Li+ cation coordination by acetonitrile—insights from crystallography†
Daniel M.
Seo
a,
Paul D.
Boyle
b,
Oleg
Borodin
c and
Wesley A.
Henderson
*a
aIonic Liquid and Electrolytes for Energy Technologies (ILEET) Laboratory, Department of Chemical & Biomolecular Engineering, North Carolina State University, Raleigh, NC 27695, United States. E-mail: wesley_henderson@ncsu.edu; Tel: 919-513-2917
bX-ray Structural Facility, Department of Chemistry, North Carolina State University, Raleigh, NC 27695, United States
cElectrochemistry Branch, Sensor & Electron Devices Directorate, U.S. Army Research Laboratory, Adelphi, MD 20783, United States
First published on 27th June 2012
Abstract
Solvation is a critical factor for determining the properties of electrolytes and lithium reagents, but only limited information is available about the coordination number for Li+ cations in solution with different solvents. The present manuscript examines the manner in which acetonitrile (AN) fully solvates Li+ cations. The results are also likely pertinent to other nitrile and dinitrile solvents. In particular, the crystal structure for a (AN)6:LiPF6 solvate is reported—this is the first 6/1 AN/Li solvate structure to be determined. The structure consists of Li+ cations fully solvated by four AN molecules (i.e., [(AN)4Li]+ species), uncoordinated PF6− anions and uncoordinated AN molecules (two per Li+ cation). This structure validates, in part, density functional theory (DFT) calculations which predict that there is little to no energetic benefit to coordinating Li+ cations with more than four AN solvent molecules.
Introduction
The manner in which cations are solvated in solution remains of interest for innumerable applications. This is particularly true for Li+ cations due to the need to refine electrolytes for new lithium battery electrode materials and the wide use of lithium reagents for organometallic chemistry. For electrolyte applications, nitrile and dinitrile solvents are one potential class of promising solvents due to their reported exceptional oxidative stability.1–5 Insight regarding how nitrile solvents coordinate Li+ cations and how this differs from the carbonate solvents current used for battery electrolytes6,7 is therefore sought. For reagent applications, the manner is which Li+ cations in lithium reagents (organolithium reagents, LiAlH4, LiEt3BH, Gilman reagents (Li+CuR2−), Li+NR2−, lithium enolates, etc.) are solvated has been widely recognized as crucial for understanding their reactivity.8–13Li+ cations can have coordination numbers from three to eight.14 Three-fold coordination is extremely rare. Eight-fold coordination may be found for ligands such as crown ethers (i.e., 12C4) (Fig. 1). The ether O–Li+ cation coordination bond lengths for (12C4)2:LiX solvates, however, are quite long (typically 2.21–2.54 Å with an average of approximately 2.37 Å from reported crystal structures), indicating relatively weak coordination bonds due to the small size of the Li+ cations and difficulty of packing eight donor atoms about the cation.15–18 In contrast, monoglyme (G1) and diglyme (G2) are able to form (G1)3:LiX and (G2)2:LiX solvates with six-fold coordination (Fig. 1) and shorter ether O–Li+ cation coordination bonds (typically 2.05–2.24 and 2.00–2.27 Å with averages of approximately 2.13 and 2.13 Å, respectively, from reported crystal structures).19–30 Six-fold and even higher coordination of Li+ cations is therefore quite favorable for multidentate solvents. Solvation is governed, however, by steric and other factors which influence the solvent's ability to approach and pack around a Li+ cation in a suitable manner to form a coordinate bond. Thus, Li+ cations fully solvated by tetrahydrofuran (THF) nearly always have four-fold coordination (Fig. 1) and short ether O–Li+ cation coordination bonds (typically 1.87–1.97 Å with an average of approximately 1.92 Å from reported crystal structures).31–36 Therefore, it remains unclear if solvents which form monodentate coordination will/can have greater than four-fold coordination to Li+ cations due to steric crowding of the solvent molecules, even for acetonitrile (AN) which is essentially a short linear rod with a coordinating end. Solvent–cation interactions have been the frequent focus of theoretical studies. One notable point from this body of work is that previous DFT studies of (AN)n–Li+ (n = 4–6) cation complexes in the gas-phase assert that Li+ cations are preferentially coordinated by four AN molecules.37–41 When a fifth or sixth AN molecule was added to the Li+ cation's coordination shell, the incremental binding energy was significantly lowered relative to the binding energy for the addition of the fourth AN molecule (from three-fold coordination). This suggests that there is little to no energetic driving force for greater solvation than four AN molecules per Li+ cation, but such solvation (five- and six-fold) is not precluded.
