A study of the role of the solvent during magnetite nanoparticle synthesis: tuning size, shape and self-assembly†
Fraser J.
Douglas
a,
Donald A.
MacLaren
*b and
Mark
Murrie
*a
aWestCHEM, School of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Mark.Murrie@glasgow.ac.uk; Fax: +44 (0)141 330 4888; Tel: +44 (0)141 330 4486
bSUPA, School of Physics and Astronomy, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: dmaclaren@physics.org; Fax: +44 (0)141 330 5886; Tel: +44 (0)141 330 4464
First published on 26th June 2012
Abstract
We present a systematic study of the role of the solvent in the autoclave-based decomposition of iron(III) acetylacetonate to synthesise iron oxide nanoparticles. Subtle variations in solvent functionality yield substantial differences in nanoparticle morphology, spanning monodisperse spheres, hexagonal platelets, compound cubes and larger hierarchical structures. Solvents capable of chelation to iron afford the greatest influence over nanoparticle growth, whilst addition of side-chains to the solvent perturb competitive adsorption on growing nanoparticles to provide a new means of morphological control.
Introduction
The synthesis of monodisperse iron oxide nanoparticles (NPs) has received much attention in both data storage and nanomedical applications.1–3 As many properties of a NP are dependent upon its size and shape, direct control over the particle morphology and uniformity is essential. A number of synthetic protocols have been described, including co-precipitation,4 thermal decomposition5 and polyol6 processes, resulting in structures including spheres,7 cubes,8 triangles,9 hexagons,10 octahedra,11 nanodisks,12 nanoprisms,13 rods,14 rings,15 and other, more complex forms.16,17 Many of these morphologies have been rationalised within the LaMer mechanism of monodisperse NP synthesis,18 which divides the nanoparticle growth process into two distinct stages: a rapid burst of nucleation events followed by slower growth and/or aggregation,19 both phases fuelled by the decomposition of a metal-containing precursor.20 A number of refinements to the simple mechanism have subsequently been proposed, including ripening through either a digestive21 or Ostwald22 mechanism, where either large or small particles, respectively, re-dissolve to contribute to the growth of a more monodisperse product. Shape control arises primarily during the growth phase, deriving either from collisions and aggregation of small NP species or from crystallographic-specific kinetics for the adsorption of molecular species on the surfaces of growing NPs. For example, iron oxide NPs grown via thermal decomposition are often stabilised using long-chain fatty acids and amines to minimise inter-particle interactions. Oleylamine shows weak, isotropic binding to iron oxide surfaces23 whereas oleic acid encourages anisotropic growth in the (100) rather than (111) direction:24 fine-tuning their ratio allows a range of shapes to be obtained.25 Nevertheless, there are numerous other influential variables and reaction optimisation remains largely by trial and error.Our aim is to study the often-overlooked influence of the solvent in NP synthesis, since solvents have a number of potentially exploitable characteristics. For example, the solvent boiling point sets the maximum attainable temperature, or pressure in the case of a sealed system, which determines the rate of decomposition of the metal-containing precursor.26 Additionally, solvent viscosity (as determined by the extent of hydrogen bonding and/or molecular weight) will determine mobility and diffusion rates of solvated ions, NP nuclei and larger NPs, thereby influencing nucleation and growth rates and the extent of aggregation, especially in unstirred systems.27 Furthermore, if the solvent has chemical functionality then it may be able to stabilise species through chelation28 or undergo direct reaction, such as acting as a reducing agent through the presence of hydroxyl groups.29 In the present study, we explore the relevance of the above factors in the synthesis of magnetite (Fe3O4) NPs. Although magnetite NPs have been synthesised within a variety of different solvents (such as high boiling point ethers,5 hydrocarbons,11 and polyols6), the rationale for a chosen solvent is not generally detailed and the profound influence of its physicochemical properties can be neglected, particularly when applying the results of one synthesis protocol to develop a protocol for another NP system. Our aim is, therefore, not to ‘improve’ upon the quality of NPs, since at least in terms of the monodispersity of spherical NPs, several excellent protocols have already been described.5,26 However, we will consider a family of related solvents to allow competing physicochemical factors to be distinguished. We demonstrate that surprisingly modest variations in solvent molecular structure – such as the length of an alkyl side-chain – can alter the competitive adsorption of molecular species on the surface of growing NPs and thereby affect NP shape, size, polydispersity and, interestingly, self-assembly.
