Mixed-ligand benzaldehyde thiosemicarbazone complexes of palladium containing N,O-donor ancillary ligands. Syntheses, structures, and catalytic application in C–C and C–N coupling reactions†
Jayita
Dutta
,
Sayanti
Datta
,
Dipravath
Kumar Seth
and
Samaresh
Bhattacharya
*
Department of Chemistry, Inorganic Chemistry Section, Jadavpur University, Kolkata - 700 032, India. E-mail: samaresh_b@yahoo.com; Fax: 91-33-24146223
First published on 2nd October 2012
Abstract
The reaction of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 4-R-benzaldehyde thiosemicarbazone [denoted in general as HL–R; where H stands for the dissociable acidic proton and R (R = OCH3, CH3, H, Cl and NO2) for the substituent] and 1-nitroso-2-naphthol (abbreviated as Hnn), with an equivalent quantity of Na2[PdCl4] in ethanolic medium affords a group of mixed-ligand complexes of the type [Pd(L–R)(nn)]. A similar reaction of a mixture of HL–R and quinolin-8-ol (Hq) with Na2[PdCl4] affords another family of mixed-ligand complexes of the type [Pd(L–R)(q)]. Crystal structures of [Pd(L–Cl)(nn)], [Pd(L–CH3)(q)] and [Pd(L–Cl)(q)] have been determined. In all the complexes the thiosemicarbazones are coordinated to the metal center, via dissociation of the acidic proton, as monoanionic bidentate N,S-donors forming five-membered chelate rings. In the [Pd(L–R)(nn)] complexes, the 1-nitroso-2-naphtholate anion is coordinated as a N,O-donor forming a five-membered chelate ring. Similarly in the [Pd(L–R)(q)] complexes, the quinolin-8-olate anion is bound to the metal center in the N,O-mode forming a five-membered chelate ring. All the [Pd(L–R)(nn)] and [Pd(L–R)(q)] complexes show characteristic 1H NMR signals, and in dichloromethane solution they all display intense absorptions in the visible and ultraviolet regions. Catalytic activities of the [Pd(L–R)(nn)] and [Pd(L–R)(q)] complexes have been examined towards some C–C and C–N coupling reactions, where both were found to show notable catalytic efficiency.
:1 mixture of 4-R-benzaldehyde thiosemicarbazone [denoted in general as HL–R; where H stands for the dissociable acidic proton and R (R = OCH3, CH3, H, Cl and NO2) for the substituent] and 1-nitroso-2-naphthol (abbreviated as Hnn), with an equivalent quantity of Na2[PdCl4] in ethanolic medium affords a group of mixed-ligand complexes of the type [Pd(L–R)(nn)]. A similar reaction of a mixture of HL–R and quinolin-8-ol (Hq) with Na2[PdCl4] affords another family of mixed-ligand complexes of the type [Pd(L–R)(q)]. Crystal structures of [Pd(L–Cl)(nn)], [Pd(L–CH3)(q)] and [Pd(L–Cl)(q)] have been determined. In all the complexes the thiosemicarbazones are coordinated to the metal center, via dissociation of the acidic proton, as monoanionic bidentate N,S-donors forming five-membered chelate rings. In the [Pd(L–R)(nn)] complexes, the 1-nitroso-2-naphtholate anion is coordinated as a N,O-donor forming a five-membered chelate ring. Similarly in the [Pd(L–R)(q)] complexes, the quinolin-8-olate anion is bound to the metal center in the N,O-mode forming a five-membered chelate ring. All the [Pd(L–R)(nn)] and [Pd(L–R)(q)] complexes show characteristic 1H NMR signals, and in dichloromethane solution they all display intense absorptions in the visible and ultraviolet regions. Catalytic activities of the [Pd(L–R)(nn)] and [Pd(L–R)(q)] complexes have been examined towards some C–C and C–N coupling reactions, where both were found to show notable catalytic efficiency.