![Li+ cation coordination in (a) [(12C4)2Li]+, (b) [(G1)3Li]+, (c) [(G2)2Li]+ and (d) [(THF)4Li]+ crystalline solvates (Li purple, O red).15–36 Note that space-fill models use spheres for atoms (and ions) with the radii proportional to the radii of the atoms. Li+ cations, however, are significantly smaller than Li atoms. Thus, while the space-fill models do provide a better understanding of steric constraints for packing of the solvates, the cation sizes are actually smaller than those shown.](/image/article/2012/RA/c2ra21290k/c2ra21290k-f1.gif) | ||
| Fig. 1 Li+ cation coordination in (a) [(12C4)2Li]+, (b) [(G1)3Li]+, (c) [(G2)2Li]+ and (d) [(THF)4Li]+ crystalline solvates (Li purple, O red).15–36 Note that space-fill models use spheres for atoms (and ions) with the radii proportional to the radii of the atoms. Li+ cations, however, are significantly smaller than Li atoms. Thus, while the space-fill models do provide a better understanding of steric constraints for packing of the solvates, the cation sizes are actually smaller than those shown. | ||
AN is of interest for exploring the solvation of Li+ cations as this is a linear solvent with essentially no extraneous bulk to the solvent which may hinder the efficient packing of the solvent molecules about the cation when coordinated. The present manuscript extends the previous theoretical DFT work and utilizes crystalline solvate structures to examine the solvation interactions of AN with Li+ (and other) cations. The prediction regarding favorable four-fold coordination is validated to some extent experimentally in the present work through an examination of solvate crystal structures with an emphasis on the (AN)6:LiPF6 solvate structure reported here. This demonstrates the value of using crystallography to aid in understanding the solvation interactions which may occur in solution.
Experimental
LiPF6 (battery grade) was purchased from Novolyte and used as-received. Anhydrous AN (Sigma-Aldrich, 99.8%) was used as-received. In a Vacuum Atmospheres inert atmosphere (N2) glove box (< 5 ppm H2O), LiPF6 (0.1 mmol) and AN (2 mmol) were sealed in a vial and the mixture (i.e., (AN)n–LiPF6 with n = 20) was heated on a hot plate to form a homogeneous solution. Upon standing in a freezer (−20 °C), colorless plate single crystals of the (AN)6:LiPF6 solvate (mp 18 °C) suitable for analysis formed in the excess AN.A single crystal of the (AN)6:LiPF6 solvate was mounted on a Mitegen polyimide micromount with a small amount of Paratone N oil. X-ray measurements were made on a Bruker-Nonius Kappa Axis X8 Apex2 diffractometer at −163 °C. Unit cell dimensions were determined from a symmetry constrained fit of 8800 reflections with 5.22° < 2θ < 57.78°. The data collection strategy was a number of ω and ϕ scans which collected data up to 68.04° (2θ). Frame integration was performed using SAINT.42 The resulting raw data was scaled and absorption corrected using a multi-scan averaging of symmetry equivalent data using SADABS.43 The structure was solved by direct methods using the SIR92 program (crystallographic data for 881135 can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif).†43 Most non-hydrogen atoms were obtained from the initial solution. The remaining non-hydrogen atomic positions were recovered from subsequent difference Fourier maps. Hydrogen atoms were introduced at idealized positions and allowed to ride on the parent atom. The two uncoordinated AN molecules are disordered and due to the crystallographic imposed nature of the disorder the occupancies for the disordered solvent molecules were presumed to be 0.50. These solvent molecules occupy channels which run in a zig–zag fashion along the [100]. The PF6− anion was also disordered over two orientations (occupancy for the primary orientation is 57%). The structural model was fit to the data using full matrix least-squares based on F2. The calculated structure factors included corrections for anomalous dispersion from the usual tabulation. The structure was refined using the XL program from SHELXTL.44 Structures were drawn with Mercury 2.3.