Experimental
Materials and instrumentation
Oleic acid 90%, oleylamine 70%, 1,2-hexadecanediol (HDD) 90%, benzyl ether (BE) 98%, diethanolamine (DEA) 98.5%, N-methyldiethanolamine (M-DEA) 98.5% and N-ethyldiethanolamine (E-DEA) 98% were purchased from Sigma-Aldrich. Fe(acac)3 97%, 3-methyl-1,3,5-pentane-triol (MPT) 80%, diethylene glycol (DEG) 98% were purchased from Fluka. All chemicals were used without further purification. Transmission electron microscopy (TEM) was performed on either a FEI Tecnai T20 instrument equipped with a LaB6 filament or an FEI Tecnai TF20 instrument fitted with a field emission gun, both operated at 200 keV. Electron energy loss spectroscopy (EELS) was performed using the latter microscope, which is also equipped with a Gatan Enfina spectrometer. TEM samples were prepared by dispersing the sample in hexane and dropping the solution onto an amorphous carbon-coated grid. TEM data were obtained and processed using either Digital Micrograph or IMAGEJ 1.41 software. Throughout this text, particle sizes are quoted as mean ± standard deviation followed by the standard deviation expressed as a percentage of the mean. Magnetic measurements were conducted at room temperature on a Lakeshore vibrating sample magnetometer using the as-obtained powders: these results are presented in the ESI.†Synthesis
We chose to focus on a solvothermal polyol protocol30 and synthesise NPs within an autoclave rather than by the more common reflux-based protocols, since a number of studies have demonstrated autoclave methods to be particularly promising for large-scale NP production: they offer a facile, relatively low-temperature route and use innocuous reagents that would be attractive for a commercially scalable process. In a typical synthesis, surfactant ratios were used as previously reported for production of magnetite NPs.5 Fe(acac)3 (0.5 mmol), 1,2-hexadecanediol (2.5 mmol), oleic acid (1.5 mmol) and oleylamine (1.5 mmol) were mixed in air with 10 mL of one of the solvents listed in Table 1,31,32 within a Teflon-lined stainless steel autoclave. Two heating protocols were adopted, both using a heating rate of 20 °C per minute: either the sample was heated in an oven at 230 °C for 6 hours; or was heated to 110 °C for 4 hours, then 230 °C for 2 hours. We refer to the latter as using a heating hold-step, which is a common protocol in reflux-based syntheses and is used to separate the nucleation and growth phases of the LaMer mechanism. Surprisingly, the use of hold-steps is not often considered in autoclave-based protocols but will be shown to profoundly affect NP morphology. The hold-temperature of 110 °C was chosen to encourage the formation of coordination complexes between the iron precursor and solvent. The maximum temperature of 230 °C was chosen as this is above the decomposition temperature of the Fe(acac)3 precursor, which thermogravimetric analysis (TGA) indicated to be ∼190 °C (see Fig. S1, ESI†). After heating, the reaction was left to cool naturally to room temperature and excess ethanol was added to precipitate the product, which was collected by centrifugation for 10 minutes at 4000 r.p.m. The black precipitate was washed with ethanol and centrifuged at least three times before being air dried at 60 °C overnight. At this temperature no additional oxidation is expected to occur, as conversion from Fe3O4 to γ-Fe2O3 usually requires temperatures above 250 °C.33 We note that the reaction temperature (220 °C) is above the boiling point of two of the solvents chosen (see Table 1), namely DEA (217 °C) and MPT (216 °C), and substantially above the boiling point of any water produced from the condensation of oleylamine and oleic acid.34 In these cases there is expected to be an elevated pressure within the autoclave, which may influence product morphology, as has been observed in other NP systems.35| Solvent | BP (°C) | Structure | Viscosity (mPa s) | Ref. |
|---|---|---|---|---|
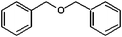
| Benzyl ether (BE) | 298 |
|
— | — |
| Diethylene glycol (DEG) | 245 |
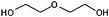
|
35.7 | 31 |
| Diethanolamine (DEA) | 217 |
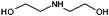
|
379.3 | 32 |
| N-Methyldiethanolamine (M-DEA) | 247 |
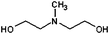
|
57.14 | 32 |
| N-Ethyldiethanolamine (E-DEA) | 252 |
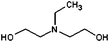
|
48.27 | 32 |
| 3-Methyl-1,3,5-pentanetriol (MPT) | 216 |
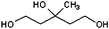
|
— | — |
Results
Representative TEM micrographs and electron diffraction (ED) patterns for NPs synthesised using each solvent shown in Table 1 are presented in Fig. 1 and 2 for protocols either omitting or including a heating hold-step, respectively. Particle size distributions for each experiment are given in Fig. 3. A summary containing particle morphologies and size distributions for each reaction condition is given in Table 2. Experimental reproducibility was confirmed through the similarity of products obtained from reactions that were repeated 2–3 times. ED patterns and lattice fringe spacings observed in individual nanoparticles are consistent with a cubic (inverse) spinel structure with measured lattice constants of 8.379 ± 0.055 Å across the data sets, in agreement with the structure of magnetite (JCPDS index card number 19-629). As the diffraction patterns of maghemite (γ-Fe2O3) and magnetite (Fe3O4) are very similar, O–K and Fe–L2,3 EELS edge profiles were used to assess the Fe oxidation state and confirmed the assignment of magnetite (see Fig. 4). That we obtained black powders in each instance also suggests that magnetite was formed, since nanoscale maghemite tends to be brown. There was one exceptional case where hematite, α-Fe2O3, is formed, as discussed below. From the figures it is apparent that particle size, shape and homogeneity vary considerably with the solvent used and so we address the results for each in turn. | ||
| Fig. 1 Overview of particles obtained using the ‘standard’ autoclave-based protocol with no heating hold-step. TEM images and electron diffraction patterns are shown for each of the solvents investigated: (a) BE, (b) DEG, (c) DEA, (d) M-DEA (e) E-DEA and (f) MPT (see Table 1). Electron diffraction patterns are indexed according to the magnetite structure with additional spots assigned to hematite in the case of BE. | ||
