Theoretical study of nitrogen-rich CN3− anion and related salts M+[CN3]− (M = Li, Na, K)†
Si-meng
Gao
and
Yi-hong
Ding
*
State Key Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130023, People's Republic of China. E-mail: yhdd@jlu.edu.cn
First published on 17th September 2012
Abstract
Hetero-doped nitrogen-rich compounds are of great interest for potential use in high energy density materials (HEDM) fields. In this paper, we report in detail the structures and stabilities of a tetra-atomic cluster CN3−, which is isoelectronic to the well-known N4. A series of higher-level energetic calculations were carried out at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d), CASPT2(12,12)/6-311+G(d)//CASSCF (12,12)/6-311+G(d) and CASPT2(12,12)/aug-cc-pVTZ//CASSCF(12,12)/aug-cc-pVTZ levels. The kinetic stability of CN3− was evaluated for the first time by studying the dissociation, isomerization and intersystem crossing barriers, as well as the Born–Oppenheimer molecular dynamic (BOMD) simulations. We found that the ground state isomer, i.e., chain-like triplet NCNN−(Cs), and a higher-energy isomer, i.e., tetrahedral-like singlet CN3−(C3v), are both kinetically stable, even when the intersystem crossing (ISC) is included. Therefore, the two isomers, 3NCNN− and 1CN3− (C3v), could be observable in future laboratory studies. By comparison with the isoelectronic N4 system, we conclude that the carbon-doping for XN3 (X = N to C−) greatly enhances the kinetic stability of the chain-like triplet and the tetrahedral-like singlet structures. Finally, for more practical use, our studies on the related salts M+[CN3]− (M = Li, Na, K) showed that both the chain-like 3NCNN− and tetrahedral-like 1CN3− could act as promising building blocks for novel HEDMs.
1. Introduction
Polynitrogen compounds are promising highly energetic materials with high energy densities that are environmentally benign. The decomposition of these materials would ideally produce only the environmentally safe dinitrogen. In the last 30 years, theoretical calculations have evaluated the structures and stabilities of numerous isomers for hypothetical Nn molecules with n ranging from 3 to 60.1–4 Although all calculations have shown their high potential energies with respect to molecular nitrogen, only selected isomers5 exhibit a unique property that makes them high energy density material (HEDM) candidates, i.e., sufficient energy barriers against dissociation to allow a relatively long lifetime. The reason is that nitrogen atoms have electron lone pairs repelling each other, which makes their single bonds much weaker than, for example, a carbon–carbon single bond. That also explains why, in more than 200 years of research since the discovery of N2, only a handful of nitrogen allotropes, like N3+, N3, N3−, N4+, N5+ and N5−,6–10 have been experimentally observed.The tetra-atomic all-nitrogen species, i.e., N4, has long been the subject of a large number of studies.1o,2i–x,3a–c,11–14 Theoretical investigations have consistently shown that the spin-allowed dissociation barrier of the high-energy tetrahedral N4 (Td) to 2N2 is around 60 kcal mol−1.2s,t Yet it has also been theoretically pointed out that the intersystem crossing would significantly reduce the kinetic stability of N4 (Td) to 28 kcal mol−1,2u which is still very considerable to stabilize N4 (Td). Thus, N4 (Td) has been long considered as one of the most powerful HEDM candidates. Unfortunately, up to now, the spectroscopic evidence for N4 (Td) is inconclusive due to the large difference between the theoretical and experimental 15N isotopic shifts.11b–11f In a 2002 experimental study, the open-chain N4 (3A′′) isomer was detected with a lifetime exceeding 1 μs based on neutralization–reionization mass spectrometry.11g,11h,13–14 The subsequent theoretical studies showed that although such a triplet chain-like structure is thermodynamically unstable with an exothermicity 22.7 kcal mol−1 to 3N2 + 1N2 , it needs to overcome a considerable barrier of 13.1 kcal mol−1, which sufficiently allows its experimental accessibility.
Hetero-doped nitrogen-rich compounds have been another recent hot topic.15–24 Compared to all-nitrogen species, hetero-doped nitrogen-rich clusters could be even more promising in synthesis. On one hand, suitable doping could reduce the strong repulsion between the neighboring N-lone pairs to increase the kinetic stability. On the other hand, such doping might help find appropriate synthetic precursors. Up to now, a series of nitrogen-rich compounds have been synthesized experimentally. For example, CN−, CN22−,17 C2N3−,18 C2N5−,20 C2N6−,21 C2N82−,22 C2N102−,23 C4N122−,24 CN7−16 and ON3+25 have been experimentally known species, while many of the corresponding pure nitrogen species remain unknown. In light of the improvement of the hetero-doping strategy on the experimental accessibility of nitrogen-rich clusters, chemists have devoted much time to the understanding of the structures and stabilities of diversely doped nitrogen-rich compounds.15
In the present work, we are interested in a nitrogen-rich tetra-atomic cluster, CN3−. It is isoelectronic to the well-known all-nitrogen N4. Bearing one negative charge, CN3− might act as a useful building block with the presence of cationic counterions, if it is intrinsically stable in kinetics. To the best of our knowledge, the structures and energetics of the CN3− isomers have been considered recently at a relatively low B3LYP/6-311+G(d)//B3LYP/6-311+G(d) level.26 Yet, the intrinsic kinetic stability of these isomers is unclear, which is vital for assessing their experimental accessibility27 and their potential as a HEDM building block. In addition, in view of the potential multireference character of CN3− isomers, precise multireference methods should be more appropriate for predicting the structures and stabilities of the related isomers of CN3−.
In this paper, we investigated the structures and stabilities of CN3− at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d), CASPT2(12,12)/6-311+G(d)//CASSCF(12,12)/6-311+G(d) and CASPT2(12,12)/aug-cc-pVTZ//CASSCF(12,12)/aug-cc-pVTZ) levels. To illustrate the kinetic stability of each CN3− isomer by evaluating the dissociation barriers and isomerization barriers, both singlet and triplet potential energy surfaces of CN3− were established at various precise levels. Both the spin-allowed and spin-forbidden potential energy surface calculations, as well as the Born–Oppenheimer molecular dynamics studies indicate that two distinct structures, i.e., chain-like 3NCNN− and tetrahedral-like 1CN3− (C3v), have good kinetic stability and have great possibility to be obtained in experiment. Finally, the stabilities of the salts M+[CN3]− (M = Li, Na, K) were investigated at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level, which leads us to conclude that the two isomers of the CN3− molecule could be promising building blocks in assembled nitrogen-rich clusters.