Density functional theory (DFT) calculations were performed using a polarized continuum model (PCM) with AN parameters as implemented in the Gaussian g09 software to account for the influence of the AN solvent present beyond the first Li+ cation solvation shell (condensed rather than gas phase). The (AN)n–Li–PF6 geometries were optimized at the M05-2X/6-31+G* level following our previous work,45,46 while single point energy calculations were performed at these geometries using Møller–Plesset perturbation theory (MP2) with aug-cc-pvDz and aug-cc-pvTz basis sets. Four MD simulations of the (AN)4–LiPF6 cluster in the gas-phase were performed with the simulation temperature decreasing from 600 K to 200 K with a cooling rate of 100 K/400 ps. Geometries were output every 100 ps. Only geometries that contained all of the AN molecules within 0.48 nm were used for future analysis. After each simulated annealing, a 0.5 ns simulation at 600–800 K was performed to generate an independent configuration. The XYZ configurations from the simulated annealing runs were used as a starting point for a molecular mechanics (MM) energy optimization that was also performed using the APPLE&P force field. The lowest energy geometries from the MM energy optimization were contact ion pair (CIP) solvates in which the anion was coordinated to the Li+ cation. The lowest three geometries were optimized using a M05-2X density functional with and without the PCM solvation model. To find the solvent-separated ion pair (SSIP) geometries in which the anion remained uncoordinated, the scan of the Li–P distance was performed starting from the CIP geometry followed by geometry optimization without any constrains with PCM included in the optimization. Standard Gaussian convergence criteria for displacement and force were used. Frequency calculations were performed to ensure that the converged complexes had no imaginary frequencies and, therefore, were minima.
Results and discussion
Crystalline solvates
Crystal structures of solvates provide tremendous insight into the optimized solvent and ion coordination and packing that is possible, as well as serving as useful models for the solvation interactions that may be present in solution. Phase diagrams have been recently prepared for AN mixtures with a variety of lithium salts. For many of these mixtures, such as (AN)n–LiClO4 and (AN)n–LiBF4, the first crystalline solvate to form from dilute mixtures has a 4/1 stoichiometry.45 This suggested that the theoretical predictions may be correct. The crystal structure for the (AN)4:LiClO4 solvate is known.47 It consists of Li+ cations fully solvated by four AN molecules and uncoordinated ClO4− anions. The same form of coordination is found for the (AN)4:LiI solvate.48 In contrast, a phase diagram for (AN)n–LiPF6 mixtures indicates that 6/1 and 5/1 AN/LiPF6 phases form from dilute mixtures.46 This appeared to contradict the theoretical predictions. The recently reported crystal structure for the (AN)5:LiPF6 solvate, however, was found to consist of Li+ cations fully solvated by four AN molecules (i.e., [(AN)4Li]+ species).49 The additional uncoordinated AN molecules (one per Li+ cation) reside within vacancies created between the uncoordinated PF6− anions and solvated cations.No information has thus far been available regarding the structure of a 6/1 crystalline solvate with a lithium salt. The (AN)6:LiPF6 solvate crystal structure is therefore reported here.50 The packing diagram for the solvate structure is shown in Fig. 2. The Li+ cation resides on a crystallographic mirror plane, while the PF6− anion sits on a crystallographic two-fold axis with the two-fold axis bisecting the F1–P1–F1 angle. The disorder in the PF6− anion is not crystallographically imposed. As for the 5/1 solvate structure, the 6/1 structure consists of Li+ cations fully solvated by four AN molecules (Fig. 3). The positioning and orientation of the solvated cations and anions results in channels within the structure (Fig. 2). These are filled with the uncoordinated AN molecules (two per Li+ cation) (Fig. 3).