Introduction
Thiosemicarbazone complexes of transition metal ions have been receiving considerable attention,1 primarily because of the bioinorganic relevance of these complexes. A large majority of the thiosemicarbazone complexes have found wide medicinal applications owing to their potentially beneficial biological (viz. antibacterial, antimalarial, antiviral and antitumor) activities.2 Systematic studies on the binding of thiosemicarbazones of selected types to different transition metal ions are of considerable importance in this respect. However, we have been exploring the chemistry of platinum metal complexes of the thiosemicarbazones,3 mainly because of the variable binding mode displayed by these ligands in their complexes and to understand the driving force behind the observed modes of binding, and the present work has emerged out of this exploration. We have recently observed that 4-R-benzaldehyde thiosemicarbazones (denoted in general as HL–R) bind to palladium as a bidentate N,S-donor forming a rather uncommon five-membered chelate ring (I).3dWith respect to the structure of the uncoordinated HL–Rligand, this mode of binding (I) was quite interesting as it involved a change in geometry around the imine (C
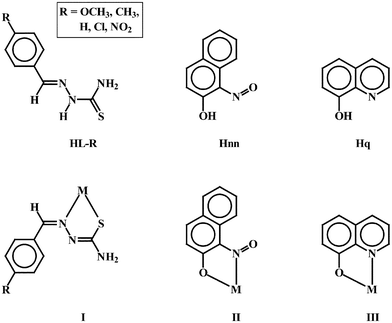
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) bond. Another interesting aspect of these complexes was their excellent catalytic efficiency towards C–C cross coupling reactions of different types. This has prompted us to study the chemistry of palladium thiosemicarbazone complexes further, with particular reference to exploration of their catalytic potential. The primary objective of the present study has been to synthesize a group of mixed-ligand complexes of palladium containing a 4-R-benzaldehyde thiosemicarbazone (HL–R) and an N,O-donor ancillary ligand, elucidate their structures, and explore their catalytic activity towards C–C and C–N coupling reactions. As ancillary ligands, we have chosen 1-nitroso-2-naphthol (abbreviated as Hnn) and quinolin-8-ol (abbreviated as Hq), each of which is known to bind to a metal center, via dissociation of the phenolic proton, as a mono-anionic bidentate N,O-donor forming stable five-membered chelate rings (II and III respectively).4,5 It may be relevant to note here that the binding of palladium simultaneously to two ligands of different types (e.g.,HL–R and, Hnn or Hq in the present case), leading to the formation of mixed-bis complex appears to have remained unexplored. For preparing the targeted mixed-bis complexes, Na2[PdCl4] has been utilized as the source of palladium. Reactions of the 4-R-benzaldehyde thiosemicarbazones (HL–R) and 1-nitroso-2-naphthol (Hnn) or quinolin-8-ol (Hq) with Na2[PdCl4] have indeed afforded two families of mixed-bis palladium complexes. This paper discusses the chemistry of these two groups of mixed-ligand palladium complexes, with special reference to their formation, structure and catalytic efficiency towards C–C and C–N coupling reactions.
N) bond. Another interesting aspect of these complexes was their excellent catalytic efficiency towards C–C cross coupling reactions of different types. This has prompted us to study the chemistry of palladium thiosemicarbazone complexes further, with particular reference to exploration of their catalytic potential. The primary objective of the present study has been to synthesize a group of mixed-ligand complexes of palladium containing a 4-R-benzaldehyde thiosemicarbazone (HL–R) and an N,O-donor ancillary ligand, elucidate their structures, and explore their catalytic activity towards C–C and C–N coupling reactions. As ancillary ligands, we have chosen 1-nitroso-2-naphthol (abbreviated as Hnn) and quinolin-8-ol (abbreviated as Hq), each of which is known to bind to a metal center, via dissociation of the phenolic proton, as a mono-anionic bidentate N,O-donor forming stable five-membered chelate rings (II and III respectively).4,5 It may be relevant to note here that the binding of palladium simultaneously to two ligands of different types (e.g.,HL–R and, Hnn or Hq in the present case), leading to the formation of mixed-bis complex appears to have remained unexplored. For preparing the targeted mixed-bis complexes, Na2[PdCl4] has been utilized as the source of palladium. Reactions of the 4-R-benzaldehyde thiosemicarbazones (HL–R) and 1-nitroso-2-naphthol (Hnn) or quinolin-8-ol (Hq) with Na2[PdCl4] have indeed afforded two families of mixed-bis palladium complexes. This paper discusses the chemistry of these two groups of mixed-ligand palladium complexes, with special reference to their formation, structure and catalytic efficiency towards C–C and C–N coupling reactions.
