, and thus fit the correlation to some degree. This issue would make it pointless to attempt to refine the correlation using alternative levels of theory and basis set, until the correct decomposition mechanisms are determined for each ionic liquid and for a wider set of structures, in general.
Predicting nucleophilicity
Predicting nucleophilicity from a simple set of parameters is a much more complicated task than the general concept of diffusivity or reactivity arising from basicity. Beyond a simple correlation with Tk(max) this is too difficult a task to claim within this publication. With ionic liquids a major difficulty is finding an accurate experimental dataset for correlation. A framework for determining the activation energies of SN2 substitution at alpha-positions, in imidazolium rings, has been laid out in an article by Hao et al.20 This involves measuring the rates of decomposition under isothermal conditions, and at several temperatures, and calculation of pre-exponential factors and activation energies from the rearrangement of the Arrhenius equation. If one were able to calculate the activation energies for all the imidazolium-based ionic liquids, the comparison of the activation energies would offer a better alternative to the relative scale of reactivity presented in this article. This could also reflect differences in cations, for the same anion. Picot et al.26 have demonstrated that ΔHPA values for thiolate ligands are proportional to the activation energies of reaction of their complexes with zinc (zinc–thiolate complexes) against the electrophile methyl iodide. If this is the case, a further refinement to our ab initio calculations might be able to calculate a range of affinities for different functionalities. This approach was adopted by Streitwieser,27 in the correlation of rate constant logarithms, with thermodynamic heats of methylation and protonation. The relevant calculations for lignocellulosic solutes should at least include enthalpies of protonation, methylation and acetylation to reflect the main kinds of electrophiles found in these materials. Differing affinities and correlations with reactivity are justified under the hard–soft acids and bases (HSAB) context of reactivity and may offer more accurate predictions of reactivity towards certain labile functionalities implicated in wood solubility or fractionation efficiency, in addition to the full array of organic reactivity.In relation to this work, by comparison with the proton affinity values, commonly used as a measure of basicity, the TGA decomposition temperatures for the neat ionic liquids can be used as a relative scale of reactivity. The less diffuse the anions are, the lower the temperature required for the thermal decomposition, into volatile species, or potential reaction with solutes. Similarly, for anions of ionic liquids decomposing via alternative mechanisms, the more diffuse the charge is, the more stable the ionic liquid is.
Stability vs. lignocellulose processing efficiency
As we have seen, essentially the less diffuse the charge is, the more nucleophilic or basic the anion is. Therefore, from the moderate to good correlation, demonstrated in Fig. 2, and the strong link between basicity and cellulose dissolution,5,6,11 the following assertion can be made: as the ability of an ionic liquid to dissolve cellulose increases, its thermal stability decreases. Moreover, as the basicity of the anion increases, the ionic liquid starts to chemically react with solutes. Until recently, these issues have been overlooked and in relation to lignocellulose processing, ionic liquids are commonly described as ‘non-derivatising solvents’. However, this concept of nucleophilicity and basicity, arising from non-diffuse anions, may be important for processing applications where the potential solutes contain labile linkages, or reactive functionalities. For example, suberin contains an abundance of acyl functionalities and is reported to be extractable with cholinium-based carboxylates.35 As we have seen previously, cellulose reacts at its reducing end groups with ionic liquids such as [emim][OAc].12,13 In the area of wood processing, native xylans in Wiley milled Eucalyptus globulus have been shown to deacetylate upon treatment with [emim][OAc] (3 h, 120 °C).14 The same has been demonstrated under milder conditions in [emim][OAc] (30 h, 100 °C).15 However, in this deacetylation phenomena, the mechanism has yet to be proven to be via the nucleophilic attack of the anion or another related mechanism. In addition to these reactive lignocellulosic functionalities, that seem to be labile under ionic liquid treatment, wood is also reported to contain LCCs.16 These are present in lignocellulosic materials either natively or occur after some kind of chemical, or perhaps even mechanical, treatment. The presence of LCCs in native wood is a disputed topic due to the difficulty of observing them in the native wood, without some potentially reactive pre-treatment to facilitate analysis. If certain ionic liquids are capable of cleaving LCCs, this could have implications for the overall solubility of wood, the efficiency of fractionation of different wood components and the sustainability of a particular process, using certain nucleophilic and basic ionic liquids. If a neat ionic liquid such as [emim][OAc] were able to cleave LCCs, the efficiency of fractionation of lignin from polysaccharides may actually be significantly higher than for other neat lignocellulose solvating ionic liquids. This is providing that the limits on the efficiency of fractionation mainly depend on LCCs, and not on the bulk diffusion of lignin fragments out of the recalcitrant wood tracheids.Ionic liquid distillation as a method of recycling