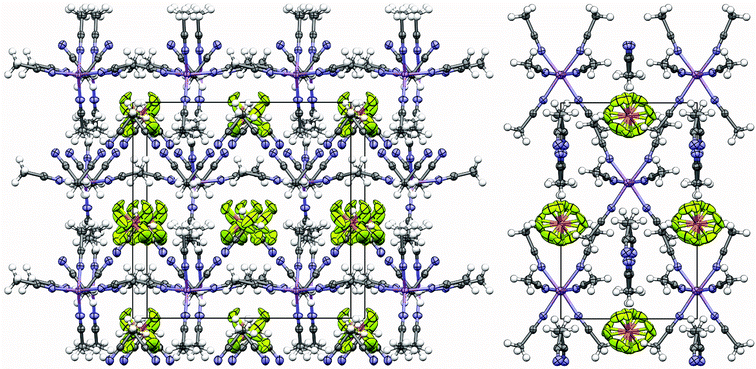 | ||
| Fig. 2 Ion and solvent packing in the (AN)6:LiPF6 solvate crystal structure (2 views) (Li purple, N blue, P orange, F light green) in which the Li+ cations are tetrahedrally coordinated to four AN molecules. | ||
![A space-fill diagram of the solvated [(AN)4Li]+ cations and head-to-tail alignment of uncoordinated AN molecules in the (AN)6:LiPF6 solvate crystal structure. The PF6− anions are disordered about one axis (Li purple, N blue, F light green).](/image/article/2012/RA/c2ra21290k/c2ra21290k-f3.gif) | ||
| Fig. 3 A space-fill diagram of the solvated [(AN)4Li]+ cations and head-to-tail alignment of uncoordinated AN molecules in the (AN)6:LiPF6 solvate crystal structure. The PF6− anions are disordered about one axis (Li purple, N blue, F light green). | ||
DFT calculations
In related work, it has been shown that the incorporation of solvent into the DFT calculations for the determination of the energetics of anion⋯Li+ cation coordination (which is typically not done) can significantly impact the results from the calculations.45,46 It is therefore possible that the inclusion of anions may also be influential for the solvent⋯Li+ cation calculations. Thus, the previously reported DFT studies with (AN)n–Li complexes37–41 have been extended here to (AN)n–Li–PF6 complexes in order to gain insight into the most stable Li+ cation coordination by the PF6− anion and AN molecules. The binding energy of the SSIP complex (B in Fig. 4) is ∼2 kcal mol−1 lower (Table 1) than the binding energy of the CIP complex (A in Fig. 4) indicating that the SSIP solvate is energetically more stable than the CIP solvate. Similar to the (AN)4–Li–PF6 results, it was found that the SSIP (AN)5–Li–PF6 solvate is more energetically stable than the corresponding CIP solvate. The focus was thus devoted to examining the Li+ cation coordination in the (AN)5–Li–PF6 SSIP solvate. Two SSIP configurations for (AN)5–Li–PF6 were considered, the first with the Li+ cation coordinated by five AN molecules (C in Fig. 4) and the second with four AN molecules strongly coordinating the Li+ cation and one AN molecule uncoordinated (D in Fig. 4). The latter was found to be more stable than the (AN)5–Li–PF6 complex with all five AN molecules coordinating the Li+ cation (Table 1), presumably due to steric hindrance between neighboring solvent molecules. Thus, the DFT calculations indicate that, for the (AN)n–Li–PF6 solvates in the condensed phase, the most energetically favorable configuration is the Li+ cation coordinated by four AN molecules with the PF6− anion uncoordinated (in agreement with the 6/1 and 5/1 solvate crystal structures). Furthermore, molecular dynamics (MD) simulations also suggest that the vast majority of the Li+ cations in AN–LiX solutions have four-fold coordination to either anions and/or AN molecules.45,46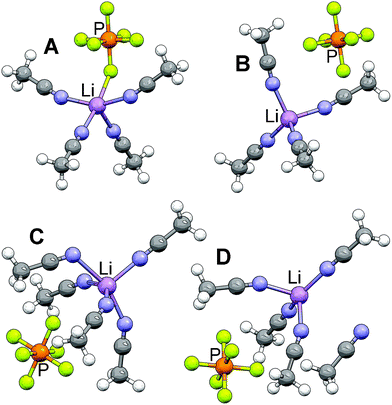 | ||
| Fig. 4 Optimized geometries (AN)n–Li–PF6 (n = 4 and 5) complexes from M05-2X/6-31+G* calculations using PCM(AN) (Li purple, N blue, P orange, F light green). | ||
| Complex | M05-2X/6-31+G* | MP2/aug-cc-pvDz | MP2/aug-cc-pvTz |
|---|---|---|---|
| (AN)4–Li–PF6E(B) − E(A) | −1.9 | −1.7 | −2.4 |
| (AN)5–Li–PF6E(D) − E(C) | −3.3 | −2.9 |
Comparison with Cu(I)+ and Ag(I)+ crystalline solvates