Results and discussion
Syntheses and structures
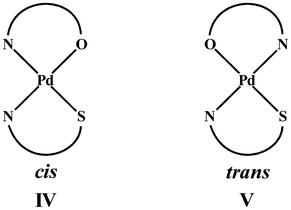
As delineated in the introduction, five 4-R-benzaldehyde thiosemicarbazones (HL–R), differing in the substituent at the para-position of the phenyl ring, have been used in the present study. As the ancillary ligand, 1-nitroso-2-naphthol (Hnn) has been first employed. Reaction of an ethanolic solution containing an equimolar quantity of a particular thiosemicarbazone and 1-nitroso-2-naphthol with an equivalent amount of Na2[PdCl4] proceeds smoothly in the presence of triethylamine to afford a group of mixed-ligand complexes of the type [Pd(L–R)(nn)] in excellent yields. Preliminary characterizations (microanalysis, NMR, IR, etc.) on these [Pd(L–R)(nn)] complexes (vide infra) agree well with the presence of a deprotonated thiosemicarbazone and a deprotonated 1-nitroso-2-naphthol in the coordination sphere. To our knowledge, the direct synthesis of such a mixed-bis complex of palladium, containing two different unsymmetric bidentate ligands, is unprecedented. Such complexes may, in principle, exist as two geometric isomers, and in view of the relative disposition of the neutral-nitrogen and anionic-sulfur/oxygen donors in the two coordinated ligands, these isomers may be labeled as cis (IV) and trans (V).In order to find out the stereochemistry of these complexes as well as the coordination mode of the thiosemicarbazones in them, the structure of one representative member of this family, viz. [Pd(L–Cl)(nn)], has been determined by X-ray crystallography. A selected view of the complex molecule is shown in Fig. 1 and some relevant bond parameters are listed in Table 1. The structure shows that the thiosemicarbazone is coordinated to palladium as a monoanionic (via loss of the hydrazinic proton) bidentate N,S-donor, forming a five-membered chelate ring (I) with a bite angle of 83.81(12)°. 1-Nitroso-2-naphthol is also coordinated (via loss of the phenolic proton) to the metal center as a monoanionic bidentate N,O-donor (II), where the nitrogen and oxygen of the 1-nitroso-2-naphtholate ligand are trans respective to the nitrogen and sulfur of the coordinated thiosemicarbazone. Thus this [Pd(L–Cl)(nn)] complex has the trans (V) stereochemistry. In this [Pd(L–Cl)(nn)] complex, palladium is nested in a N2OS core, which is distorted from the ideal square planar geometry, as manifested in the bond parameters around the metal center. The Pd–N, Pd–O, Pd–S bond distances are all quite normal, as observed in structurally characterized complexes of palladium containing these bonds.6 The structure of the [Pd(L–Cl)(nn)] complex shows that an interesting change in geometry around the C
 N bond, with respect to the uncoordinated HL–Rligand,3d,n has occurred during the synthesis of this complex, resulting in the formation of a rather unusual five-membered ring.3d,7 In view of the similarity in the method of synthesis of the [Pd(L–R)(nn)] complexes, as well as the similarity in their properties (vide infra), the remaining four [Pd(L–R)(nn)] (R = OCH3, CH3, H and NO2) complexes are assumed to have structures similar to [Pd(L–Cl)(nn)].
N bond, with respect to the uncoordinated HL–Rligand,3d,n has occurred during the synthesis of this complex, resulting in the formation of a rather unusual five-membered ring.3d,7 In view of the similarity in the method of synthesis of the [Pd(L–R)(nn)] complexes, as well as the similarity in their properties (vide infra), the remaining four [Pd(L–R)(nn)] (R = OCH3, CH3, H and NO2) complexes are assumed to have structures similar to [Pd(L–Cl)(nn)].
![Structure of [Pd(L–Cl)(nn)].](/image/article/2012/RA/c2ra21078a/c2ra21078a-f1.gif) | ||
| Fig. 1 Structure of [Pd(L–Cl)(nn)]. | ||
| [Pd(L–Cl)(nn)] | ||||
|---|---|---|---|---|
| Bond distances (Å) | ||||
| Pd(1)–N(1) | 2.028(6) | N(1)–C(2) | 1.293(9) | |
| Pd(1)–N(4) | 1.993(8) | N(1)–N(2) | 1.378(8) | |
| Pd(1)–O(1) | 2.066(5) | C(1)–S(1) | 1.735(8) | |
| Pd(1)–S(1) | 2.241(2) | C(9)–N(4) | 1.359(10) | |
| N(4)–O(2) | 1.236(9) | C(10)–O(1) | 1.283(11) | |
| Bond angles (°) | ||||
| N(1)–Pd(1)–N(4) | 178.5(3) | N(1)–Pd(1)–S(1) | 83.52(15) | |
| O(1)–Pd(1)–S(1) | 177.34(19) | N(4)–Pd(1)–O(1) | 80.6(2) | |
| [Pd(L–CH3)(q)] | ||||
|---|---|---|---|---|
| Bond distances (Å) | ||||
| Pd(1)–N(1) | 2.008(4) | N(1)–C(2) | 1.292(6) | |
| Pd(1)–N(4) | 2.015(3) | N(1)–N(2) | 1.380(4) | |
| Pd(1)–O(1) | 2.065(3) | C(1)–S(1) | 1.726(5) | |
| Pd(1)–S(1) | 2.2342(12) | C(17)–O(1) | 1.310(6) | |
| Bond angles (°) | ||||
| N(1)–Pd(1)–N(4) | 177.16(14) | N(1)–Pd(1)–S(1) | 83.75(10) | |
| O(1)–Pd(1)–S(1) | 176.38(9) | N(4)–Pd(1)–O(1) | 81.95(13) | |
Formation of the [Pd(L–R)(nn)] complexes, containing two different unsymmetric bidentate ligands, by a rather simple method was quite intriguing. To examine whether such a method is applicable, in general, for the synthesis of similar mixed-bis complexes of palladium, reactions of the same series of 4-R-benzaldehyde thiosemicarbazones and quinolin-8-ol (Hq) have also been carried out similarly as before with the same palladium starting material, viz. Na2[PdCl4]. These reactions indeed have afforded another set of five mixed-ligand complexes of the desired type, viz. [Pd(L–R)(q)] in decent yields. The structure of a selected member of this family, viz. [Pd(L–CH3)(q)], has been determined by X-ray crystallography to ascertain the binding mode of the thiosemicarbazone and also to find out the stereochemistry. The structure (Fig. 2) shows that the thiosemicarbazone is coordinated to palladium in the same manner as before, forming five-membered chelate rings (I), and the quinolinolate ligand is also coordinated in the expected N,O-mode (III). The two coordinated nitrogens are trans and so are the oxygen and sulfur, and hence these [Pd(L–R)(q)] complexes also have the trans stereochemistry (V).