2. Computational methods
All of the calculations were carried out using the Gaussian03,28 Gaussian0929 and Molpro 200830 program packages. The optimized geometries and harmonic frequencies of minimum isomers and transition states were initially calculated using the B3LYP method31a–d with 6-311+G(d)32a–j basis sets. To check the connection of each transition state, the intrinsic reaction coordinate (IRC) calculations were carried out using the same theoretical methodology that was employed in the geometry optimization. Single-point calculations for all isomers and transition states were performed at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level.33–35 The simulations conducted in this study use Born–Oppenheimer molecular dynamics (BOMD), as implemented within the Gaussian series of electronic structure codes.36a–d These simulations utilize the B3LYP hybrid density functional with double-ζ polarized-diffused 6-31G(d) basis, as suggested from previous studies on similar systems.36e–i In light of the potential multireference character, all isomers and a portion of important transition state structures were re-optimized using the multi-configurational CASSCF method,38–40 in conjunction with the 6-311+G(d) and aug-cc-pVTZ37b,c basis set. A correct characterization of the CN3− potential energy surface requires that all isomers and transition states are treated equivalently. The active space in the CASSCF calculations was selected to fulfill this requirement, and included all molecular orbitals arising from the valence atomic carbon and nitrogen p-orbital. As a result, 12 electrons were distributed over 12 active orbitals, namely, CAS(12,12). The 8 inner orbitals were inactive and held doubly occupied in the CASSCF wave function.The harmonic vibrational frequencies were also obtained at the CASSCF(12,12)/6-311+G(d) and aug-cc-pVTZ level. To get more reliable relative energies, we further applied a modified version of CASPT2 (Complete Active Space with Second-order Perturbation Theory, developed by Celani and Werner,41a referred to as “RS2C” in Molpro), which accounts for dynamic correlation, using the CASSCF wavefunctions as references in the RS2C calculation. The two singlet–triplet PES conical intersection points were located by performing CASSCF(8,8)/6-311+G(d) and CASSCF(12,12)/6-311+G(d) calculations, respectively, using the Molpro 2008 program. Spin–orbit coupling (SOC) calculations were performed at the CASSCF(8,8)/6-311+G(d) level with the CASSCF wave function and Pauli–Breit Hamiltonian, including rigorous one and two-electron terms.41b–d
3. Results and discussions
We have performed a global isomeric search for CN3− in both the singlet and triplet electronic states using the grid-based comprehensive isomeric search42 strategies at the B3LYP/6-311+G(d) level. 8 CN3− minimum isomers (3 for singlet and 5 for triplet) (sm) and 10 interconversion transition states (5 for singlet and 5 for triplet) (sTSm/n) were obtained at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d). Note that the top left corner number s denotes the electronic state (1 for singlet and 3 for triplet) of the isomers or transition states. The optimized structures of all of the singlet and triplet CN3− species calculated using the B3LYP/6-311+G(d), CASSCF(12,12)/6-311+G(d) and CASSCF(12,12)/aug-cc-pVTZ methods are illustrated in Fig. 1. For comparison, the geometrical parameters obtained at the three levels are given in the notations. The energetic properties of CN3− species are listed in Table S1, ESI†. Finally, the schematic potential energy surfaces (PESs) showing the isomerization and dissociation of CN3− are presented in Fig. 2 (for triplet) and Fig. 3 (for singlet).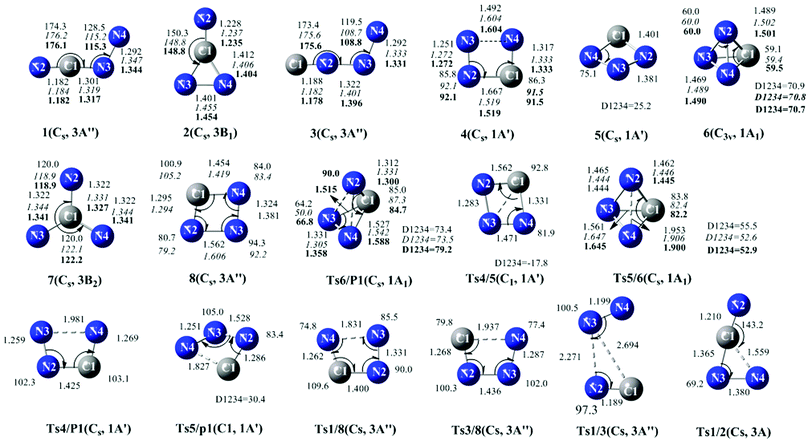 | ||
| Fig. 1 Optimized structures of isomers and interconversion transition states at the B3LYP/6-311+G(d) level. Parameters in italic are at the CASSCF(12,12)/6-311+G(d) level and parameters in bold are at the CASSCF(12,12)/aug-cc-pVTZ. The symmetry of Ts1/P1 is C1 at the CASSCF(12,12)/aug-cc-pVTZ level, while at the B3LYP/6-311+G(d) and CASSCF(12,12)/6-311+G(d) levels, both are in Cs symmetry. Bond lengths are in angstroms and angles in degrees. | ||
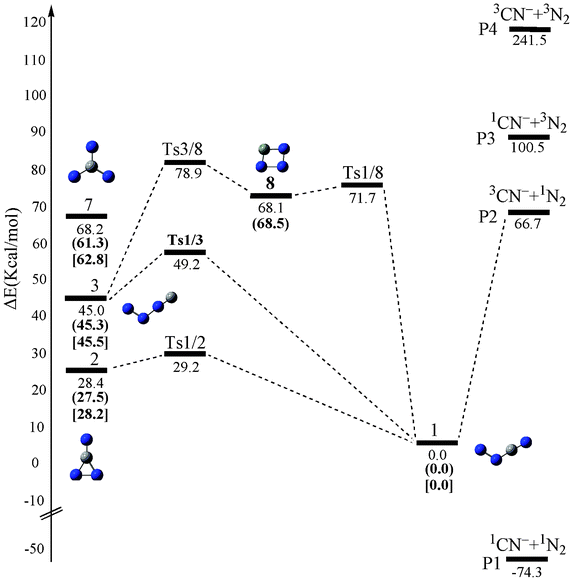 | ||
| Fig. 2 The schematic potential-energy surface of triplet CN3− at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level. The relative values in parentheses are at the CASPT2 (12,12)/6-311+G(d)//CASSCF(12,12)/6-311+G(d) level, the values in brackets are at the CASPT2(12,12)/aug-cc-pVTZ//CASSCF(12,12)/aug-cc-pVTZ level. | ||
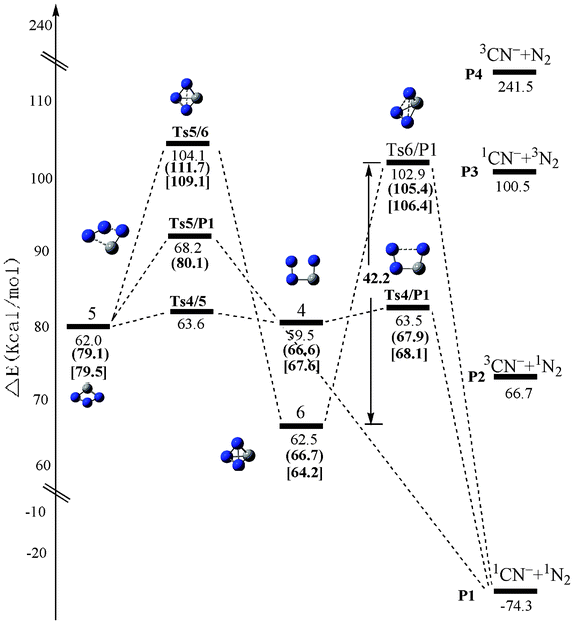 | ||
| Fig. 3 The schematic potential-energy surface of singlet CN3− at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level. The relative values in parentheses are at the CASPT2 (12,12)/6-311+G(d)//CASSCF(12,12)/6-311+G(d) level, the values in brackets are at the CASPT2(12,12)/aug-cc-pVTZ//CASSCF(12,12)/aug-cc-pVTZ level. | ||
3.1 Structures and energetics of CN3− isomers
The eight isomers are of the chain-like, three membered, four membered ring, tetrahedral-like forms, as shown in Fig. 1. As indicated in Table 1S, ESI†, CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d), CASPT2(12,12)/6-311+G(d)//CASSCF/(12,12)/6-311+G(d) and CASPT2(12,12)/aug-cc-pVTZ//CASSCF(12,12)/aug-cc-pVTZ used in this paper all predict 31 to be the ground state with the Cs symmetry. The structure of chain-like 31 can be described as resonating between the valence structures [·N2![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C1–N3
C1–N3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N4·]− and [·N2C1N3–N4·]−, which is consistent with the calculated Wiberg bond index (WBI) and bond distances, i.e.,WBIN2–C1, C1–N3, N3–N4 = 2.544, 1.331, 1.427 and RN2–C1, C1–N3, N3–N4 = 1.182, 1.301, 1.292 Å. Like the ground state of N4 of Cs symmetry, NCNN 31 also has two sets of delocalized π-orbitals on internal C–N bonding and has two sets of localized π-orbitals within N–C and N–N. As a result, the shorter inner C–N bond (1.301 Å), compared to the typical C–N single bond value 1.465 Å in H3CNH2, is due to the delocalized π-orbitals at the B3LYP/6-311+G(d) level. The N–N distance lies in between the typical NN double bond in HNNH (1.236 Å in cis and 1.238 Å in trans form) and N–N single bond in H2NNH2 (1.482 Å).
N4·]− and [·N2C1N3–N4·]−, which is consistent with the calculated Wiberg bond index (WBI) and bond distances, i.e.,WBIN2–C1, C1–N3, N3–N4 = 2.544, 1.331, 1.427 and RN2–C1, C1–N3, N3–N4 = 1.182, 1.301, 1.292 Å. Like the ground state of N4 of Cs symmetry, NCNN 31 also has two sets of delocalized π-orbitals on internal C–N bonding and has two sets of localized π-orbitals within N–C and N–N. As a result, the shorter inner C–N bond (1.301 Å), compared to the typical C–N single bond value 1.465 Å in H3CNH2, is due to the delocalized π-orbitals at the B3LYP/6-311+G(d) level. The N–N distance lies in between the typical NN double bond in HNNH (1.236 Å in cis and 1.238 Å in trans form) and N–N single bond in H2NNH2 (1.482 Å).