[emim][OAc] is now regarded as one of the best, if not the best ionic liquid for the dissolution of cellulose. Quantitative methods for establishing the ability of an ionic liquid to dissolve cellulose, for comparison purposes, are elusive. A more rigorous method would lie within the realm of rheometry. Rheological measurements on cellulose–ionic liquid solutions are however tedious and no broad comparisons have been made across all the different classes of ionic liquids known to dissolve cellulose. [emim][OAc] however is a noticeably superior solvent to most of the competition. Due to its solvating capabilities [emim][OAc] is being promoted for use as a dissolving media for regenerated cellulose applications (e.g. Lyocell) and for wood fractionation.36 It is therefore important that there are efficient methods to recycle the spent ionic liquid after several processing cycles. Distillation would be an obvious choice. BASF have patented a procedure whereby ionic liquids, such as [emim][OAc], are distilled using specialised short-path distillation equipment.37 The typical pressure ranges quoted are 0.05 mbar, which is verging on ‘high-vacuum’ conditions, under which short-path ‘molecular’ distillation occurs. Under these conditions there is a negligible gas-phase pressure and the mean free path of individual molecules rapidly becomes bigger than the ‘short-path’ (distillation apparatus length), even for clusters of molecules. It is not clear however whether molecular distillation is occurring under these conditions. Due to the elevated pressure it is more likely medium-vacuum distillation. To further our understanding of this distillation we repeated the distillation of [emim][OAc] using two different short-path distillation apparatus: (1) a Kugelrohr and (2) a sublimation apparatus containing boiling stones (Fig. 3).![Short-path distillation apparatus; Kugelrohr (a) and sublimation apparatus (b) for the distillation of [emim][OAc].](/image/article/2012/RA/c2ra21287k/c2ra21287k-f3.gif) | ||
| Fig. 3 Short-path distillation apparatus; Kugelrohr (a) and sublimation apparatus (b) for the distillation of [emim][OAc]. | ||
Apparatus 1 (Kugelrohr) achieved pressures of ∼0.3 mbar during the distillation and apparatus 2 (sublimation apparatus) achieved pressures of ∼0.03 mbar during the distillation. In the Kugelrohr, the solution started to boil significantly at ∼170 °C–180 °C. When the material collected in the distillation bulb it was analysed by 1H NMR, a mixture of predominantly 1-methylimidazole, 1-ethylimidazole and traces of [emim][OAc] were found, along with other minor impurities. Despite the TGA curve demonstrating decomposition onset at >200 °C for [emim][OAc], decomposition and vaporization is already beginning to occur quite rapidly at ∼175 °C, under high-vacuum. This is fairly strong evidence for SN2 decomposition, in addition to the already documented C2–H deprotonation occurring even at lower temperatures (even at 25 °C).21,22 However, when the distillation was performed using the sublimation apparatus (∼30 ml volume, Fig. 3b), with boiling stones, slow distillation started to occur even at ∼140 °C and increased in rate up to 160 °C. This temperature was not exceeded and when the distillate was analysed, it was observed to be mainly [emim][OAc]. Despite the BASF patent, to the best of our knowledge, this is the first academic report to describe the distillation of [emim][OAc]. In both cases, splashing of the ionic liquid was observed to occur part way through the vessels, followed by immediate even condensation onto the furthest surfaces. The boiling stones seemed to nucleate the bubbling, suggesting the use of large surface areas with nucleation sites as being the most ideal for the distillation of thermally unstable ionic liquids, such as [emim][OAc]. Under these conditions, this is suggestive of the molecular or vacuum distillation of clusters of molecules. However, vaporization of neutral species into the gas-phase and reformation to give [emim][OAc] cannot be excluded to a certain degree. Indeed, with the molecular distillation of single ion pairs, any of the anions shown in Table 1 should deprotonate the imidazolium species to give [emim]: (1-ethyl-3-methylimidazol-2-ylidene) in the gas-phase. This is now supported in the literature experimentally for the molecular distillation of [emim][OAc].21 It is not however known if this deprotonation would occur in small clusters or on the surface of clusters, where ion pairs are stabilised by the additional solvation from adjacent ion pairs. From this simple experimental setup, the degree to which 1-ethyl-3-methylimidazol-2-ylidene (carbene) and acetic acid is distilling across is unknown. Despite the important result from Rodríguez et al.22 identifying room temperature C2–H deprotonation in the solution-phase, clearly more needs to be done to compare, measure or predict the rates and extents of equilibration (acid–base and nucleophile–electrophile) in solution, for these classes of ionic liquids, over a range of temperatures.