![Structure of [Pd(L–CH3)(q)].](/image/article/2012/RA/c2ra21078a/c2ra21078a-f2.gif) | ||
| Fig. 2 Structure of [Pd(L–CH3)(q)]. | ||
For a proper comparison of the bond parameters with those observed in the [Pd(L–Cl)(nn)] complex, the structure of the corresponding [Pd(L–Cl)(q)] complex was solved first. As expected, its structure (Fig. S1, ESI†) was found to be similar to that of the [Pd(L–CH3)(q)] complex, however, due to the poor quality of the crystal, the structure did not refine well (Table S1, ESI†) and hence the bond parameters (Table S2, ESI†) were not very precise. On the other hand, the observed bond parameters in [Pd(L–CH3)(q)] (Table 1), compare well with those found for the [Pd(L–Cl)(nn)] complex. As all the [Pd(L–R)(q)] complexes were obtained similarly and they show similar properties (vide infra), the other three [Pd(L–R)(q)] (R = OCH3, H and NO2) complexes are assumed to have the same structure as their R = CH3 or Cl analogue.
Spectral properties
1H NMR spectra of both the [Pd(L–R)(nn)] and [Pd(L–R)(q)] complexes, recorded in CDCl3 solutions, show all the expected signals. From the coordinated thiosemicarbazone, signals for the NH2protons and azomethineproton could be easily detected in most of the complexes. Signals for the OCH3 and CH3 groups, in complexes having one of these substituents in the coordinated thiosemicarbazone, are observed in the expected positions. Out of the expected signals from the aryl rings of the coordinated thiosemicarbazone and the N,O-donor ligand, most are clearly observed in the aromatic region, while few could not be detected due to overlap problems.Infrared spectra of the [Pd(L–R)(nn)] and [Pd(L–R)(q)] complexes show many bands of varying intensities within the 4000–400 cm−1 range, which are attributable to the coordinated thiosemicarbazone and the N,O-coordinated co-ligand. Both complexes show sharp bands near 1600, 1505, 1316, 820, 749 and 663 483 cm−1, and in comparison with the spectra of the [Pd(L–R)2] complexes,3d these bands are attributed to the coordinated thiosemicarbazone ligand. Among these bands, the one near 749 cm−1 is believed to be due to the C–S stretching.8 In all the [Pd(L–R)(nn)] complexes, a sharp band near 1260 cm−1 could be identified as the νNO vibration, and another sharp band near 1093 cm−1 is assignable to the phenolic C–O stretch. In the [Pd(L–R)(q)] complexes, the phenolic C–O stretch is observed around 1110 cm−1 as a sharp band. The 1H NMR and infrared spectral data of the [Pd(L–R)(nn)] and [Pd(L–R)(q)] complexes are therefore in good agreement with their compositions.
Both the [Pd(L–R)(nn)] and [Pd(L–R)(q)] complexes are soluble in common organic solvents, such as methanol, ethanol, acetone, acetonitrile, dichloromethane, chloroform, etc., producing intense red and yellow solutions respectively. Electronic spectra of all these complexes have been recorded in dichloromethane solutions. Spectral data are presented in Table 2. Each complex shows several intense absorptions within the 500–250 nm range. While the absorptions in the ultraviolet region are believed to be due to transitions within the ligand orbitals, the origin of those in the visible region was unclear. To gain insight into the nature of the absorptions in the visible region, DFT calculations have been performed on all the complexes. The results are found to be qualitatively similar for all five members of each group. Compositions of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are given in Table 3. Contour plots of these molecular orbitals for [Pd(L–Cl)(nn)] is shown in Fig. 3 and those for [Pd(L–Cl)(q)] are deposited as Fig. S2 (ESI†). For the [Pd(L–R)(nn)] complexes the HOMO is delocalized mostly (>76%) over the thiosemicarbazone ligand, whereas the LUMO is concentrated mostly (>80%) on the 1-nitroso-2-naphtholate ligand. Hence, the lowest energy absorption (near 475 nm) in these complexes is assignable to a transition from a filled thiosemicarbazone orbital to a vacant orbital of the 1-nitroso-2-naphtholate ligand. DFT calculations on the [Pd(L–R)(q)] complexes show that the HOMO has a major (>67%) contribution from the coordinated quinolin-8-olate ligand and the LUMO is delocalized almost equally over both the coordinated thiosemicarbazone and quinolin-8-olate. Therefore, the lowest energy absorption (near 426 nm) in these complexes is attributable to a transition taking place from the filled quinolin-8-olate orbital to the vacant orbital spread over both the thiosemicarbazone and quinolin-8-olate ligands.