The structure of tetrahedral-like 16 (C3v) is very similar to the analogue N4 with Td symmetry. As shown in Fig. 1, for 16, the distance between the carbon atom and the three nitrogen atoms is 1.489 Å at the B3LYP/6-311+G(d) level; a little longer than the typical C–N single bond in H3CNH2 (1.465 Å) at the same level. The distance between N–N bond is 1.469 Å, which is a little shorter than the typical N–N single bond in H2NNH2 (1.482 Å). As shown in Fig. 1, geometrical parameters obtained using B3LYP method are close to those obtained at the CASSCF(12,12)/6-311+G(d) and CASSCF(12,12)/aug-cc-pVTZ level. Besides, the basis set between 6-311+G(d) and aug-cc-pVTZ have very little influence on the geometrical parameters of CN3− isomers. Usually, CASSCF overestimates the equilibrium distances. Yet luckily, this might be different here due to the rather large active space. The ground state of the chain-like 3NCNN− is mainly characterized by the open-shell electronic configuration. In the construction of CASSCF wave functions, the active spaces, including 12 electrons in 8a′ (9a′–16a′) and 4a′′ (1a′′–4a′′) orbitals, have been selected. The calculated value of the CI vector is 0.96 around the equilibrium position. The construction of the CASSCF wave function for the tetrahedral-like 16 includes 12 electrons in 8a′ (7a′–14a′) and 4a′′ (3a′′–6a′′) orbitals. The corresponding CI vector is 0.94 at the equilibrium position, which demonstrates that the multi-reference description is warranted.
As is well known, the high dissociation barrier of tetrahedral N4 originates from its inherent orbital nature. The decomposition route to 2N2 is prohibited by the orbital symmetry rule, which means that a lot more energy should be required for the reaction.2s,r For CN3− (C3v), its configuration from CASSCF calculations in Cs symmetry is:
1A′: 1a′1a′′2a′3a′4a′2a′′5a′6a′7a′3a′′8a′9a′10a′4a′′.
This clearly differs from the configuration of 1CN−+N2, i.e., 1A′: 1a′2a′3a′4a′5a′6a′7a′8a′9a′10a′1a′′11a′2a′′12a′.
The configuration of the associated spin-allowed dissociation transition state, i.e., 1A′: 1a′1a′′2a′3a′4a′5a′2a′′6a′7a′8a′3a′′9a′4a′′10a′, is the mixing of the reactant (CN3− (C3v)) and the product (1CN−+N2). Thus, the dissociation from 1 to 1CN−+N2 is an orbital symmetry forbidden process.
3.2 Stability of ground state 3CN3− (Cs)
The schematic potential energy surface (PES) of the triplet CN3− system is depicted in Fig. 2. Different from the potential energy surfaces of N4, the dissociation of the ground state 31 is found to be an endothermic process with reaction energy of 87.4 kcal mol−1. This clearly contrasts with the exothermic fragmentation of 3NNNN→ 3N2 + 1N2. Thus, the hetero-doping of N by C− can increase the thermodynamic stability of NXNN.| NCNN 31 → 3CN− + 1N2 | (a) |
This process corresponds to a simple bond cleavage without a transition structure.
As shown in Fig. 2, the remaining three isomers, i.e., 32, 33 and 38, on the triplet potential energy surface each have a small energy barrier towards the isomerization to the ground isomer, i.e., less than 5 kcal mol−1 at CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level. Thus, they could be viewed as kinetically unstable. For the higher-energy branched isomer 37, we did not locate any transition state connecting it. Yet, our BOMD calculations (see Fig. 4) showed that 37 should also be kinetically unstable with its lifetime being less than 0.2 ps.
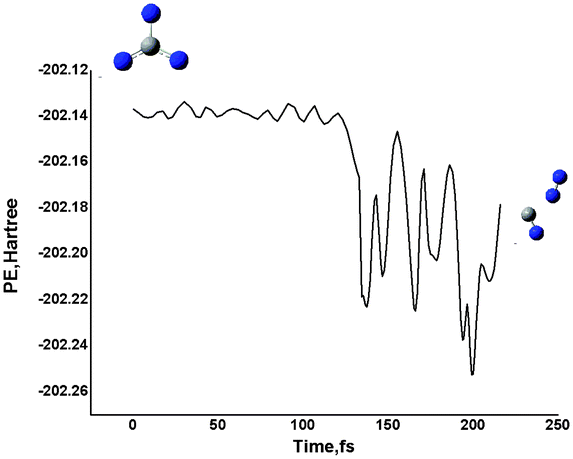 | ||
| Fig. 4 BOMD simulation of 7 CN3−(Cs) at 300 K at the B3LYP/6-31G(d) level. Potential energy (in au) versus time (in fs). | ||
It’s of interest to inspect the singlet dissociation products 1CN− + 1N2, which have lower energies than the triplet 1 (i.e., −74.3 kcal mol−1 at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level). To reach the singlet product, a crossing and spin inversion process needs to take place in the reaction pathway. Spin inversion is a nonadiabatic process and we need to inspect a crossing seam on the singlet and triplet PESs to determine the mechanism of intersystem crossing (ISC).
We first carried out point-wise calculations for both the 1A′ and 3A′′ surfaces of NCNN− in Cs symmetry at a series of the R(C1–N3) bond values (a set of partial geometry optimization calculations at the fixed R(C1–N3) values). We found that at the R(C1–N3) value of 1.451 Å, the 1A′ and 3A′′ reaction systems have equal CASSCF(8,8)/6-311+G(d) energy values of −201.8019 a.u. Thus, at this point of geometry, we performed CASSCF(8,8)/6-311+G(d) conical intersection calculations. The conical intersection region of the reaction system from 3NCNN− to 3CN−+N2 was located at the following crossing point geometry (see Table S3, ESI†) and energy at the conical intersection was −201.7769 a.u. at the CASPT2(8,8)/6-311+G(d)//CASSCF(8,8)/6-311+G(d) level (the energy difference between the singlet and triplet states being 0.43 × 10−7 kcal mol−1). At the same level, the energy of NCNN (3A˝) was calculated to be −201.8097 a.u., which is lower than the energy at the conical intersection of 20.6 kcal mol−1. Therefore, the reaction system will access a relatively lower energy pathway by changing its spin multiplicity via the minimum energy crossing point (MECP). However, the kind of mechanism (non-adiabatic or adiabatic) involved will depend on the magnitude of the transition probability. Among the factors that affect the magnitude of the transition probability is the spin–orbit coupling (SOC) interaction between the two states. Therefore, the strength of SOC is the key to understanding the mechanism. Although the crossing has an energy of 20.6 kcal mol−1 relative to that of 3NCNN−, the corresponding SOC constant is comparatively small (23.9 cm−1). Thus, ISC at chain-like geometries seems unlikely because of the small SOC involved. The situation of 3NCNN− contrasts sharply with that of NNNN (3A′′), the ground isomer of N4, for which 1) the fragmentation is exothermic by 22.7 kcal mol−1, 2) there is a fragmentation barrier of 13.1 kcal mol−1 and 3) there is no ISC. Thus, we can say that in comparison to NNNN (3A′′), the stability of 3NCNN− is much increased.
The Born–Oppenheimer molecular dynamic (BOMD)36a–d calculations at the B3LYP/6-31G(d) level further supported the good stability of the chain-like CN3−. As shown in Fig. 5, we recorded a movie of the MD simulation at 300 K. The simulation indicates that NCNN− (3A′′) could keep the original structure well for at least 100 ps, sufficiently allowing for future spectroscopic detection. Thus, we conclude that NCNN− (Cs) should be considered as thermally stable.