 | ||
| Fig. 2 Overview of particles obtained using the ‘standard’ autoclave-based protocol when a 110 °C heating hold-step was used. TEM images and electron diffraction patterns are shown for each of the solvents investigated: (a) BE, (b) DEG, (c) DEA, (d) M-DEA (e) E-DEA and (f) MPT (see Table 1). Electron diffraction patterns are indexed according to the magnetite structure. For particles obtained using M-DEA a bright-field TEM image and the corresponding composite dark-field TEM image are shown. Particles obtained when a 180 °C step was used are shown in (f), as no particles were obtained using a 110 °C step. | ||
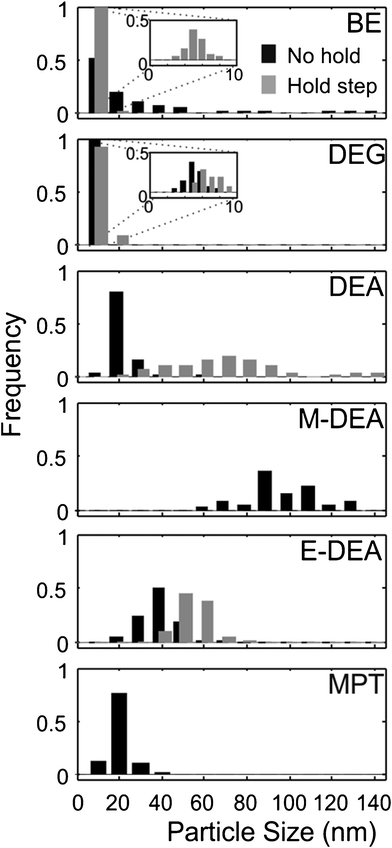 | ||
| Fig. 3 Size distributions for various reaction conditions. Black histograms show size distributions obtained when no heating step was used. Grey histograms show size distributions obtained when the 110 °C heating step was used. The same scale is used for each distribution. No data are given for the M-DEA 110 °C hold-step reaction as larger, micron-sized agglomerates were obtained (see text). No particles were obtained for the 110 °C hold-step conditions when MPT was used as a solvent. Distributions derive from typically 100 particles, measured across 5 discrete areas of the sample grid. | ||
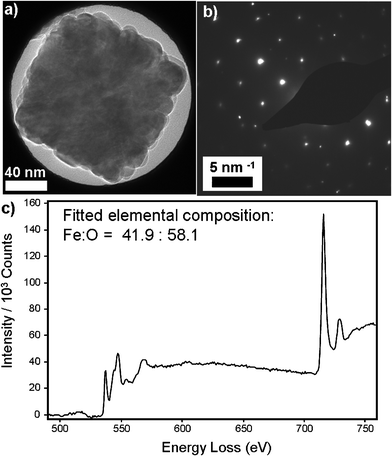 | ||
| Fig. 4 (a) Bright-field TEM image and (b) corresponding selected area electron diffraction pattern, showing a distinct cubic spot pattern. (c) An averaged EELS spectrum collected from a typical compound cube particle, after subtraction of a fitted power-law background function. The elemental composition was calculated assuming Hartree–Slater cross-sections within the Digital Micrograph software package. | ||
| No hold-step | Hold-step | |||
|---|---|---|---|---|
| Solvent | Morphology | Particle size | Morphology | Particle size |
| BE | Spherical, hexagonal platelet | 10.2 ± 5.0 nm (49%) | Spherical | 5.4 ± 0.9 nm (17%) |
| DEG | Spherical | 4.9 ± 1.1 nm (22%) | Spherical | 6.7 ± 2.0 nm (30%) |
| DEA | Spherical, rounded octahedral | 16.6 ± 6.4 nm (39%) | Compound | 75 ± 27 nm (36%) |
| M-DEA | Compound cubic | 98 ± 20 nm (20%) | Linear assemblies | Multiple sub-units |
| E-DEA | Spherical | 34.2 ± 8.5 nm (25%) | Compound | 49 ± 7 nm (14%) |
| MPT | Spherical | 12.0 ± 6.6 nm (54%) | Spherical | 14.7 ± 4.6 nm (31%) |
Benzyl ether
Benzyl ether (BE) was selected because, unlike the other solvents in Table 1, it does not contain hydroxyl groups to chelate to iron and therefore provides a useful benchmark of an inactive, spectator solvent. Binding to iron may be possible through the central ether oxygen, though this is sterically hindered by the two benzyl groups and should therefore be weak. The absence of hydroxyl groups also means that the only reducing agent present is 1,2-hexadecanediol. In terms of physical characteristics, the boiling point of BE is 298 °C, which is above the reaction temperature, so there will be little solvent contribution to the reaction pressure. The relatively low viscosity of BE will also improve the mobility of all reaction species and in comparision with the other solvents, can therefore influence reaction kinetics by increasing the frequency of the interactions/collisions between species.TEM images for the particles obtained using BE are given in Fig. 1(a) and 2(a). Additional TEM images are given in Fig. S2, ESI.† When no heating hold-step is used, polydisperse and inhomogeneous particles are formed [Fig. 1(a) and 3]. The TEM images reveal at least two NP populations: small, roughly spherical particles, 10.2 ± 5.0 nm (49%) in diameter; and large hexagonal and triangular plates with edges up to 100 nm long. ED analysis reveals the presence of a mixture of iron oxide species – hematite (α-Fe2O3) and magnetite (Fe3O4). Formation of this mixture suggests that some NPs were able to grow without reduction by HDD and indicates an absence of strong driving forces to select a particular morphology. Analysis of the diffraction fringes within the smaller particles identifies them as magnetite [Fig. 1(a)] whilst the diffraction fringes evident in the image of the platelets in Fig. S2(c) are consistent with the (104) reflection of hematite. The formation of hexagonal platelets agrees with the crystal habit of hematite36 and similar hexagonal iron oxide platelets (albeit of magnetite) have been observed in previous syntheses using non-chelating solvents, such as super-critical CO2.37 Our own experiments using non-chelating, non-planar squalene and octyl ether solvents also yield platelets; these additional measurements are illustrated in Fig. S3(a–c), ESI.†
In contrast, the inclusion of a heating hold-step resulted in small magnetite NPs of 5.4 ± 0.9 nm (17%) diameter: a relatively narrow particle size distribution and much more like the NPs produced when using BE within a reflux-based protocol.5 Also present were easily separable, micron-sized agglomerated particles. Importantly, no platelets were observed, implying that their formation, or the formation of their precursors, is hindered at lower temperatures and probably requires the direct decomposition of the Fe(acac)3 precursor. Since platelets are not obtained using any of the other solvents presented in Table 1, we conclude that their synthesis is relatively slow. This is shown by time-dependent reactions [see Fig. S4(a–c), ESI†] and is only apparent if competing reactions have been suppressed. All the reaction precursors are instead exhausted during the heating hold-step by the synthesis of spherical NPs, which is facile below the Fe(acac)3 decomposition temperature. NP synthesis presumably initiates via ligand exchange to form reaction intermediates; e.g. iron oleate/olyelamine complexes formed by ligand exchange between acetylacetonate and the surfactants. The narrow size distribution of the small NPs highlights the crucial role of the hold-step in forming NP nuclei that will go on to grow uniformly.