Other crystal structures of solvates further support this conclusion. Ionic radii are determined by the distances obtained from crystal structures of salts and solvates. Ionic radii are reported to vary based upon several factors including the coordination number.51,52 But a comparison of the relative sizes of ions can be made by comparing similar solvates. Numerous solvates are now known which contain [(AN)4M]+ (M = Cu(I)+, Li+, Ag(I)+) solvated cations. Such solvates consistently indicate that the Cu(I)+ and Ag(I)+ cations are slightly smaller and larger, respectively, than Li+ cations, as is evident from the N⋯cation bond lengths found in the reported 4/1 and 5/1 solvate crystal structures with ClO4−, BF4− and PF6− anions (Table 2).53–64 These solvates are all isostructural with the corresponding lithium salts.47,49 In all cases, the cations have four-fold (tetrahedral) coordination.| Solvates | T (K) | Bond lengths (Å) | |||
|---|---|---|---|---|---|
| (AN)4:CuClO4 | 173 | 1.979 | 1.991 | 1.994 | 2.005 |
| 1.957 | 1.976 | 1.976 | 2.003 | ||
| 1.978 | 2.001 | 2.001 | 2.004 | ||
| (AN)4:LiClO4 | 173 | 1.990 | 2.016 | 2.017 | 2.020 |
| 2.007 | 2.027 | 2.041 | 2.043 | ||
| 2.015 | 2.031 | 2.039 | 2.045 | ||
| (AN)4:AgClO4 | 173 | 2.240 | 2.247 | 2.251 | 2.273 |
| 2.228 | 2.274 | 2.289 | 2.293 | ||
| 2.248 | 2.254 | 2.276 | 2.314 | ||
| (AN)4:CuBF4 | 173 | 1.972 | 2.000 | 2.013 | 2.026 |
| 1.972 | 1.983 | 1.998 | 2.029 | ||
| 1.964 | 1.977 | 1.977 | 2.011 | ||
| (AN)4:AgBF4 | 178 | 2.249 | 2.257 | 2.276 | 2.316 |
| 2.231 | 2.244 | 2.262 | 2.295 | ||
| 2.229 | 2.278 | 2.298 | 2.300 | ||
| (AN)5:CuPF6 | 150 | 1.968 | 2.003 | 2.019 | 2.019 |
| 1.997 | 2.000 | 2.005 | 2.018 | ||
| 1.993 | 2.004 | 2.017 | 2.030 | ||
| (AN)5:LiPF6 | 110 | 2.009 | 2.021 | 2.038 | 2.048 |
| 2.012 | 2.017 | 2.025 | 1.032 | ||
| 2.007 | 2.012 | 2.036 | 2.063 | ||
But a further scrutiny of the known solvate structures between AN and copper salts somewhat confounds the coordination arguments made. A 4/1 crystalline solvate structure has been reported with the salt CuB(C6F5)4, but so has a 6/1 crystalline solvate structure (Fig. 5).65,66 The Cu(I)+ cations in the 6/1 crystalline solvate are fully coordinated by six AN molecules in an octahedral arrangement. Given that the Cu(I)+ cations are slightly smaller than Li+ cations (Table 2), this suggests that 6/1 solvation by AN should indeed be possible for Li+ cations. Closer scrutiny of the 6/1 crystalline solvate with CuB(C6F5)4, however, reveals that the N⋯Cu(I)+ bond lengths are 1.987, 1.987, 1.999, 1.999, 2.386 and 2.386 Å.66 Thus, two of the solvating (axial) AN molecules have very weak coordinate bonds to the Cu+ cations relative to the other four (equatorial) AN molecules.
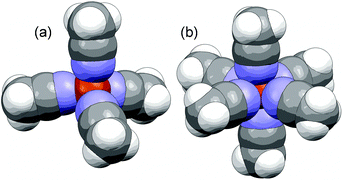 | ||
| Fig. 5 A space-fill diagram of the Cu(I)+ cation coordination in the (a) (AN)4:CuB(C6F5)4 and (b) (AN)6:CuB(C6F5)4 solvate crystal structures (Cu red, N blue).65,66 | ||
One explanation for the formation of the 6/1 crystalline solvate with Cu(I)+ cations with six-fold coordination may be that while there is little to no energetic benefit to incorporating more than four AN molecules in the coordination shell of a single Li+ cation (and presumably a Cu(I)+ cation), this may not be the case for crystalline solvates where optimization of the packing of the solvated cations and uncoordinated anions may impact the solvation energetics (system energetics rather than solvated cation energetics). The 6/1 crystalline solvate for CuB(C6F5)4 may therefore be an exception to the typical solvation interactions which favor four-fold coordination. Such factors associated with crystal packing, however, would not be relevant for solvated cations in electrolyte solutions. Furthermore, thermal motion of the solvent molecules in the coordination shell of cations in solution would also hinder the packing of more than four AN molecules about the cations.