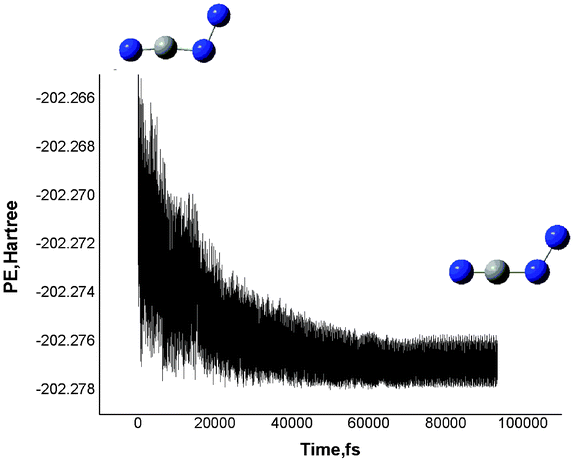 | ||
| Fig. 5 The BOMD simulation of ground state 1 CN3−(Cs) at 300 K at the B3LYP/6-31G(d) level. Potential energy (in au) versus time (in fs). | ||
3.3 Stability of 1CN3− (C3v)
The schematic potential energy surface (PES) of the singlet CN3− system is depicted in Fig. 3. The corresponding total energies and relative energies are also listed in Table S1, ESI†. Isomer 6 can release a large amount of heat of 136.8 kcal mol−1 when the fragmentation is considered (1CN−+N2). Such a large heat is indicative of the potential of tetrahedral-like 6 as a HEDM candidate, if it is kinetically stable. As shown in Fig. 3, isomer 6 can convert to a four-membered ring isomer 5 by breaking one N–N bond via the transition state 1Ts5/6. This process needs to overcome a high barrier of 45.0 kcal mol−1 at the CASPT2(12,12)/6-311+G(d)//CASSCF(12,12)/6-311+G(d) level. Alternatively, isomer 6 can directly dissociate to the fragments 1CN−+N2via a concerted transition state 1Ts6/P1 by breaking two N–N bonds at the same time. The corresponding barrier is 38.6 kcal mol−1 at this level. Thus, the fragmentation is the rate-determining step of the tetrahedral-like isomer 6 on the singlet PES. At our highest CASPT2(12,12)/aug-cc-pVTZ//CASSCF(12,12)/aug-cc-pVTZ level, the dissociation barrier is increased to be as considerable as 42.2 kcal mol−1. Such a value should sufficiently stabilize CN3− (C3v).The BOMD calculations at the B3LYP/6-31G(d) level further supported the good kinetic stability of the tetrahedral-like CN3−. As shown in Fig. 6, we recorded a movie of the MD simulation at 300 K. The simulation indicates that CN3− (C3v) could keep the original structure well for at least 25 ps, sufficiently allowing for future spectroscopic detection. Thus, we conclude that CN3− (C3v) should be considered as kinetically stable.
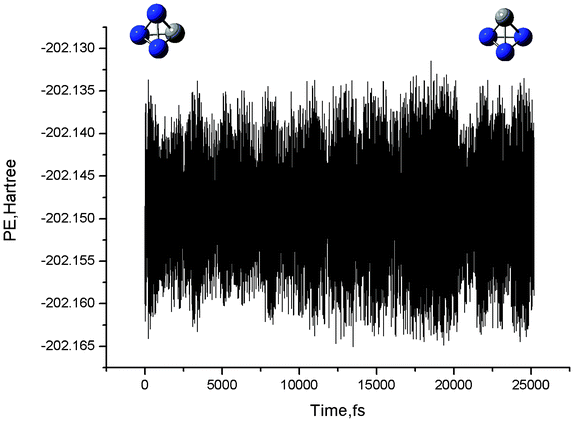 | ||
| Fig. 6 BOMD simulation of 6 CN3−(C3v) at 300 K at the B3LYP/6-31G(d) level. Potential energy (in a.u.) versus time (in fs). | ||
It’s of interest to inspect whether the tetrahedral-like isomer 6 can undergo the intersystem crossing like N4. To our surprise, different from the potential energy surfaces of N4, the triplet dissociation products 3CN− + 1N2 have higher energies than the singlet 6 (i.e., 4.2 kcal mol−1 at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level). We carried out point-wise calculations for both the 1A′ and 3A′′ surfaces of CN3− in C3v symmetry at a series of ∠(N2–C1–N4) and ∠(N2–C1–N3) values (two sets of partial geometry optimization calculations at the fixed angle ∠(N2–C1–N4)). The result showed that for CN3− (C3v), the singlet–triplet crossing point is very close to the singlet transition state. Thus, for exploring the 1A′–3A′′ conical intersection, we performed CASSCF(12,12)/6-311+G(d) conical intersection calculations and the CASSCF(12,12)/6-311+G(d) Ts6/P1 geometry was taken as a starting geometry. The conical intersection region of the reaction system from 1CN3− to 1CN−+N2 was located at the following CN3− geometry (see Table S4, ESI†) and the energy at the conical intersection was −201.643024 a.u. at the CASPT2(12,12)/6-311+G(d)//CASSCF(12,12)/6-311+G(d) level (the energy difference between the singlet and triplet states is 0.4 × 10−7 a.u.). At the same level, the energy of Ts6/P1 (1A′) was calculated to be −201.645160 a.u., which is lower than the CASPT2(12,12)/6-311+G(d)//CASSCF(12,12)/6-311+G(d) energy at the conical intersection 1.3 kcal mol−1. The ∠(N2–C1–N4) and ∠(N2–C1–N3) angle values in the CASSCF(12,12)/6-311+G(d) geometry of Ts6/P1 (1A′) are 87.3° and 90.0°, respectively, which are both a little larger than the value of 80.2 in the geometry for the conical intersection. Hence, we conclude that the conical intersection of CN3− PES occurs before Ts6/P1 (1A′), but away from the singlet minimum energy path. As a result, the singlet–triplet intersystem crossing (6→P23CN−+N2) does not influence the kinetic stability of isomer 6. We performed CASSCF(8,8)/6-311+G(d) calculations for the 1A′–3A′′ spin–orbital coupling constant at the conical intersection geometry and the obtained spin–orbital coupling constant values 18.5 cm−1 (very small). Since the energy of Ts6/P1 is lower than that of the 1A′–3A′′ conical intersection and the spin–orbit coupling between the 1A′ and 3A′′ states (at the conical intersection geometry) is rather small, there is little possibility for a process from 1A′ PES to 3A′′ PES via an intersystem transition. We concluded that the intersection between 1A′ and 3A′′ PESs does not cause a predissociation of CN3− into the product 3CN−+1N2.
It's of interest to compare with the well-known N4 (Td), for which the intersystem crossing can significantly (by half) reduce the dissociation barrier to 28 kcal mol−1. Thus, though the spin-allowed barrier of CN3− (C3v) (around 42 kcal mol−1) is much smaller than that (around 60 kcal mol−12s,t) of N4 (Td), the overall kinetic stability (42 kcal mol−1) of the former is much larger than that (28 kcal mol−1) of the latter when the intersystem crossing is included. So the singlet–triplet intersystem crossing does not have an effect on the dissociation stability of isomer 6. Overall, we can say that due to the C-doping, the kinetic stability of the tetrahedral-like structure CN3− is raised relative to N4.