Diethylene glycol
Derivatives of ethylene glycol have long been considered versatile reagents to use in NP synthesis since the hydroxyl groups allow for both chelation to, and reduction of, reaction intermediates28,38,39 and growing NPs. In other words, DEG is influential as it can mediate both stages of the LaMer mechanism. TEM images for the particles obtained using DEG are given in Fig. 1(b) and 2(b). The images show that small, spherical particles have been generated in each instance, with diameters of 4.9 ± 1.1 nm (22%) and 6.7 ± 2.0 nm (30%) (without and with the heating hold-step, respectively). Spherical NPs can be rationalised if DEG adsorbs isotropically on the growing magnetite, in direct comparison to the cases of BE (above), where little adsorption is expected, and M-DEA (below), where the product morphology indicates anisotropic adsorption.In the context of the LaMer mechanism, a well-defined burst of nucleation events is essential for product monodispersity, and the inclusion of a hold-step should encourage the formation of NP seeds during the nucleation stage, thereby increasing the number but reducing the size of product NPs. Interestingly, the results show the reverse: slightly larger and more polydisperse NPs are formed; the relative particle size distribution, which would remain constant if the process is self-similar, increases from 22% to 30%. The increases of size and relative polydispersity that arise with the inclusion of a heating step suggest either an ill-defined nucleation/growth threshold or a poorly chosen hold-step. To investigate this, two variables were altered: the heating rate and the hold-step temperature. Firstly, the heating rate was reduced from 20 °C min−1 to 5 °C min−1 to increase the time between NP nucleation and growth stages. This resulted in larger particles (up to 30 nm) and a broader size distribution [see Fig. S5(a,b), ESI†]. Secondly, the hold-step temperature was increased to 180 °C. The particles obtained at this elevated hold-step temperature remain similar to those observed for the 110 °C hold-step, with a size distribution of 4.8 ± 1.1 nm (23%) [Fig. S5(c), ESI†]. The results from these two reaction conditions strengthen the theory that the nucleation/growth threshold for synthesis using DEG is poorly defined.
The small size of the NPs derived using DEG under standard reaction conditions implies that the iron–DEG chelate is labile, since a large number of NP nuclei are formed early on in the reaction. A large number of nucleation events will then limit the amount of iron–DEG chelate available in the reaction mixture to contribute to NP growth, resulting in smaller particles.28 The lack of stirring in an autoclave seems ideal for NP synthesis using DEG, since we find that using the same reagents under stirred reflux conditions produces large, poorly defined micron-sized particles [representative results are presented in Fig. S5(d), ESI†]. In the autoclave case, interactions between Fe–DEG chelates and NP nuclei are minimised and NPs grow slowly and independently. In contrast, stirring during reflux increases collisions and interactions between species. As the DEG chelate is labile it does not protect NP seeds from coalescence and they readily combine to produce large, poorly defined NPs.
Diethanolamine
DEA presents an interesting comparison with DEG as the two molecules are structurally similar and have the same number of hydroxyl groups, so can be expected to undergo similar chemical interactions with NP species. However, changing the ether linkage for a secondary amine enhances intermolecular hydrogen bonding and substantially increases solvent viscosity, by an order of magnitude, whilst only slight depressing the boiling point (Table 1). As the boiling point is below the usual temperature used for reactions, a control reaction was performed with an open, unpressurised autoclave. This trial yielded identical products, leading us to conclude that elevated pressure does not influence results in this case.TEM images for the NPs obtained using DEA as a solvent are given in Fig. 1(c) and 2(c). We find that the large change in viscosity between DEG and DEA does not affect NP shape, at least when no hold-step was used, in agreement with the similar chelation chemistry expected from these two solvents. However, particle size has increased considerably, with the DEA reaction yielding mostly spherical particles with rounded octahedral particles also present. The particles are 16.6 ± 6.4 nm (39%) diameter, over 10 nm larger than those obtained using DEG. The emergence of a mixture of spherical and rounded octahedral particles suggests that DEA perturbs the adsorption dynamics of oleic acid, oleylamine and HDD at the particle surface, since previous reflux-based studies have shown that the production of cuboidal over spherical NPs can be manipulated through the ratio of adsorbed oleic acid and oleylamine.40 The increase in particle size between use of DEG and DEA suggests that the amine group does not contribute substantially to the initial reduction of precursor, since this would manifest as a larger number of smaller NPs. Conversely, the increase in particle size is consistent with the increase in solvent viscosity, since greater viscosity limits species mobility and hence favours slow nucleation of NP nuclei, which rely on iron–DEA chelate–chelate collisions to grow. Slow seed nucleation means that fewer nuclei form, leaving more iron–DEA chelates in solution for subsequent particle growth.