Conclusions
In conclusion, the relative inherent reactivities of imidazolium-based ionic liquids can be predicted from their TGA decomposition temperatures. This relation to lignocellulose processing translates into solvation capabilities and covalent interaction with solutes. The latter may include reactivity resulting from the basicity of the anion, nucleophilicity of the anion or potentially through other decomposition mechanisms. From this study and previous literature accounts, the major pathway for irreversible decomposition, for the most basic ionic liquids, occurs via SN2 attack at the alpha positions on the imidazolium ring. The less basic structures obviously do not decompose via this mechanism, however a correlation between their decomposition temperatures and proton affinities, as a measure of anion diffusivity, still exists. It is also now obvious that the better the ability of an imidazolium-based ionic liquid to dissolve cellulose (and wood), the more unstable it is. In some cases the instability, due to the inherent basicity and potentially nucleophilicity, can allow for reaction of the ionic liquid with lignocellulosic substrates at common processing temperatures. It is important to note however that deacetylation of lignocellulosic functionalities has not yet been proven to occur via a mechanism whereby a moiety derived from the ionic liquid (e.g. acetyl or carbene) acts as a nucleophile towards the acetyl carbonyl. This is crucial to prove/disprove in the future, as this will undoubtedly affect process sustainabilities and likely efficiencies. Recycling of the most unstable ionic liquids, by distillation, is also predicted to be problematic due to their lower decomposition temperatures, with [emim][OAc] rapidly decomposing at ∼170 °C. However, we have demonstrated for the first time in an academic report that it is possible to distil this material to yield a pure ionic liquid.Acknowledgements
The authors wish to acknowledge Forest Cluster Ltd and TEKES (The Finnish Funding Agency for Technology and Innovation) for sponsorship, under the FuBio (Future Biorefinery) project. The authors also wish to acknowledge Dr Jan-Albert van den Berg (Sasol), Dr Ekaterina L. Izgorodina Pas (Monash University) and Dr Gordon W. Driver (Umeå University) for their discussions on the ab initio calculations.References
- J. P. Hallett, C. L. Liotta, G. Ranieri and T. Welton, J. Org. Chem., 2009, 74, 1864–1868 CrossRef CAS.
- G. Ranieri, J. P. Hallett and T. Welton, Ind. Eng. Chem. Res., 2008, 47, 638–644 CrossRef CAS.
- R. Bini, C. Chiappe, E. Marmugi and D. Pieraccini, Chem. Commun., 2006, 897–899 RSC.
- D. Landini and A. Maia, Tetrahedron Lett., 2005, 46, 3961–3963 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS.
- Y. Fukaya, K. Hayashia, M. Wada and H. Ohno, Green Chem., 2008, 10, 44–46 RSC.
- J. Wu, J. Zhang, H. Zhang, J. He, Q. Ren and M. Guo, Biomacromolecules, 2004, 5, 266–268 CrossRef CAS.
- A. W. T. King, J. Asikkala, I. Mutikainen, P. Järvi and I. Kilpeläinen, Angew. Chem., Int. Ed., 2011, 50, 6301–6305 CrossRef CAS.
- I. Kilpeläinen, H. Xie, A. W. T. King, M. Granström, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef.
- A. W. T. King, L. Zoia, I. Filpponen, A. Olszewska, H. Xie, I. Kilpeläinen and D. S. Argyropoulos, J. Agric. Food Chem., 2009, 57, 8236–8243 CrossRef CAS.
- A. Brandt, J. P. Hallett, D. J. Leak and T. Welton, Chem. Commun., 2010, 12, 672–679 Search PubMed.