Conclusions
The crystal structure of the reported (AN)6:LiPF6 solvate, which contains solvated [(AN)4Li]+ cations, and refined DFT calculations provide strong credence for the full solvation of Li+ cations in solution, in general, by four AN molecules. The formation of solvates with greater than four-fold (tetrahedral) coordination is not precluded, but such solvates are not energetically favorable.Acknowledgements
The authors wish to express their gratitude to the U.S. Department of Energy (DOE) Office of Basic Energy Science-Division of Materials Sciences and Engineering which fully supported the experimental research under Award DE-SC0002169. Modeling work was partially supported by an Interagency Agreement between the U.S. DOE and the U.S. Army Research Laboratory (ARL) under DE-IA01-11EE003413 for the Office of Vehicle Technologies Programs including the Batteries for Advanced Transportation Technologies (BATT) Program. The authors also wish to thank the Department of Chemistry of North Carolina State University and the State of North Carolina for funding the purchase of the Apex2 diffractometer.References
- Q. Wang, P. Pechy, S. M. Zakeeruddin and M. Grätzel, J. Power Sources, 2005, 146, 813, DOI:10.1016/j.jpowsour.2005.03.157.
- Y. Abu-Lebdeh and I. Davidson, J. Electrochem. Soc., 2009, 156, A60, DOI:10.1149/1.3023084.
- Y. Abu-Lebdeh and I. Davidson, J. Power Sources, 2009, 189, 576, DOI:10.1016/j.jpowsour.2008.09.113.
- M. Nagahama, N. Hasegawa and S. Okada, J. Electrochem. Soc., 2010, 157, A748, DOI:10.1149/1.3417068.
- P. Isken, C. Dippel, R. Schmitz, R. W. Schmidtz, M. Kunze, S. Passerini, M. Winter and A. Lex-Balducci, Electrochim. Acta, 2011, 56, 7530, DOI:10.1016/j.electacta.2011.06.095.
- K. Xu, Chem. Rev., 2004, 104, 4303, DOI:10.1021/cr030203g.
- A. Lex-Balducci, W. Henderson and S. Passeriniin, Lithium-Ion Batteries: Advanced Materials and Technologies, ed. X. Yuan, H. Liu and J. Zhang, CRC Press: Boca Raton, FL, 2012 Search PubMed.
- C. Reichert, Solvents and Solvent Effects in Organic Chemistry, VCH: Weinheim, Germany, 1990 Search PubMed.
- B. L. Lucht and D. B. Collum, Acc. Chem. Res., 1999, 32, 1035, DOI:10.1021/ar960300e.
- K. W. Henderson, A. E. Dorigo, Q.-Y. Liu and P. G. Williard, J. Am. Chem. Soc., 1997, 119, 11855, DOI:10.1021/ja971920t.
- H. Nöth, S. Thomas and M. Schmidt, Chem. Ber., 1996, 129, 451, DOI:10.1002/cber.19961290414.
- M. John, C. Auel, C. Behrens, M. Marsch, K. Harms, F. Bosold, R. M. Gschwind, P. R. Rajamohanan and G. Boche, Chem.–Eur. J., 2000, 6, 3060, DOI:10.1002/1521-3765(20000818)6:16<3060::AID-CHEM3060>3.0.CO;2-M.
- R. P. Davies, Coord. Chem. Rev., 2011, 255, 1226, DOI:10.1016/j.ccr.2011.01.011.
- U. Olsher, R. M. Izatt, J. S. Bradshaw and N. K. Dalley, Chem. Rev., 1991, 91, 137, DOI:10.1021/cr00002a003.
- R. A. Lewis, G. Wu and T. W. Hayton, Inorg. Chem., 2011, 50, 4660, DOI:10.1021/ic200490v.
- H. Hope, M. M. Olmstead, P. P. Power and X. Xu, J. Am. Chem. Soc., 1984, 106, 819, DOI:10.1021/ja00315a075.
- J. C. Gallucci, M. R. Sivik, L. A. Paquette, F. Zaegel, P. Meunier and B. Gautheron, Acta Crystallogr., 1996, C52, 1673, DOI:10.1107/S0108270196002454.