A more interesting comparison is between Fig. 1(c) and 2(c). The inclusion of a heating hold-step drastically broadens the size distribution and results in the formation of ‘compound’ NPs: particles that do not exist as discrete single crystals but rather as fused agglomerations comprising several crystalline sub-units. The average agglomerate size was 75 ± 27 nm (36%) with sub-units of varying shape and with diameters typically ranging between 10 and 20 nm, similar to the sizes of individual particles obtained when no heating step was used. We rationalise this dramatic difference in NP morphology as follows. The inclusion of a hold-step exhausts iron–DEA chelate supplies in the formation of a larger number of nuclei, which subsequently coalesce during the elevated temperature phase. Gao et al. recently reported that lowering the reaction temperature in solvothermal Fe3O4 particle synthesis limits the diffusion of active species and therefore induces disproportionation and aggregation.38 In our system, the final temperature has not changed but the use of a heating hold-step extends the time that the reaction is held at a lower temperature, again decreasing the reaction rate and slowing diffusion, thus broadening the size distribution. The observed agglomeration may occur because competitive adsorption of DEA, oleylamine, oleic acid and HDD destabilises the protective co-ordinating shell on the NP surface, so that collisions lead to fusion of individual NPs (similar trends are seen for both methyl-DEA and ethyl-DEA, discussed below). This is plausible since the formation of compound NPs is known to be favoured when surfactant concentration is low.41
Methyldiethanolamine
M-DEA differs from DEA by the inclusion of a methyl group on the central N atom, which reduces the solvent viscosity by limiting the amount of hydrogen bonding. Although the presence of two hydroxyl units suggests the chemical activity to be similar to that of DEA and DEG, it is worth noting that the bulkier M-DEA molecules may not pack as densely in an adsorbed layer, thereby affecting lability of the solvent and, indeed, of the other surfactants present in the reaction mixture.TEM images for the particles obtained using M-DEA as a solvent are given in Fig. 1(d) and 2(d). Notably, changing the solvent from DEA to M-DEA causes a dramatic change in particle morphology. Comparison between Fig. 1(c) and 1(d) reveals that the particles formed using M-DEA are larger [98 ± 20 nm (20%)] and are a mixture of single crystalline and ‘compound’ particles, with a preference for cuboid morphology. Compound particles dominate, although the proportion of large single crystal cubes increased when the heating rate was increased and no single crystal cubes were obtained when the rate is reduced to 5 °C min−1. Analysis of the fringes observed by TEM, ED data and EELS data (discussed below) confirms that all the particles in Fig. 1(d) are magnetite. The sub-units that combine to form the assembled, compound cubes were typically pseudo-cubic particles ∼20 nm in side-length, similar to the sub-units seen when DEA was used with a hold-step. The more cuboid character of these NP sub-units is interesting, as previous studies have shown that cuboid NPs can be produced when the ratio of oleylamine to oleic acid is perturbed towards increased acid.22 Our results therefore suggest that M-DEA adsorption again perturbs the balance of oleylamine and oleic acid adsorption. The comparison with DEA is then of particular interest since it implies that the methyl side-chain of M-DEA has a profound effect on the competitive adsorption of other species onto growing NPs and that such functionalisation of the solvent provides a new route to NP shape optimisation and self-assembly.
We also find there to be a preferred orientation of sub-units within each compound cube assembly. There was consistency in the number of sub-units – typically ∼5 – comprising the edge of each cube. Perhaps oriented attachment proceeds until enough surfactant is present to stabilise the exposed NP surfaces. Preferred orientation was also revealed through selected area ED (SAED) experiments. SAED of a single compound particle revealed a distinct spot pattern [Fig. 4(a,b)] which indicates a preferred crystalline orientation since randomly oriented sub-units would yield a ring pattern. The formation of compound particles of defined shape and uniform intra-particle crystallinity implies that anisotropic self-assembly is occurring, which is specific to M-DEA, as similar self-assembly was not seen for any of the other solvents.
To investigate the chemical composition of the compound particles, scanning TEM (STEM) EELS experiments were performed. EELS spectra were averaged across a number of points within a particle, and a typical spectrum, yielding an elemental composition of Fe:O = 41.9:58.1, is displayed in Fig. 4(c). The composition is close to that of magnetite (Fe:O = 42.9:57.1) but suggests the presence of a shell of maghemite (Fe:O = 40:60), which is corroborated by examination of the iron and oxygen elemental intensities at various points across a particle, showing an increase in oxygen concentration at the particle edge (Fig. S6, ESI†). In our experience, mild oxidation of magnetite to maghemite can occur as a consequence of the repeated washing cycles required to ensure the samples are clean enough for STEM experiments; a thin shell of maghemite would be insufficient to yield distinguishable diffractive features in Fig. 4(b).