3.4 Stability of M+[CN3]− (M = Li, Na, K)
The good kinetic stability and the anionic nature give us hope that two isomers of CN3− could serve as a promising building block for complexes. To test this hypothesis, we studied the salt-like complexes M+[CN3]− (X = Li, Na, K) following the “assembly” strategy43 at the B3LYP/6-311+G(d) level, which allows the M-atom to position at various directions of the designated isomer. The located isomeric structures of M = Li, Na and K and their relative energies are all illustrated in Fig. 7, 8 and 9, respectively. It can be seen that with presence of three types of counterions, the triplet NCNN− skeleton can still persist as the global minima. Moreover, the decomposition of the triplet complexed NCNN− is still an endothermic process, though the endothermicity becomes less than that of the naked NCNN−. Thus, the triplet NCNN− has good stability with the presence of counterions.![Optimized structures of Li+[CN3]− at the B3LYP/6-311+G(d) level. The values in parentheses (kcal mol−1) are the relative energies calculated at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level.](/image/article/2012/RA/c2ra21250a/c2ra21250a-f7.gif) | ||
| Fig. 7 Optimized structures of Li+[CN3]− at the B3LYP/6-311+G(d) level. The values in parentheses (kcal mol−1) are the relative energies calculated at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level. | ||
![Optimized structures of Na+[CN3]− at the B3LYP/6-311+G(d) level. The values in parentheses (kcal mol−1) are the relative energies calculated at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level.](/image/article/2012/RA/c2ra21250a/c2ra21250a-f8.gif) | ||
| Fig. 8 Optimized structures of Na+[CN3]− at the B3LYP/6-311+G(d) level. The values in parentheses (kcal mol−1) are the relative energies calculated at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level. | ||
![Optimized structures of K+[CN3]− at the B3LYP/6-311+G(d) level. The values in parentheses (kcal mol−1) are the relative energies calculated at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level.](/image/article/2012/RA/c2ra21250a/c2ra21250a-f9.gif) | ||
| Fig. 9 Optimized structures of K+[CN3]− at the B3LYP/6-311+G(d) level. The values in parentheses (kcal mol−1) are the relative energies calculated at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level. | ||
For the tetrahedral-like CN3−, we focus on its fragmentation and isomerization stability with the counterion-complexation, whose channels are presented in Fig. 10, 11 and 12. It can be found that various isomeric forms can be formed when the counterions attach to the tetrahedra with close energies. Besides, they could easily undergo the interconversion to each other via the appropriate M+-migration (for example see Fig. 10 with M = Li). Taking Li+[CN3]− as an example (see Fig. 10), at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level, the conversion from the tetrahedral-like structure to the four-membered ring structure (1Li-15→1Li-13) and to the fragments (1Li-18→1LiCN + N2 and 1Li-20→1LiCN + N2) have to consume high barriers. The associated barriers are as considerable as 33.5, 35.6 and 36.8 kcal mol−1. Clearly, the salt-like form M+[CN3]− (M = Li) has good kinetic stability. The main features of the PESs of Na+[CN3]− (Fig. 11) and K+[CN3]− (Fig. 12) are similar to that of Li+[CN3]−. The Na+ and K+ can easily migrate around the CN3− (C3v) moiety. The lowest fragmentation barrier is 32.7 kcal mol−1 and 35.4 kcal mol−1, respectively.
![The schematic potential-energy surface of singlet Li+[CN3]− at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level. The relative values are at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level.](/image/article/2012/RA/c2ra21250a/c2ra21250a-f10.gif) | ||
| Fig. 10 The schematic potential-energy surface of singlet Li+[CN3]− at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level. The relative values are at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level. | ||
![The schematic potential-energy surface of singlet Na+[CN3]− at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) +ZPVE level. The relative values are at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level.](/image/article/2012/RA/c2ra21250a/c2ra21250a-f11.gif) | ||
| Fig. 11 The schematic potential-energy surface of singlet Na+[CN3]− at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) +ZPVE level. The relative values are at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level. | ||
![The schematic potential-energy surface of singlet K+[CN3]− at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level. The relative values are at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level.](/image/article/2012/RA/c2ra21250a/c2ra21250a-f12.gif) | ||
| Fig. 12 The schematic potential-energy surface of singlet K+[CN3]− at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level. The relative values are at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d)+ZPVE level. | ||
Conclusions
At various levels, including the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d), CASPT2(12,12)/6-311+G(d)//CASSCF(12,12)/6-311+G(d) and CASPT2(12,12)/aug- cc-pVTZ//CASSCF(12,12)/aug-cc-pVTZ, we for the first time theoretically investigate the structures and stability of a tetra-atom molecule CN3− which is isoelectronic to the well-known N4. Our results showed that two isomers 3NCNN− and 1CN3− (C3v) have great possibility to be obtained in experiment. Besides the global minimum 3NCNN−, the tetrahedral-like isomer of CN3− (C3v) has very high barriers, both for dissociation and isomerization. Although the spin-allowed barrier (around 42 kcal mol−1) of CN3− (C3v) is much lower than that of N4 (around 60), the overall kinetic stability (around 42 kcal mol−1) is greatly improved when the intersystem crossing is included. Similar to the case of the CN3− unit, the rate-determining step of the salt-like M+[CN3]− is associated with the fragmentation to MCN+N2. The corresponding barriers are 33.5, 32.7 and 35.4 kcal mol−1 for M = Li, Na and K, respectively. These large values show that the CN3−-based salts are kinetically very stable. Future laboratory studies on the tetra-atom CN3− are greatly desired.Acknowledgements
This work was funded by the National Key Basic Research and Development Program of China under Grant No. 2010CB327701, the National Natural Science Foundation of China (No. 20773054, 60977024, 21073074), Doctor Foundation by the Ministry of Education (20070183028), Excellent Young Teacher Foundation of Ministry of Education of China, Excellent Young People Foundation of Jilin Province (20050103) and Program for New Century Excellent Talents in University (NCET).References
- (a) T. W. Archibald and J. R. Sabin, J. Chem. Phys., 1971, 55, 1821 CrossRef CAS; (b) J. M. Dyke, N. B. H. Jonathan, A. E. Lewis and A. Morris, Mol. Phys., 1982, 47, 1231 CrossRef CAS; (c) J. N. Murrell, O. Novaro, S. Castillo and V. Saunders, Chem. Phys. Lett., 1982, 90, 421 CrossRef CAS; (d) C. Petrongolo, J. Mol. Struct., 1988, 175, 215 CrossRef CAS; (e) J. M. L. Martin, J. P. Francois and R. Gijbels, J. Chem. Phys., 1989, 90, 6469 CrossRef CAS; (f) D. Yu, A. Rauk and D. A. Armstrong, J. Phys. Chem., 1992, 96, 6031 CrossRef CAS; (g) P. Zhang, K. Morokuma and A. M. Wodtke, J. Chem. Phys., 2005, 122, 014106 CrossRef; (h) D. Babikov, P. Zhang and K. Morokuma, J. Chem. Phys., 2004, 121, 6743 CrossRef CAS; (i) M. Bittererova, H. Ostmark and T. Brinck, J. Chem. Phys., 2002, 116, 9740 CrossRef CAS; (j) R. Prasad, J. Chem. Phys., 2003, 119, 9549 CrossRef CAS; (k) M. J. Pellerite, R. L. Jackson and J. I. Brauman, J. Phys. Chem., 1981, 85, 1624 CrossRef CAS; (l) D. Babikov, V. A. Mozhayskiy and A. I. Krylov, J. Chem. Phys., 2006, 125, 084306 CrossRef; (m) V. A. Mozhayskiy, D. Babikov and A. I. Krylov, J. Chem. Phys., 2006, 124, 224309 CrossRef; (n) Z. L. Cai, Y. F. Wang and H. M. Xiao, Chem. Phys., 1992, 164, 377 CrossRef CAS; (o) T. J. Venanzi and J. M. Schulman, Mol. Phys., 1975, 30, 281 CrossRef CAS; (p) J. S. Wright, J. Am. Chem. Soc., 1974, 96, 4753 CrossRef CAS; (q) R. Tarroni and P. Tosi, Chem. Phys. Lett., 2004, 389, 274 CrossRef CAS; (r) Y. G. Byun, S. Saebo and C. U. Pittman, J. Am. Chem. Soc., 1991, 113, 3689 CrossRef CAS.