- S. T. Liddle and W. Clegg, Polyhedron, 2003, 22, 3507, DOI:10.1016/j.poly.2003.09.011.
- A. N. Kornev, N. V. Belina, V. V. Sushev, G. K. Fukin, E. V. Baranov, Y. A. Kurskiy, A. I. Poddelskii, G. A. Abakumov, P. Lönnecke and E. Hey-Hawkins, Inorg. Chem., 2009, 48, 5574, DOI:10.1021/ic900135b.
- S. Wang, Q. Yang, T. C. W. Mak and Z. Xie, Organometallics, 2000, 19, 334, DOI:10.1021/om990648o.
- L. Zhou, Y. Yao, C. Li, Y. Zhang and Q. Shen, Organometallics, 2006, 25, 2880, DOI:10.1021/om060055v.
- Z. Qiu and Z. Xie, J. Am. Chem. Soc., 2009, 131, 2084, DOI:10.1021/ja809389k.
- H. Link and D. Fenske, Z. Anorg. Allg. Chem., 1999, 625, 1878, DOI:10.1002/(SICI)1521-3749(199911).
- M. Saito, S. Imaizumi, T. Tajima, K. Ishimura and S. Nagase, J. Am. Chem. Soc., 2007, 129, 10974, DOI:10.1021/ja072478+.
- W. A. Henderson, N. R. Brooks, W. W. Brennessel and V. G. Young Jr., J. Phys. Chem. A, 2004, 108, 225, DOI:10.1021/jp035836d.
- M. W. Esterhuysen and H. G. Raubenheimer, Eur. J. Inorg. Chem., 2003, 3861, DOI:10.1002/ejic.200300334.
- J. Pauls, E. Iravani, P. Köhl, B. Neumüller and Z. Anorg, Z. Anorg. Allg. Chem., 2004, 630, 876, DOI:10.1002/zaac.200400061.
- V. Seneviratne, R. Frech, J. E. Furneaux and M. Khan, J. Phys. Chem. B, 2004, 108, 8124, DOI:10.1021/jp031128g.
- W. Palitzsch, U. Böhme, C. Beyer and G. Roewer, Organometallics, 1998, 17, 2965, DOI:10.1021/om971061h.
- Y. G. Andreev, V. Seneviratne, M. Khan, W. A. Henderson, R. E. Frech and P. G. Bruce, Chem. Mater., 2005, 17, 767, DOI:10.1021/cm048310u.
- G. T. Sazama and T. A. Betley, Inorg. Chem., 2010, 49, 2512, DOI:10.1021/ic100028y.
- N. Nakata, R. Izumi, V. Y. Lee, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2004, 125, 5058, DOI:10.1021/ja040001h.
- C. B. Serrano, R. J. Less, M. McPartlin, V. Naseri and D. S. Wright, Organometallics, 2010, 29, 5754, DOI:10.1021/om100838u.
- S. Wang, H.-W. Li and Z. Xie, Organometallics, 2004, 23, 2469, DOI:10.1021/om0498807.
- G. B. Nikiforov, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Polyhedron, 2004, 23, 561, DOI:10.1016/j.poly.2003.10.008.
- I. Fernández, E. Martinez-Viviente, F. Breher and P. S. Pregosin, Chem.–Eur. J., 2005, 11, 5149, DOI:10.1002/chem.200400867.
- W. Kunz, J. Barthel, L. Klein, T. Cartailler, P. Turq and B. Reindl, J. Solution Chem., 1991, 20, 875, DOI:10.1007/BF01074950.
- E. M. Cabaleiro-Lago and M. A. Ríos, Chem. Phys., 2000, 254, 11, DOI:10.1016/S0301-0104(00)00007-0.
- X. Xuan, H. Zhang, J. Wang and H. Wang, J. Phys. Chem. A, 2004, 108, 7513, DOI:10.1021/jp047313r.
- D. Spångberg and K. Hermansson, Chem. Phys., 2004, 300, 165, DOI:10.1016/j.chemphys.2004.01.011.
- E. Pasgreta, R. Puchta, A. Zahl and R. van Eldik, Eur. J. Inorg. Chem., 2007, 13, 1815, DOI:10.1002/ejic.200600930.
- Bruker.SAINT and SADABS. 2009, Bruker AXS Inc., Madison, WI, USA Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435, DOI:10.1107/S002188989400021X.
- Bruker-AXSXL version 2009.9, Bruker-AXS Inc., Madison, WI, USA Search PubMed.