![Contour plots of the HOMO and LUMO of [Pd(L–Cl)(nn)].](/image/article/2012/RA/c2ra21078a/c2ra21078a-f3.gif) | ||
| Fig. 3 Contour plots of the HOMO and LUMO of [Pd(L–Cl)(nn)]. | ||
| Complex | λ max, nm (ε, M−1 cm−1) |
|---|---|
| a Shoulder. | |
| [Pd(L–OCH3)(nn)] | 480a (6400), 441(6100), 407a (9500), 355(18500), 305(22200), 256(23000) |
| [Pd(L–CH3)(nn)] | 478a (8600), 446(8700), 409a (12000), 357(20000), 301(31600), 257(30500) |
| [Pd(L–H)(nn)] | 475a (8500), 442(8800), 408a (11000), 358(12000), 310(20000), 258(18000) |
| [Pd(L–Cl)(nn)] | 473a (5600), 439(8000), 409a (8000), 360(11500), 300(19400), 258(18500) |
| [Pd(L–NO2)(nn)] | 469a (7400), 392(13500), 298(15000), 260(20200) |
| [Pd(L–OCH3)(q)] | 431a (7000), 370a (17000), 319(23700), 265a (33000), 246(37500) |
| [Pd(L–CH3)(q)] | 426a (7000), 369a (12800), 314(21000), 265a (33200), 247(36700) |
| [Pd(L–H)(q)] | 432a (6000), 370a (11600), 306(21400), 265a (31200), 245(37300) |
| [Pd(L–Cl)(q)] | 429a (5300), 375a (8300), 311(15400), 264a (28400), 247(29500) |
| [Pd(L–NO2)(q)] | 437a (5400), 339(4900), 271a (21200), 251(30200) |
| Complex | Contributing fragments | % Contribution of fragments to | |
|---|---|---|---|
| HOMO | LUMO | ||
| [Pd(L–OCH3)(nn)] | Pd | 11.12 | 6.54 |
| L–OCH3 | 75.23 | 11.80 | |
| nn | 13.65 | 81.66 | |
| [Pd(L–CH3)(nn)] | Pd | 12.66 | 6.77 |
| L–CH3 | 71.99 | 10.14 | |
| nn | 15.35 | 83.09 | |
| [Pd(L–H)(nn)] | Pd | 10.13 | 8.12 |
| L–H | 76.17 | 12.51 | |
| nn | 13.70 | 79.37 | |
| [Pd(L–Cl)(nn)] | Pd | 10.23 | 8.34 |
| L–Cl | 76.21 | 11.15 | |
| nn | 13.56 | 80.51 | |
| [Pd(L–NO2)(nn)] | Pd | 12.49 | 5.89 |
| L–NO2 | 63.12 | 18.26 | |
| nn | 24.39 | 75.85 | |
| [Pd(L–OCH3)(q)] | Pd | 9.25 | 3.76 |
| L–OCH3 | 25.53 | 43.95 | |
| q | 65.22 | 52.29 | |
| [Pd(L–CH3)(q)] | Pd | 12.72 | 4.01 |
| L–CH3 | 20.12 | 46.76 | |
| q | 67.16 | 49.23 | |
| [Pd(L–H)(q)] | Pd | 9.22 | 2.96 |
| L–H | 21.61 | 46.69 | |
| q | 69.17 | 50.35 | |
| [Pd(L–Cl)(q)] | Pd | 11.42 | 3.14 |
| L–Cl | 19.63 | 51.67 | |
| q | 68.95 | 45.19 | |
| [Pd(L–NO2)(q)] | Pd | 8.83 | 2.85 |
| L–NO2 | 17.80 | 55.21 | |
| q | 73.37 | 41.94 | |
Catalytic activity
As described in the introduction, the primary objective of the present study has been to explore the catalytic properties of the palladium complexes towards C–C and C–N coupling reactions. The fact that palladium complexes are well known for serving as efficient catalysts in bringing about C–C and C–N coupling reactions,9 has led us to explore such catalytic properties in the present series of [Pd(L–R)(nn)] and [Pd(L–R)(q)] complexes. The catalytic activities of all the newly prepared complexes of the two series have been examined towards Suzuki type C–C coupling and Buchwald type C–N coupling reactions.Initially all the [Pd(L–R)(nn)] complexes were tested as catalysts in the Suzuki coupling of phenylboronic acid and p-bromoacetophenone to yield the biphenyl product. All these complexes showed notable catalytic efficiency. For example, when using [Pd(L–OCH3)(nn)] and [Pd(L–NO2)(nn)] as catalyst with a 0.001% loading, this particular Suzuki coupling was achieved, under relatively mild experimental conditions, in high yield (entries 1 and 4, Table 4). When the amount of catalyst was reduced by ten times, the yield becomes significantly lower (entry 2, Table 4). By changing the base from Cs2CO3 to NaOH a similar yield was obtained in much less time (entries 1 and 5, Table 4). With an electron-withdrawing substituent (e.g., CHO, CN) in the arylbromides, longer times (relative to the COCH3 substituent) were required to achieve full conversion (entries 6 and 7, Table 4). The observed ease of this catalytic coupling reaction, with particular reference to the mild reaction conditions and low catalyst loading, prompted us to examine whether the same