- (a) F. M. Bickelhaupt, R. Hoffmann and R. D. Levine, J. Phys. Chem. A, 1997, 101, 8255 CrossRef CAS; (b) S. C. Decastro and H. F. Schaefer, J. Chem. Phys., 1981, 74, 550 CrossRef CAS; (c) L. B. Knight, K. D. Johannessen, D. C. Cobranchi, E. A. Earl, D. Feller and E. R. Davidson, J. Chem. Phys., 1987, 87, 885 CrossRef CAS; (d) S. Williams, R. A. Dressler and Y. H. Chiu, J. Chem. Phys., 1999, 111, 9634 CrossRef CAS; (e) D. C. Conway, J. Chem. Phys., 1975, 63, 2219 CrossRef CAS; (f) J. H. Langenberg, I. B. Bucur and P. Archirel, Chem. Phys., 1997, 221, 225 CrossRef CAS; (g) C. Leonard, P. Rosmus, S. Carter and N. C. Handy, J. Phys. Chem. A, 1999, 103, 1846 CrossRef CAS; (h) I. Carmichael, J. Phys. Chem., 1994, 98, 5044 CrossRef CAS; (i) M. F. Guest, I. H. Hillier and V. R. Saunders, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 2070 RSC; (j) G. Trinquier, J. P. Malrieu and J. P. Daudey, Chem. Phys. Lett., 1981, 80, 552 CrossRef CAS; (k) S. Elbel, J. Kudnig, M. Grodzicki and H. Lempka, Chem. Phys. Lett., 1984, 109, 312 CrossRef CAS; (l) A. Y. Shibaev and Y. V. Puzanov, J. Struct. Chem., 1986, 27, 472 CrossRef; (m) M. N. Glukhovtsev, B. Y. Simkin and V. I. Minkin, J. Struct. Chem., 1987, 28, 483 CrossRef; (n) B. Y. Simkin, M. N. Glukhovtsev and V. I. Minkin, Zh. Org. Khim., 1988, 24, 24 Search PubMed; (o) J. M. Seminario and P. Politzer, Chem. Phys. Lett., 1989, 159, 27 CrossRef CAS; (p) M. M. Francl and J. P. Chesick, J. Phys. Chem., 1990, 94, 526 CrossRef CAS; (q) T. J. Lee and J. E. Rice, J. Chem. Phys., 1991, 94, 1215 CrossRef CAS; (r) W. J. Lauderdale, J. F. Stanton and R. J. Bartlett, J. Phys. Chem., 1992, 96, 1173 CrossRef CAS; (s) K. M. Dunn and K. Morokuma, J. Chem. Phys., 1995, 102, 4904 CrossRef CAS; (t) A. A. Korkin, A. Balkova, R. J. Bartlett, R. J. Boyd and P. V. Schleyer, J. Phys. Chem., 1996, 100, 5702 CrossRef CAS; (u) D. R. Yarkony, J. Am. Chem. Soc., 1992, 114, 5406 CrossRef CAS; (v) M. N. Glukhovtsev and P. V. Schleyer, Int. J. Quantum Chem., 1993, 46, 119 CrossRef CAS; (w) M. N. Glukhovtsev and S. Laiter, J. Phys. Chem., 1996, 100, 1569 CrossRef CAS; (x) M. Bittererova, T. Brinck and H. Ostmark, J. Phys. Chem. A, 2000, 104, 11999 CrossRef CAS.
- (a) M. T. Nguyen, T. L. Nguyen, A. M. Mebel and R. Flammang, J. Phys. Chem. A, 2003, 107, 5452 CrossRef CAS; (b) M. Bittererova, H. Ostmark and T. Brinck, Chem. Phys. Lett., 2001, 347, 220 CrossRef CAS; (c) S. Evangelisti, Int. J. Quantum Chem., 2004, 96, 598 CrossRef CAS; (d) M. T. Nguyen and T. K. Ha, Chem. Phys. Lett., 2000, 317, 135 CrossRef CAS; (e) X. Wang, H. R. Hu, A. M. Tian, N. B. Wong, S. H. Chien and W. K. Li, Chem. Phys. Lett., 2000, 329, 483 CrossRef CAS; (f) M. T. Nguyen and T. K. Ha, Chem. Phys. Lett., 2001, 335, 311 CrossRef CAS; (g) M. R. Manaa, Chem. Phys. Lett., 2000, 331, 262 CrossRef CAS; (h) M. T. Nguyen, M. A. Mc Ginn and A. F. Hegarty, Polyhedron, 1985, 4, 1721 CrossRef CAS; (i) M. T. Nguyen, M. Sana and G. Leroy, J. Can. Chem. (Rev. Can. Chim.), 1983, 61, 1435 CrossRef; (j) M. T. Nguyen and T. K. Ha, Chem. Ber., 1996, 129, 1157 CrossRef CAS; (k) T. K. Ha, R. Cimiraglia and M. T. Nguyen, Chem. Phys. Lett., 1981, 83, 317 CrossRef CAS; (l) P. Saxe and H. F. Schaefer, J. Am. Chem. Soc., 1983, 105, 1760 CrossRef CAS; (m) H. Huber, T. K. Ha and M. T. Nguyen, J. Mol. Struct.: THEOCHEM, 1983, 14, 351 CrossRef CAS; (n) M. Ramek, J. Mol. Struct.: THEOCHEM, 1984, 18, 391 CrossRef CAS; (o) R. Engelke, J. Phys. Chem., 1989, 93, 5722 CrossRef CAS; (p) R. Engelke, J. Phys. Chem., 1990, 94, 6924 CrossRef CAS; (q) M. T. Nguyen, J. Phys. Chem., 1990, 94, 6923 CrossRef; (r) T. K. Ha and M. T. Nguyen, Chem. Phys. Lett., 1992, 195, 179 CrossRef CAS; (s) R. Engelke, J. Phys. Chem., 1992, 96, 10789 CrossRef CAS; (t) B. M. Gimarc and M. Zhao, Inorg. Chem., 1996, 35, 3289 CrossRef CAS; (u) M. Bittererova, T. Brinck and H. Ostmark, Chem. Phys. Lett., 2001, 340, 597 CrossRef CAS.
- (a) M. Tobita and R. J. Bartlett, J. Phys. Chem. A, 2001, 105, 4107 CrossRef CAS; (b) M. N. Glukhovtsev and P. V. Schleyer, Chem. Phys. Lett., 1992, 198, 547 CrossRef CAS; (c) T. M. Klapotke, J. Mol. Struct.: THEOCHEM, 2000, 499, 99 CrossRef CAS; (d) L. Gagliardi, S. Evangelisti, V. Barone and B. O. Roos, Chem. Phys. Lett., 2000, 320, 518 CrossRef CAS; (e) L. J. Wang, P. Warburton and P. G. Mezey, J. Phys. Chem. A, 2002, 106, 2748 CrossRef CAS; (f) Q. S. Li and Y. D. Liu, J. Phys. Chem. A, 2002, 106, 9538 CrossRef CAS; (g) H. H. Michels, J. A. Montgomery, K. O. Christe and D. A. Dixon, J. Phys. Chem., 1995, 99, 187 CrossRef CAS; (h) C. K. Law, W. K. Li, X. Wang, A. Tian and N. B. Wong, J. Mol. Struct.: THEOCHEM, 2002, 617, 121 CrossRef CAS; (i) Y. D. Liu, J. F. Zhao and Q. S. Li, Theor. Chem. Acc., 2002, 107, 140 CrossRef CAS; (j) J. F. Zhao and Q. S. Li, Chem. Phys. Lett., 2003, 368, 12 CrossRef CAS; (k) Q. S. Li, X. Hu and W. Xu, Chem. Phys. Lett., 1998, 287, 94 CrossRef CAS; (l) X. Wang, Y. Ren, M. B. Shuai, N. B. Wong, W. K. Li and A. M. Tian, J. Mol. Struct.: THEOCHEM, 2001, 538, 145 CrossRef CAS; (m) X. Wang, A. M. Tian, N. B. Wong, C. K. Law and W. K. Li, Chem. Phys. Lett., 2001, 338, 367 CrossRef CAS; (n) R. Engelke and J. R. Stine, J. Phys. Chem., 1990, 94, 5689 CrossRef CAS; (o) M. W. Schmidt, M. S. Gordon and J. A. Boatz, Int. J. Quantum Chem., 2000, 76, 434 CrossRef CAS.
- M. N. Glukhovtsev, H. J. Jiao and P. V. Schleyer, Inorg. Chem., 1996, 35, 7124 CrossRef CAS.
- (a) T. Curtius, Berichte, 1890, 23, 3023 CrossRef; (b) B. A. Thrush, Proc. R. Soc. London, Ser. A, 1956, 235, 143 CrossRef; (c) A. E. Douglas and W. J. Jones, Can. J. Phys., 1965, 43, 2216 CrossRef CAS; (d) M. Winnewisser and R. L. Cook, J. Chem. Phys., 1964, 41, 999 CrossRef CAS; (e) C. R. Brazier, P. F. Bernath, J. B. Burkholder and C. J. Howard, J. Chem. Phys., 1988, 89, 1762 CrossRef CAS.