- T. Liebert and T. Heinze, Bioresources, 2008, 3, 576–601 Search PubMed.
- G. Ebner, S. Schiehser, A. Potthast and T. Rosenau, Tetrahedron Lett., 2008, 49, 7322–7324 CrossRef CAS.
- Ö. P. Çetinkol, D. C. Dibble, G. Cheng, M. S. Kent, B. Knierim, M. Auer, D. E. Wemmer, J. G. Pelton, Y. B. Melnichenko, J. Ralph, B. A. Simmons and B. M. Holmes, Biofuels, 2010, 1, 33–46 Search PubMed.
- L. K. J. Hauru, M. Hummel and H. Sixta, Books of Abstracts, 241st ACS National Meeting, Anaheim, CA, March 27-31, 2011; American Chemical Society: Washington, DC, 2011, CELL-297 Search PubMed.
- T. Koshijima and T. Watanabe, Association between Lignin and Carbohydrates in Wood and Other Plant Tissues, Springer, Heidelberg, 2003 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Mongomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- N. Meine, F. Benedito and R. Rinaldi, Green Chem., 2010, 12, 1711–1714 RSC.
- H. Ohtani, S. Ishimura and M. Kumai, Anal. Sci., 2008, 24, 1335–1340 CrossRef CAS.
- Y. Hao, J. Peng, S. Hu, J. Li and M. Zhai, Thermochim. Acta, 2010, 501, 78–83 CrossRef CAS.
- O. Hollóczki, D. Gerhard, K. Massone, L. Szarvas, B. Németh, T. Veszprémi and L. Nyulászi, New J. Chem., 2010, 34, 3004–3009 RSC.
- R. Rodríguez, G. Gurau, J. D. Holbrey and R. D. Rogers, Chem. Commun., 2011, 47, 3222–3224 RSC.
- T k(max) was obtained by taking the 1st derivative of the TGA trace and using the temperature at which the lowest value on the 1st derivative curve was observed.
- E. I. Izgorodina, M. Forsyth and D. R. MacFarlane, Aust. J. Chem., 2007, 60, 15–20 CrossRef CAS.
- L. M. Mihichuk, G. W. Driver and K. E. Johnson, ChemPhysChem, 2011, 12, 1622–1632 CrossRef CAS.
- D. Picot, G. Ohanessian and G. Frison, C. R. Chim., 2009, 12, 546–553 CrossRef CAS.
- A. Streitwieser Jr., Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 8288–8290 Search PubMed.
- S. Köhler, T. Liebert, M. Schöbitz, J. Schaller, F. Meister, W. Günther and T. Heinze, Macromol. Rapid Commun., 2007, 28, 2311–2317 CrossRef.
- B. Zhao, L. Greiner and W. Leitner, RSC Adv., 2012, 2, 2476–2479 RSC.
- S. Barthel and T. Heinze, Green Chem., 2006, 8, 301–306 RSC.
- M. Abe, Y. Fukaya and H. Ohno, Green Chem., 2010, 12, 1274–1280 RSC.
- M. Hummel, C. Froschauer, G. Laus, T. Röder, H. Kopacka, L. K. J. Hauru, H. K. Weber, H. Sixta and H. Schottenberger, Green Chem., 2011, 13, 2507–2517 RSC.
- J. Vitz, T. Erdmenger, C. Haensch and U. S. Schubert, Green Chem., 2009, 11, 417–424 RSC.
- The authors were made aware of this phenomenon by Dr Ekaterina L. Izgorodina Pas (Monash University) in a private communication.
- H. Garcia, R. Ferreira, M. Petkovic, J. L. Ferguson, M. C. Leitão, H. Q. N. Gunaratne, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Green Chem., 2010, 12, 367–369 RSC.
- N. Sun, X. Jiang, M. L. Maxim, A. Metlen and R. D. Rogers, ChemSusChem, 2011, 4, 65–73 CrossRef CAS.
- K. Massonne, M. Siemer, W. Mormann and W. Leng, WO Pat., 027250, A3, 2009 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: full equipment details, computational details (including optimised geometries) and experimental are included. See DOI: 10.1039/c2ra21287k |
| This journal is © The Royal Society of Chemistry 2012 |