For the M-DEA protocol the inclusion of a 110 °C heating hold-step yields compound particles of similar size to those described above, but also induces a remarkable, second level of self-assembly. The compound cubes form into chains, as shown in Fig. 2(d), which were confirmed to retain the magnetite phase by HRTEM and ED characterisation (Fig. S7, ESI†). The chain agglomerates were the dominant species observed on the TEM grid, and isolated compound cube particles (like those observed when no heating step was used) were not observed. The fact that the cubes obtained (either isolated or as part of the chain agglomerates) are consistently the same size, regardless of the reaction conditions, implies that the iron precursor is completely converted to NP nuclei during the early stages of the reaction. If a higher temperature hold-step (180 °C) was used, a mixture of single crystalline and compound cubes was obtained, though self-assembly was minimal (see Fig. S8, ESI†). Thus, chain formation is favoured by a slow reaction rate at a low temperature.
As mentioned above, SAED of the compound cube NPs obtained in Fig. 1(d) revealed that there is alignment of individual sub-units within a compound cube. To determine the nature of attachment of these compound cubes within a larger self-assembled chain, dark-field (DF) TEM imaging was used, as illustrated in Fig. 2(d). The DF TEM image has been false-coloured to highlight variations in crystallographic orientation within the chain, with each colour representing an image collected under unique diffraction conditions and therefore indicating different crystal orientation. That the colour varies in sections along the chain implies that these hierarchical assemblies form by face-to-face alignment of individual pre-formed compound cubes, as opposed to continuous chain growth along a specific crystal axis at an end of one cube. Similar ‘hierarchical assembly’ of NPs has been observed previously, including formation of supercrystals42 and helical chains.43 Literature examples17,44 of chain growth suggest the alignment of magnetic dipoles as a viable anisotropic driving force towards chain formation since dipole interactions could encourage end-to-end addition of sub-units and still allow the rotational disorder evidenced by the colour variation along the chain in Fig. 2(d). Face-to-face contact of cuboids provides a greater van der Waals force to direct assembly than that between two spherical NPs and so facilitates self-assembly, perhaps explaining why no hierarchical structures are observed between the particles obtained using DEA.
Ethyldiethanolamine
The TEM images for those particles obtained using E-DEA as a solvent are given in Fig. 1(e) and 2(e). E-DEA has a similar boiling point and viscosity to M-DEA, and differs structurally only by the length of the alkyl chain. When no hold-step was present, rounded particles of size 34.2 ± 8.5 nm (25%) were formed. These particles are different to those obtained from M-DEA and in fact are more similar to those obtained using DEA. Thus, there is no simple trend between N-substituent chain length and final NP morphology. The lower viscosity of E-DEA compared to M-DEA means that iron/E-DEA chelates and the subsequent NP nuclei are slightly more mobile. The number of hydroxyl groups is the same for all the DEA derivatives, so the reductive power of each solvent should be similar. The longer alkyl substituent will sterically hinder chelate packing between E-DEA and iron seed particles, worsening the ability of E-DEA to stabilise NP nuclei – which could explain why larger and more polydisperse particles are obtained using E-DEA than DEA.The inclusion of a 110 °C heating hold-step resulted in the formation of popcorn-like compound particles of size 49.2 ± 7.0 nm (14%), shown in Fig. 2(e). These particles bear resemblance to other ‘nano-flower’ species reported previously,45–47 in which radial epitaxial growth of iron oxide occurs from a central nanoparticle core. In our system, TEM and lattice spacing analysis reveal each sub-unit of the compound particles is a single crystalline ∼20 nm magnetite particle. If the temperature of the heating hold-step was raised to 180 °C, no compound particles and only spherical particles were obtained, implying that popcorn-like particles only seed at low temperatures. At lower temperatures viscosity will be increased and if E-DEA is unable to fully stabilise NP nuclei (due to more labile chelation as a result of increased steric hindrance from the N-ethyl group), then agglomeration will occur. The radial nature of the sub-units suggests that agglomeration followed by further growth of these sub-units, i.e. “epitaxial overgrowth” is responsible for the popcorn-like morphology.
3-Methyl-1,3,5-pentane-triol
MPT was included in this study to ascertain the effects of a triol chelating agent. Based on the number of hydroxyl groups a triol would be expected to possess a greater reducing ability than a diol, which may promote formation of Fe3O4 seeds and in turn facilitate the growth of well-defined Fe3O4 NPs. This assumption was based on the observation that long-chain monoalcohols worsen NP yield and quality compared with long-chain diols.5 Hence, a triol could improve the quality of the NP product. As the boiling point of MPT is below the usual reaction temperature, a control reaction was again conducted using an unpressurised autoclave, and no substantial differences in product were noted, indicating that elevated pressures do not affect NP morphology here.The TEM images obtained for MPT are given in Fig. 1(f) and 2(f). When no heating step was present, mostly spherical Fe3O4 particles were obtained with sizes 12.0 ± 6.6 nm (54%). The majority of particles were small, though some larger, poorly defined particles were also present. No NPs were obtained using a 110 °C hold-step, only micrometre sized agglomerates. It was only after increasing the hold-temperature to 180 °C that spherical 14.7 nm ± 4.6 (31%) particles were obtained. This represents a slight increase in size but a marked narrowing of the size distribution compared to when no hold-step was used. The spherical NPs are similar to those seen when using DEG, DEA and E-DEA: slightly larger than for DEG but smaller than those obtained using DEA derivatives. This implies that the NP nucleation is easier using MPT than it is using DEA derivatives and therefore suggests that the ether link of DEG contributes more to the reductive power of the solvent than the amino links of DEA derivatives.