coupling reaction of phenylboronic acid with p-bromoacetophenone, using [Pd(L–OCH3)(nn)] as the catalyst, can be carried out at room temperature. The attempted reaction proceeded very smoothly at room temperature, showing similar yields with greater catalyst loading (entries 1 and 8, Table 4). Encouraged by the facile C–C coupling of aryl bromides with phenylboronic acid, catalyzed by the [Pd(L–R)(nn)] complexes, similar coupling reactionviaactivation of the C–Cl bond of aryl chlorides was also attempted. These [Pd(L–R)(nn)] complexes were indeed found to bring about C–Cl bond activation of aryl chlorides and thus promote their coupling with phenylboronic acid (entries 9–11, Table 4). However, compared to the corresponding aryl bromides, a much larger amount of catalyst was needed for these reactions to obtain similar yields (entries 5 and 11, Table 4). Even with a greater catalyst loading, a significant reduction in yield was observed upon changing the substituent from COCH3 to CHO to CN in the aryl chloride moiety (entries 12 and 13, Table 4). We have also examined a similar coupling of aryl iodide and aryl fluoride, and as expected, the coupling reaction via C–I bond activation was found to be very facile (entry 14, Table 4) and that via C–F bond activation was equally difficult (entry 15, Table 4). In order to examine whether replacement of the 1-nitroso-2-naphtholate ligand with the quinolin-8-olate ligand has been effective in changing the catalytic efficiency, the [Pd(L–R)(q)] complexes were also tested as catalysts towards the Suzuki coupling reactions, and the results are included in Table 4 (entries 16–21). Here also, all five [Pd(L–R)(q)] complexes showed similar and mutually comparable catalytic efficiency (entries 16 and 17, Table 4). Again NaOH was found to be a better base than Cs2CO3 (entry 18, Table 4). The reactivity of the [Pd(L–R)(q)] complexes towards C–X (X = F, Cl, Br and I) bonds (entries 18–21, Table 4) was found to follow a similar trend to that of the [Pd(L–R)(nn)] complexes. Thus the catalytic efficiency of the [Pd(L–R)(q)] complexes is found to be similar to that of the [Pd(L–R)(nn)] complexes.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | X | Catalystb | Base | T (°C) | Time (h) | Amt of cat., mol (%) | Yieldc (%) | TON |
| a Reaction conditions: aryl halide (1.0 mmol), phenylboronic acid (1.2 mmol), Cs2CO3 (2.5 mmol) or NaOH (1.7 mmol), Pd catalyst, polyethyleneglycol (4 mL). b 1–R means [Pd(L–R)(nn)] and 2–R means [Pd(L–R)(q)]. c Determined by GC-MS. | |||||||||
| 1 | COCH3 | Br | 1–OCH3 | Cs2CO3 | 120 | 24 | 0.001 | 100 | 100000 |
| 2 | COCH3 | Br | 1–OCH3 | Cs2CO3 | 120 | 24 | 0.0001 | 20 | 200000 |
| 3 | COCH3 | Br | 1–OCH3 | NaOH | 120 | 24 | 0.0001 | 47 | 470000 |
| 4 | COCH3 | Br | 1–NO2 | Cs2CO3 | 120 | 24 | 0.001 | 97 | 97000 |
| 5 | COCH3 | Br | 1–OCH3 | NaOH | 120 | 6 | 0.001 | 100 | 100000 |
| 6 | CHO | Br | 1–OCH3 | NaOH | 120 | 24 | 0.001 | 100 | 100000 |
| 7 | CN | Br | 1–OCH3 | NaOH | 120 | 24 | 0.001 | 100 | 100000 |
| 8 | COCH3 | Br | 1–OCH3 | NaOH | Room temp. | 24 | 0.1 | 100 | 1000 |
| 9 | COCH3 | Cl | 1–OCH3 | NaOH | 120 | 24 | 0.001 | 25 | 25000 |
| 10 | COCH3 | Cl | 1–OCH3 | NaOH | 120 | 24 | 0.01 | 65 | 6500 |
| 11 | COCH3 | Cl | 1–OCH3 | NaOH | 120 | 24 | 0.1 | 100 | 1000 |
| 12 | CHO | Cl | 1–OCH3 | NaOH | 120 | 24 | 1 | 55 | 55 |
| 13 | CN | Cl | 1–OCH3 | NaOH | 120 | 24 | 1 | 52 | 52 |
| 14 | COCH3 | I | 1–OCH3 | NaOH | 120 | 4 | 0.001 | 100 | 100000 |
| 15 | COCH3 | F | 1-OCH3 | NaOH | 120 | 24 | 1 | 10 | 10 |
| 16 | COCH3 | Br | 2–OCH3 | Cs2CO3 | 120 | 24 | 0.001 | 100 | 100000 |
| 17 | COCH3 | Br | 2–NO2 | Cs2CO3 | 120 | 24 | 0.001 | 100 | 100000 |
| 18 | COCH3 | Br | 2–OCH3 | NaOH | 120 | 6 | 0.001 | 100 | 100000 |
| 19 | COCH3 | Cl | 2–OCH3 | NaOH | 120 | 24 | 0.1 | 100 | 1000 |