- (a) F. Carnovale, J. B. Peel and R. G. Rothwell, J. Chem. Phys., 1988, 88, 642 CrossRef CAS; (b) K. Norwood, G. Luo and C. Y. Ng, J. Chem. Phys., 1989, 91, 849 CrossRef CAS; (c) V. Aquilanti, M. Bartolomei, D. Cappelletti, E. Carmona-Novillo and F. Pirani, Phys. Chem. Chem. Phys., 2001, 3, 3891 RSC; (d) V. Aquilanti, M. Bartolomei, D. Cappelletti, E. Carmona-Novillo and F. J. Pirani, Chem. Phys., 2002, 117, 615 CAS; (e) L. B. Knight, K. D. Johannessen, D. C. Cobranchi, E. A. Earl, D. Feller and E. R. Davidson, J. Chem. Phys., 1987, 87, 885 CrossRef CAS; (f) S. Williams, R. A. Dressler and Y. H. Chiu, J. Chem. Phys., 1999, 111, 9634 CrossRef CAS; (g) I. Carmichael, J. Phys. Chem., 1994, 98, 5044 CrossRef CAS; (h) M. Saporoschenko, Phys. Rev., 1958, 111, 1550 CrossRef CAS; (i) W. E. Thompson and M. E. Jacox, J. Chem. Phys., 1990, 93, 3856 CrossRef CAS; (j) T. Ruchti, T. Speck, J. P. Connelly, E. J. Bieske, H. Linnartz and J. P. Maier, J. Chem. Phys., 1996, 105, 2591 CrossRef CAS; (k) H. Bohringer, F. Arnold, D. Smith and N. G. Adams, Int. J. Mass Spectrom. Ion Phys., 1983, 52, 25 CrossRef; (l) L. G. McKnight, K. B. McAfee and D. P. Sipler, Phys. Rev., 1967, 164, 62 CrossRef CAS; (m) M. Whitaker, M. A. Biondi and R. Johnsen, Phys. Rev. A, 1981, 24, 743 CrossRef CAS; (n) M. F. Jarrold, A. J. Illies and M. T. Bowers, J. Chem. Phys., 1984, 81, 214 CrossRef CAS; (o) T. F. Magnera, D. E. David and J. Michl, Chem. Phys. Lett., 1986, 123, 327 CrossRef CAS; (p) G. P. Smith and L. C. Lee, J. Chem. Phys., 1978, 69, 5393 CrossRef CAS; (q) S. C. Ostrander and J. C. Weisshaar, Chem. Phys. Lett., 1986, 129, 220 CrossRef CAS.
- (a) R. Huisgen, Angew. Chem., 1956, 68, 705 CrossRef CAS; (b) J. D. Wallis and J. D. Dunitz, J. Chem. Soc., Chem. Commun., 1983, 910 RSC; (c) R. N. Butlers, S. Collier and A. F. M. Fleming, J. Chem. Soc., Perkin Trans. 2, 1996, 801 RSC; (d) R. N. Butler, A. Fox, S. Collier and L. A. Burke, J. Chem. Soc., Perkin Trans. 2, 1998, 2243 RSC; (e) M. Witanowski, L. Stefaniak, H. Januszewski, K. Bahadur and G. A. Webb, J. Cryst. Mol. Struct., 1975, 5, 137 CrossRef CAS; (f) R. Muller, J. D. Wallis and W. Vonphilipsborn, Angew. Chem., Int. Ed., 1985, 24, 513 CrossRef; (g) P. Carlqvist, H. Ostmark and T. Brinck, J. Phys. Chem. A, 2004, 108, 7463 CrossRef CAS.
- (a) K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, J. Fluorine Chem., 2000, 101, 151 CrossRef CAS; (b) K. O. Christe, X. Z. Zhang, J. A. Sheehy and R. Bau, J. Am. Chem. Soc., 2001, 123, 6338 CrossRef CAS; (c) R. Haiges, S. Schneider, T. Schroer and K. O. Christe, Angew. Chem., Int. Ed., 2004, 43, 4919 CrossRef CAS; (d) K. O. Christe, W. W. Wilson, J. A. Sheehy and J. A. Boatz, Angew. Chem., Int. Ed., 1999, 38, 2004 CrossRef CAS.
- P. C. Samartzis and A. M. Wodtke, Int. Rev. Phys. Chem., 2006, 25, 527 CrossRef CAS.
- (a) J. P. Zheng, J. Waluk, J. Spanget-Larsen, D. M. Blake and J. G. Radziszewski, Chem. Phys. Lett., 2000, 328, 227 CrossRef CAS; (b) T. J. Lee and J. M. L. Martin, Chem. Phys. Lett., 2002, 357, 319 CrossRef CAS; (c) S. A. Perera and R. J. Bartlett, Chem. Phys. Lett., 1999, 314, 381 CrossRef CAS; (d) M. Bittererova, T. Brinck and H. Ostmark, Chem. Phys. Lett., 2001, 340, 597 CrossRef CAS; (e) H. Ostmark, O. Launila, S. Wallin and R. Tryman, J. Raman Spectrosc., 2001, 32, 195 CrossRef CAS; (f) T. J. Lee and C. E. Dateo, Chem. Phys. Lett., 2001, 345, 295 CrossRef CAS; (g) F. Cacace, Chem. – Eur. J., 2002, 8, 3839 Search PubMed; (h) F. Cacace, G. de Petris and A. Troiani, Science, 2002, 295, 480 CrossRef CAS.
- M. T. Nguyen, T. L. Nguyen, A. M. Mebel and R. Flammang, J. Phys. Chem. A, 2003, 107, 5452 CrossRef CAS.
- E. E. Rennie and P. M. Mayer, J. Chem. Phys., 2004, 120, 10561 CrossRef CAS.
- J. Barber, D. E. Hof, C. A. Meserole and D. J. Funk, J. Phys. Chem. A, 2006, 110, 3853 CrossRef CAS.
- (a) X. L. Zhu, X. H. Lu and X. Feng, Spectrochim. Acta, Part A, 2007, 67, 756 CrossRef; (b) A. Hammerl and T. M. Klapotke, Inorg. Chem., 2002, 41, 906 CrossRef CAS; (c) L. J. Wang, M. Z. Zgierski and P. G. Mezey, J. Phys. Chem. A, 2003, 107, 2080 CrossRef CAS; (d) L. J. Wang and M. Z. Zgierski, J. Phys. Chem. A, 2004, 108, 4679 CrossRef CAS; (e) Q. S. Li and L. P. Cheng, J. Phys. Chem. A, 2003, 107, 5561 CrossRef CAS; (f) K. J. Wilson, S. A. Perera and R. J. Bartlett, J. Phys. Chem. A, 2001, 105, 7693 CrossRef CAS; (g) G. Chaban, D. R. Yarkony and M. S. Gordon, J. Chem. Phys., 1995, 103, 7983 CrossRef CAS; (h) M-J. Crawford, A. Ellern, K. Karaghiosoff, F. Martin and P. Mayer, Inorg. Chem., 2010, 49, 2674 CrossRef CAS; (i) L. P. Cheng and W. Q. Cao, J. Mol. Model., 2007, 13, 1073 CrossRef CAS; (j) G. H. Zhang, Y. F. Zhao, F. Y. Hao, P. X. Zhang and X. D. Song, Int. J. Quantum Chem., 2009, 109, 226 CrossRef CAS; (k) L. P. Cheng and X. Q. Li, J. Mol. Model., 2006, 12, 805 CrossRef CAS; (l) E. P. F. Lee, J. M. Dyke and R. P. Claridge, J. Phys. Chem. A, 2002, 106, 8680 CrossRef CAS.
- T. M. Klapötke and J. Stierstorfer, J. Am. Chem. Soc., 2009, 131, 1122 CrossRef.
- (a) R. Riedel, E. Kroke, A. Greiner, A. O. Gabriel, L. Ruwisch and J. Nicolich, Chem. Mater., 1998, 10, 2964 CrossRef CAS; (b) M. A. Bredig, J. Am. Chem. Soc., 1942, 64, 1730 CrossRef CAS; (c) N. G. Vannerberg, Acta Chem. Scand., 1962, 16, 2263 CrossRef CAS; (d) M. J. Cooper, Acta Crystallogr., 1964, 17, 1452 CrossRef CAS; (e) M. G. Down, M. J. Haley, P. Hubberstey, R. J. Pulham and A. E. Thunder, J. Chem. Soc., Chem. Commun., 1978,(2), 52 RSC.