Discussion
We have provided a systematic study of the often-overlooked role of solvent in the autoclave-based decomposition of iron(III) acetylacetonate to synthesise iron oxide nanoparticles. Our results demonstrate that the solvent is active and as influential as all other reagents in determining NP product characteristics. It should not be overlooked in experimental design: its functionality can determine the decomposition rate of precursors; its viscosity determines the diffusion rate of reaction intermediates; and its structure can profoundly alter product morphology and assembly. The sequential variations in solvent structure studied above allow some general observations to be drawn. Firstly, choosing a solvent with functionality – particularly primary alcohols in the case of magnetite NP synthesis – leads to far greater control over the NP product and is far more important than a solvent's physical characteristics. For example, although a solvent's viscosity will influence the number of interactions and collisions between reactive species, the concentration of those reactive species depends strongly on the solvent's ability to reduce the precursor in the first place. Thus, unstirred BE was shown to produce large, heterogeneous NPs because the increased collision rate arising from a relatively low viscosity is unimportant if formation of reaction intermediates has been limited by slow decomposition of the precursor. A second observation is to consider the molecular packing of adsorbates on growing NPs. This observation has previously been made in the context of competition just between oleylamine and oleic acid,40 where NP shape selectivity is driven by the adsorption dynamics of oleic acid on specific crystallographic surfaces. In our study, the ratio of these surfactants is unchanged, yet the product morphology, including the production of cuboidal NPs, can still be manipulated via the solvent. A useful conceptual image (that would merit a detailed theoretical study) is that of monodentate adsorption and parallel alignment of the hydrocarbon chains of surfactant and solvent molecules about growing NPs, forming a densely packed solvation sphere. This image helps to explain why there is no simple trend with an increase in the ‘reducing power’ (i.e. the number of hydroxyl units) of a solvent: although additional hydroxyl units may speed precursor decomposition and the rate of formation of seeds during the first stages of reaction, only the terminal group is in physical contact with the growing NP during NP growth phase. Similarly, changes to secondary functionality – for example, changing from DEG to DEA – have little effect during the growth phase and the differences observed in NP morphology seem more consistent with changes in solvent viscosity. Further, the addition of bulky side-chains on the solvent backbone will disrupt the densely packed adsorption layer, with two results. Disrupted packing will make the growing NP more susceptible to attack and coalescence and therefore facilitate the formation of compound structures (as seen using M-DEA and E-DEA). Steric effects will also alter the competitive adsorption kinetics of surfactants, as seen in production of cuboidal particles using M-DEA. Prediction of specific changes to NP morphology for a particular side-chain is difficult, because morphology arises from a delicate balance of several factors, but we have demonstrated that it merits further study as a new means of control of NP morphology and self-assembly.Conclusions
In conclusion, we have demonstrated a simple, reliable protocol for the production of monodisperse spheres, hexagonal platelets and compound cubes. We have demonstrated the versatility of autoclave-based protocols, particularly with the inclusion of hold-temperatures, but anticipate the more general conclusions above to have relevance to other protocols. Careful tuning of the reaction heating conditions and solvent functionality permits the formation of self-assembled, hierarchical structures, all tuned through subtle variations in solvent chemistry.Acknowledgements
Financial support from the University of Glasgow to FJD is gratefully acknowledged. The authors wish to thank Professor Shouheng Sun (Brown University) for the use of the VSM.References
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133 CrossRef CAS.
- A. Heymer, D. Haddad, M. Weber, U. Gbureck, P. M. Jakob, J. Eulert and U. Nöth, Biomaterials, 2008, 29, 1473 CrossRef CAS.
- T. Neuberger, B. Schopf, H. Hofmann, M. Hofmann and B. von Rechenberg, J. Magn. Magn. Mater., 2005, 293, 483 CrossRef CAS.
- R. Massart, IEEE Trans. Magn., 1981, 17, 1247 CrossRef.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273 CrossRef CAS.
- L. Zhao, H. Zhang, Y. Xing, S. Song, S. Yu, W. Shi, X. Guo, H. Yang, Y. Le and F. Cao, Chem. Mater., 2008, 20, 198 CrossRef CAS.
- S. Xuan, F. Wang, Y. X. Wang, J. C. Yu and K. C. Leung, J. Mater. Chem., 2010, 20, 5086 RSC.
- S.-B. Wang, Y.-L. Min and S.-H. Yu, J. Phys. Chem. C, 2007, 111, 3551 CAS.
- X. Li, Z. Si, Y. Lei, J. Tang, S. Wang, S. Su, S. Song, L. Zhao and H. Zhang, CrystEngComm, 2010, 12, 2060 RSC.
- H. Zhou, R. Yi, J. Li, Y. Su and X. Liu, Solid State Sci., 2010, 12, 99 CrossRef CAS.
- L. Zhang, J. Wu, H. Liao, Y. Hou and S. Gao, Chem. Commun., 2009, 29, 4378 RSC.
- A. Yan, X. Liu, G. Qiu, N. Zhang, R. Shi, R. Yi, M. Tang and R. Che, Solid State Commun., 2007, 144, 315 CrossRef CAS.
- Y. Zeng, R. Hao, B. Xing, Y. Hou and Z. Xu, Chem. Commun., 2010, 46, 3920 RSC.
- L. Liu, H.-Z. Kou, W. Mo, H. Liu and Y. Wang, J. Phys. Chem. B, 2006, 110, 15218 CrossRef CAS.