| 20 | COCH3 | I | 2–OCH3 | NaOH | 120 | 4 | 0.001 | 100 | 100000 |
| 21 | COCH3 | F | 2–OCH3 | NaOH | 120 | 24 | 1 | 12 | 12 |
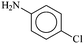
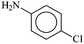
Encouraged by the facile Suzuki coupling reactions catalyzed by the present series of mixed-ligand Pd complexes, we have also investigated catalytic activity of these complexes towards Buchwald type C–N coupling reactions between aryl halides and aromatic amines. At first, C–N coupling between primary amines and aryl halides was attempted, and the targeted products could be achieved under relatively mild experimental conditions in reasonably high yield (Table 5). The [Pd(L–OCH3)(nn)] catalyzed coupling of p-chloroaniline and p-iodoacetophenone using Diphos as an additive resulted in moderate yield, whereas, the use of XPhos enhanced the yield significantly (entries 1 and 2, Table 5). A decrease in catalyst loading brought about an appreciable decrease in the yield (entry 3, Table 5). With an electron-withdrawing substituent in the aryl halide fragment, the yield decreased significantly (entry 4, Table 5). Similarly, with an electron-donating substituent in the aryl amine moiety, a decrease in the yield was observed (entries 5 and 6, Table 5). The use of p-bromoacetophenone or p-chloroacetophenone, instead of p-iodoacetophenone, gradually lowered the yield to a great extent (entries 7 and 8, Table 5). When the same coupling reactions were performed with the [Pd(L–R)(q)] complexes as catalyst, a similar trend was observed as in the case of [Pd(L–R)(nn)] complexes (entries 9–15, Table 5).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | X | Amine | Catalystb | Amount of cat. (mol%) | Additive added | Amount of add. (eq.) | Yieldc (%) | TON |
| a Reaction conditions: aryl halide (1.0 mmol), amines (1.0 mmol), NaOt-Bu (1.7 mmol), Pd catalyst, polyethyleneglycol (2 mL). b 1–R means [Pd(L–R)(nn)] and 2–R means [Pd(L–R)(q)]. c Determined by GC-MS. | |||||||||
| 1 | COCH3 | I |
|
1–OCH3 | 1 | Diphos | 1 | 67 | 67 |
| 2 | COCH3 | I |
|
1–OCH3 | 0.1 | XPhos | 0.1 | 100 | 1000 |
| 3 | COCH3 | I |
|
1–OCH3 | 0.01 | XPhos | 0.1 | 65 | 650 |
| 4 | CHO | I |
|
1–OCH3 | 0.5 | XPhos | 0.1 | 55 | 550 |
| 5 | COCH3 | I |
|
1–OCH3 | 1 | XPhos | 0.1 | 77 | 770 |
| 6 | COCH3 | I |
|
1–OCH3 | 1 | XPhos | 0.1 | 35 | 350 |
| 7 | COCH3 | Br |
|
1–OCH3 | 1 | XPhos | 0.1 | 65 | 650 |
| 8 | COCH3 | Cl |
|
1–OCH3 | 1 | XPhos | 0.1 | 10 | 100 |
| 9 | COCH3 | I |
|
2–OCH3 | 0.1 | XPhos | 0.1 | 100 | 1000 |
| 10 | COCH3 | I |
|
2–OCH3 | 0.01 | XPhos | 0.1 | 62 | 620 |
| 11 | CHO | I |
|
2–OCH3 | 0.5 | XPhos | 0.1 | 53 | 530 |
| 12 | COCH3 | I |
|
2–OCH3 | 1 | XPhos | 0.1 | 80 | 800 |
| 13 | COCH3 | I |
|
2–OCH3 | 1 | XPhos | 0.1 | 42 | 420 |
| 14 | COCH3 | Br |
|
2–OCH3 | 1 | XPhos | 0.1 | 55 | 550 |
| 15 | COCH3 | Cl |
|
2–OCH3 | 1 | XPhos | 0.1 | 6 | 60 |
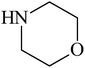
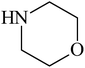
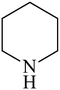
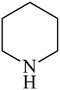
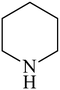
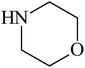
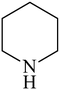
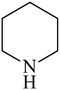
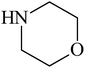
Besides primary amines, the catalytic efficiency of all complexes was also tested towards C–N coupling reactions for secondary amines. Two secondary amines, viz.morpholine and piperidine, were selected for this purpose. The coupling of these amines with the same set of aryl halides was scrutinized using the same two complexes, viz. [Pd(L–OCH3)(nn)] and [Pd(L–OCH3)(q)] as catalysts. The general features of these reactions were found to be similar to those observed in the coupling of primary amines, and the results are summarized in Table 6. Similar results were obtained for both morpholine and piperidine (entries 7–12, Table 6).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | X | Amine | Catalystb | Amount of cat. (mol%) | Additive | Amount of add. (eq.) | Yieldc (%) | TON |