- (a) B. Jürgens, E. Irran and W. Schnick, J. Solid State Chem., 2001, 157, 241 CrossRef; (b) M. Becker, M. Jansen, A. Lieb, W. Milius and W. Z. Schnick, Z. Anorg. Allg. Chem., 1998, 624, 113 CrossRef CAS.
- (a) J. R. Witt and D. Britton, Acta Crystallogr., 1971, B27, 1835 Search PubMed; (b) L. Jäger, M. Kretschmann and H. Köhler, Z. Anorg. Allg. Chem., 1992, 611, 68 CrossRef; (c) H. Köhler, M. Jeschke and V. I. Nefedov, Z. Anorg. Allg. Chem., 1987, 552, 210 CrossRef; (d) P. Andersen, B. Klewe and E. Thom, Acta Chem. Scand., 1967, 21, 1530 CrossRef CAS.
- (a) H. P. H. Arp, A. Decken, J. Passmore and D. J. Wood, Inorg. Chem., 2000, 39, 1840 CrossRef CAS; (b) E. J. Graeber and B. Morosin, Acta Crystallogr., 1983, C39, 567 CAS.
- (a) T. M. Klapötke, C. Kuffer, P. Mayer, K. Polborn, A. Schulz and J. J. Weigand, Inorg. Chem., 2005, 44, 5949 CrossRef; (b) W. P. Norris and R. J. Henry, J. Am. Chem. Soc., 1963, 29, 650 Search PubMed.
- (a) R. Reed, V. L. Brady and J. M. Hitner, Proceedings of the 18th International Pyrotechnics Seminar, 1992, 13, 701 Search PubMed; (b) Y. Akutsu and M. J. Tamura, J. Energ. Mater., 1993, 11, 205 CrossRef CAS.
- (a) M. A. Hiskey, D. E. Chavez, D. L. Naud, S. F. Son, H. L. Berghout and C. A. Bolme, Proc. Int. Pyrotech. Semin., 2000, 27, 3 Search PubMed; (b) M. A. Hiskey, N. Goldman and J. R. Stine, J. Energ. Mater., 1998, 16, 119 CrossRef CAS.
- (a) J. Lifschitz, Chem. Ber., 1915, 48, 410 CrossRef CAS; (b) T. Curtius, A. Darapsky and E. Müller, Chem. Ber., 1915, 48, 1614 CrossRef CAS; (c) J. Lifschitz, Chem. Ber., 1916, 49, 489 CrossRef CAS; (d) J. Lifschitz and W. F. Donath, Recl. Tra. Chim. Pays-Bas., 1918, 37, 270 Search PubMed.
- R. Engelke, N. C. Blais and R. K. Sander, J. Phys. Chem. A, 1999, 103, 5611 CrossRef CAS.
- M. D. Chen, J. W. Liu, L. Dang and Q. E. Zhang, J. Chem. Phys., 2004, 121, 11661 CrossRef CAS.
- R. Hoffmann, P. R. Schleyer and H. F. Schaefer, Angew. Chem., Int. Ed., 2008, 47, 7164 CrossRef.
- Gaussian 03, Revision C.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004.
- Gaussian 09, Revision A.1, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz and J. Cioslowski, D. J. Fox, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- MOLPRO, version 2008.1, a package of abinitio programs , Werner, H.-J.; Knowles, P. J.; Lindh, R.; Manby, F. R.; Schütz, M.; Celani, P.; Korona, T.; Mitrushenkov, A.; Rauhut, G.; Adler, T. B.; Amos, R. D.; Bernhardsson, A.; Berning, A.; Cooper, D. L.; Deegan, M. J. O.; Dobbyn, A. J.; Eckert, F.; Goll, E.; Hampel, C.; Hetzer, G.; Hrenar, T.; Knizia, G.; Köppl, C.; Liu, Y.; Lloyd, A. W.; Mata, R. A.; May, A. J.; McNicholas, S. J.; Meyer, W.; Mura, M. E.; Nicklass, A.; Palmieri, P.; Pflüger, K.; Pitzer, R.; Reiher, M.; Schumann, U.; Stoll, H.; Stone, A. J.; Tarroni, R.; Thorsteinsson, T.; Wang, M.; Wolf, A., see http://www.molpro.net Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS; (d) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- (a) J.-P. Blaudeau, M. P. McGrath, L. A. Curtiss and L. Radom, J. Chem. Phys., 1997, 107, 5016 CrossRef CAS; (b) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS; (c) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (d) A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033 CrossRef CAS; (e) P. J. Hay, J. Chem. Phys., 1977, 66, 4377 CrossRef CAS; (f) K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062 CrossRef; (g) M. P. McGrath and L. Radom, J. Chem. Phys., 1991, 94, 511 CrossRef CAS; (h) R. C. Binning Jr. and L. A. Curtiss, J. Comput. Chem., 1990, 11, 1206 CrossRef; (i) T. Clark, J. G. Chandrasekhar, W. Spitznagel and P. v. R. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS; (j) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS.
- J. Cizek, Adv. Chem. Phys., 1969, 14, 35 CrossRef CAS.
- G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910 CrossRef CAS.
- (a) G. E. Scuseria, C. L. Janssen and H. F. Schaefer III, J. Chem. Phys., 1988, 89, 7382 CrossRef CAS; (b) G. E. Scuseria and H. F. Schaefer III, J. Chem. Phys., 1989, 90, 3700 CrossRef CAS; (c) J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968 CrossRef CAS.
- (a) I. S. Y. Wang and M. Karplus, J. Am. Chem. Soc., 1973, 95, 8160 CrossRef CAS; (b) C. Leforestier, J. Chem. Phys., 1978, 68, 4406 CrossRef CAS; (c) K. Bolton, W. L. Hase and G. H. Peslherbe, Direct Dynamics of ReactiVe Systems; World Scientific: Singapore, 1998; p 143 Search PubMed; (d) M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS; (e) S. S. Iyengar, M. K. Petersen, T. J. F. Day, C. J. Burnham, V. E. Teige and G. A. Voth, J. Chem. Phys., 2005, 123, 084309 CrossRef; (f) S. S. Iyengar and M. J. Frisch, J. Chem. Phys., 2004, 121, 5061 CrossRef CAS; (g) S. S. Iyengar, T. J. F. Day and G. A. Voth, Int. J. Mass Spectrom., 2005, 241, 197–204 CrossRef CAS; (h) D. Svozil and P. Jungwirth, J. Phys. Chem. A, 2006, 110, 9194 CrossRef CAS; (i) S. Sadhukhan, D. Munoz, C. Adamo and G. E. Scuseria, Chem. Phys. Lett., 1999, 306, 83 CrossRef CAS.
- (a) H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef; (b) R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS; (c) J. E. Del Bene, J. Phys. Chem., 1993, 97, 107 CrossRef CAS.
- H. -J. Werner and P. J. Knowles, J. Chem. Phys., 1985, 82, 5053 CrossRef CAS.
- P. J. Knowles and H. -J. Werner, Chem. Phys. Lett., 1985, 115, 259 CrossRef CAS.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157 CrossRef CAS.
- (a) P. Celani and H. -J. Werner, J. Chem. Phys., 2000, 112, 5546 CrossRef CAS; (b) D. G. Fedorov and M. S. Gordon, J. Chem. Phys., 2000, 112, 5611 CrossRef CAS; (c) A. Berning, M. Schweizer, H.-J. Werner, P. J. Knowles and P. Palmieri, Mol. Phys., 2000, 98, 1823 CrossRef CAS; (d) D. G. Fedorov, S. Koseki, M. W. Schmidt and M. S. Gordon, Int. Rev. Phys. Chem., 2003, 22, 551 CrossRef CAS.
- C. B. Shao and Y. H. Ding, Grid-Based Comprenhensive Isomeric Search Algorithm; Jilin University: Changchun, China, 2010 Search PubMed.
- J. Xu and Y.-H. Ding, Assembly code, Jilin University, Changchun, P. R. China, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The total energies (a.u.) and relative energies of singlet and triplet isomers, the transition states and the dissociation products P1, P2, P3, P4 at four levels. The structure parameters and total energies of the conical intersections between singlet and triplet potential energy surfaces. See DOI: 10.1039/c2ra21250a |
| This journal is © The Royal Society of Chemistry 2012 |