- C.-J. Jia, L.-D. Sun, F. Luo, X.-D. Han, L. J. Heyderman, Z.-G. Yan, C.-H. Yan, K. Zheng, Z. Zhang, M. Takano, N. Hayashi, M. Eltschka, M. Klaeui, U. Ruediger, T. Kasama, L. Cervera-Gontard, R. E. Dunin-Borkowski, G. Tzvetkov and J. Raabe, J. Am. Chem. Soc., 2008, 130, 16968 CrossRef CAS.
- G. Zou, K. Xiong, C. Jiang, H. Li, T. Li, J. Du and Y. Qian, J. Phys. Chem. B, 2005, 109, 18356 CrossRef CAS.
- H. Bi, X. Wang, H. Li, B. Xi, Y. Zhu and Y. Qian, Solid State Commun., 2009, 149, 2115 CrossRef CAS.
- V. LaMer and R. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847 CrossRef CAS.
- T. Davis, T. Drews, H. Ramanan, C. He, J. Dong, H. Schnablegger, M. Katsoulakis, E. Kokkoli, A. McCormick, R. Penn and M. Tsapatsis, Nat. Mater., 2006, 5, 400 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2008, 47, 2 CrossRef.
- S. Stoeva, K. J. Klabunde, C. M. Sorensen and I. Dragieva, J. Am. Chem. Soc., 2002, 124, 2305 CrossRef CAS.
- H. G. Yang and H. C. Zeng, J. Phys. Chem. B, 2004, 108, 3492 CrossRef CAS.
- H. Yang, T. Ogawa, D. Hasegawa and M. Takahashi, J. Appl. Phys., 2008, 103, 07D526 CrossRef.
- Y. Hou, Z. Xu and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 6329 CrossRef CAS.
- Q. Song, Y. Ding, Z. L. Wang and Z. J. Zhang, J. Phys. Chem. B, 2006, 110, 25547 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J. Park, H. Noh, J. Kim, J. Park, N. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS.
- X. Liang, X. Wang, J. Zhuang, Y. Chen, D. Wang and Y. Li, Adv. Funct. Mater., 2006, 16, 1805 CrossRef CAS.
- J. Wang, M. Yao, G. Xu, P. Cui and J. Zhao, Mater. Chem. Phys., 2009, 113, 6 CrossRef CAS.
- L.-P. Zhu, H.-M. Xiao, W.-D. Zhang, G. Yang and S.-Y. Fu, Cryst. Growth Des., 2008, 8, 957 CAS.
- Z. Ai, K. Deng, Q. Wan, L. Zhang and S. Lee, J. Phys. Chem. C, 2010, 114, 6237 CAS.
- C. Gotaas, P. Havelka, H. A. Jakobsen, H. F. Svendsen, M. Hase, N. Roth and B. Weigand, Phys. Fluids, 2007, 19, 102106 CrossRef.
- R. M. DiGuilio, R. J. Lee, S. T. Schaeffer, L. L. Brasher and A. S. Teja, J. Chem. Eng. Data, 1992, 37, 239 CrossRef CAS.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204 CrossRef CAS.
- H. Wu, Y. Yang and Y. C. Cao, J. Am. Chem. Soc., 2006, 128, 16522 CrossRef CAS.
- Y. Zhu, T. Mei, Y. Wang and Y. Qian, J. Mater. Chem., 2011, 21, 11457 RSC.
- R. D. Rodriguez, D. Demaille, E. Lacaze, J. Jupille, C. Chaneac and J.-P. Jolivet, J. Phys. Chem. C, 2007, 111, 16866 CAS.
- Z. Li, J. F. Godsell, J. P. O'Byrne, N. Petkov, M. A. Morris, S. Roy and J. D. Holmes, J. Am. Chem. Soc., 2010, 132, 12540 CrossRef CAS.
- G. Gao, R. Shi, W. Qin, Y. Shi, G. Xu, G. Qiu and X. Liu, J. Mater. Sci., 2010, 45, 3483 CrossRef CAS.
- J. Wan, W. Cai, X. Meng and E. Liu, Chem. Commun., 2007, 47, 5004 RSC.
- Y. Hou, Z. Xu and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 6329 CrossRef CAS.
- A. Naravanaswamy, H. Xu, N. Pradhan and X. Peng, Angew. Chem., Int. Ed., 2006, 45, 5361 CrossRef.
- A. Demortiere, P. Launois, N. Goubet, P. A. Albouy and C. Petit, J. Phys. Chem. B, 2008, 112, 14583 CrossRef CAS.
- K. Cho, D. Talapin, W. Gaschler and C. Murray, J. Am. Chem. Soc., 2005, 127, 7140 CrossRef CAS.
- L. Guo, F. Liang, X. Wen, S. Yang, L. He, W. Zheng, C. Chen and Q. Zhong, Adv. Funct. Mater., 2007, 17, 425 CrossRef CAS.
- S.-H. Choi, H. Bin Na, Y. I. Park, K. An, S. G. Kwon, Y. Jang, M.-H. Park, J. Moon, J. S. Son, I. C. Song, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2008, 130, 15573 CrossRef CAS.
- F. Song, J. Guan, X. Fan and G. Yan, J. Alloys Compd., 2009, 485, 753 CrossRef CAS.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. Sun, Nano Lett., 2005, 5, 379 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: additional TEM images, thermogravimetric analysis (TGA) information and magnetic measurements. See DOI: 10.1039/c2ra20494k |
| This journal is © The Royal Society of Chemistry 2012 |