- D. M. Seo, O. Borodin, S.-D. Han, Q. Ly, P. D. Boyle and W. A. Henderson, J. Electrochem. Soc., 2012, 159, A553, DOI:10.1149/2.jes112264.
- D. M. Seo, O. Borodin, S.-D. Han, P. D. Boyle and W. A. Henderson, J. Electrochem. Soc., 2012, 159(9), A1–A12 Search PubMed.
- Y. Yokota, V. G. Young Jr. and J. G. Verkade, Acta Crystallogr., 1999, C55, 196, DOI:10.1107/S0108270198011834.
- C. L. Raston, C. R. Whitaker and A. H. White, Aust. J. Chem., 1989, 42, 201, DOI:10.1071/CH9890201.
- D. M. Seo, P. D. Boyle and W. A. Henderson, Acta Crystallogr., 2011, E67, m1148, DOI:10.1107/S1600536811027528.
- Crystal data for (AN)6:LiPF6: FW = 398.23, C12H18F6LiN6P, colorless, prism (0.46 × 0.30 × 0.27 mm), orthorhombic, space group Ama2, a = 14.830(3) Å, b = 9.2250(16) Å, c = 14.606(2) Å, α = β = γ = 90°, V = 1998.2(6) Å3, ρcalc = 1.324 g cm-3, Z = 4, 2θmax = 68.04°, MoKα radiation (λ = 0.71073 Å), T = 110 K. Converged with R1 = 0.0402 and wR2 = 0.1010 for 3787 independent reflections with GOF =1.019.
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751, DOI:10.1107/S0567739476001551.
- P. L. Lang and B. C. Smith, Dalton Trans., 2010, 39, 7786, 10.1039/C0DT00401D.
- I. Csöregh, P. Kierkegaard and K. Norrestam, Acta Crystallogr., 1975, B31, 314, DOI:10.1107/S0567740875002634.
- G. A. Bowmaker, D. S. Gill, B. W. Skelton, N. Somers and A. H. White, Z. Naturforsch., 2004, B59, 1307 Search PubMed.
- E. K. Beloglazkina, A. V. Shimorsky, A. G. Mazhuga, O. V. Shilova, V. A. Tafeenko and N. V. Zyk, Russ. J. Gen. Chem., 2009, 79, 1504, DOI:10.1134/S1070363209070172.
- H.-G. Hao, X.-D. Zheng and T.-B. Lu, Angew. Chem., Int. Ed., 2010, 49, 8148, DOI:10.1002/anie.201004928.
- M. Sharma and B. Mondal, Inorg. Chem., 2011, 50, 3206, DOI:10.1021/ic1011988.
- K. Nilsson and Å. Oskarsson, Acta Chem. Scand., Ser. A, 1984, 38, 79, DOI:10.3891/acta.chem.scand.38a-0079.
- P. G. Jones and E. Bembenek, Z. Kristallogr., 1993, 208, 213, DOI:10.1524/zkri.1993.208.Part-2.213.
- P. G. Jones and O. Crespo, Acta Crystallogr., 1998, C54, 18, DOI:10.1107/S0108270197013322.
- J. W. Bats, T. Kretz and H.-W. Lerner, Acta Crystallogr., 2009, C65, m94, DOI:10.1107/S0108270109001139.
- A. A. M. Aly, B. Walfort and H. Lang, Z. Kristallogr. NCS, 2004, 219, 489 Search PubMed.
- J. R. Black, W. Levason and M. Webster, Acta Crystallogr., 1995, C51, 623, DOI:10.1107/S0108270194012527.
- L. A. Dakin, P. C. Ong, J. S. Panek, R. J. Staples and P. Stavrospoulos, Organometallics, 2000, 19, 2896, DOI:10.1021/om0003786.
- A. K. Hijazi, H. Y. Yeong, Y. Zhang, E. Herdtweck, O. Nuyken and F. E. Kuhn, Macromol. Rapid Commun., 2007, 28, 670, DOI:10.1002/marc.200600139.
- Y. Zhang, W. Sun, C. Freund, A. M. Santos, E. Herdtweck, J. Mink and F. E. Kuhn, Inorg. Chim. Acta, 2006, 359, 4723, DOI:10.1016/j.ica.2005.12.044.
Footnote |
| † CCDC reference numbers 881135. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21290k |
| This journal is © The Royal Society of Chemistry 2012 |