| a Reaction conditions: aryl halide (1.0 mmol), amines (1.0 mmol), NaOt-Bu (1.7 mmol), Pd catalyst, polyethyleneglycol (2 mL). b 1–R means [Pd(L–R)(nn)] and 2–R means [Pd(L–R)(q)]. c Determined by GC-MS. | |||||||||
| 1 | COCH3 | I |
|
1–OCH3 | 1 | Diphos | 1 | 65 | 65 |
| 2 | COCH3 | I |
|
1–OCH3 | 0.1 | XPhos | 0.1 | 100 | 1000 |
| 3 | COCH3 | I |
|
1–OCH3 | 0.01 | XPhos | 0.1 | 62 | 620 |
| 4 | CHO | I |
|
1–OCH3 | 0.5 | XPhos | 0.1 | 52 | 520 |
| 5 | COCH3 | Br |
|
1–OCH3 | 1 | XPhos | 0.1 | 62 | 620 |
| 6 | COCH3 | Cl |
|
1–OCH3 | 1 | XPhos | 0.1 | 4 | 40 |
| 7 | COCH3 | I |
|
1–OCH3 | 1 | XPhos | 0.1 | 100 | 1000 |
| 8 | COCH3 | I |
|
1–OCH3 | 0.1 | XPhos | 0.1 | 100 | 1000 |
| 9 | COCH3 | I |
|
1–OCH3 | 0.01 | XPhos | 0.1 | 62 | 620 |
| 10 | CHO | I |
|
1–OCH3 | 0.5 | XPhos | 0.1 | 67 | 670 |
| 11 | COCH3 | Br |
|
1–OCH3 | 1 | XPhos | 0.1 | 55 | 550 |
| 12 | COCH3 | Cl |
|
1–OCH3 | 1 | XPhos | 0.1 | 3 | 30 |
| 13 | COCH3 | I |
|
2–OCH3 | 0.1 | XPhos | 0.1 | 100 | 1000 |
| 14 | CHO | I |
|
2–OCH3 | 0.5 | XPhos | 0.1 | 57 | 570 |
| 15 | COCH3 | I |
|
2–OCH3 | 0.1 | XPhos | 0.1 | 100 | 1000 |
| 16 | CHO | I |
|
2–OCH3 | 0.5 | XPhos | 0.1 | 55 | 550 |
| 17 | COCH3 | Br |
|
2–OCH3 | 1 | XPhos | 0.1 | 50 | 500 |
| 18 | COCH3 | Cl |
|
2–OCH3 | 1 | XPhos | 0.1 | 4 | 40 |
The present studies thus demonstrate that the mixed-ligand palladium thiosemicarbazone complexes of both types, [Pd(L–R)(nn)] and [Pd(L–R)(q)], are efficient catalysts for C–C and C–N coupling reactions. The C–C coupling reactions took place in high yield with very low catalyst loading, and without any additional ligand, which is relatively less common.10 A plausible mechanism for the observed catalytic C–C cross-coupling reactions is presented in Scheme 1. As no additional ligand was necessary for these reactions, it is believed that the protonated thiosemicarbazone and N,O-donor ligands remain coordinated to the palladium(0) center, which is formed in situ. In addition to activating C–I and C–Br bonds with notable efficiency, these complexes could successfully activate C–Cl bonds in aryl chlorides, affording the C–C coupled product in 100% yield, which is rarely observed.11 The complexes could also catalyze Suzuki coupling reactions at room temperature. The catalytic efficiency of the two types of mixed-bis palladium complexes is found to compare well with that of the mono- and bis-thiosemicarbazone complexes of palladium.3d However, the observed efficiency is much better than that displayed by many such molecular palladium complexes under similar ligand-free conditions.12 Though, compared to C–C couplings, the C–N coupling reactions needed slightly more catalyst, even then the C–N coupling with such efficiency is not very common.9a,c,d The mechanism for the observed C–N coupling reaction is shown in Scheme 2. As the use of XPhos was a must for these reactions, it is assumed that under the experimental conditions, the coordinated thiosemicarbazone and N,O-donor ligands get dissociated during the reduction of the palladium center, and the reduced metal center is stabilized only by XPhos coordination. The catalytic efficiency of the present group of complexes in C–N coupling reactions is comparable to that of several other palladium complexes under similar experimental conditions.13 The almost comparable catalytic activity of both series of mixed-ligand complexes may be attributed to the similar coordination environment in them, which probably supported the in situ generation of the palladium(0) center with comparable efficiency.
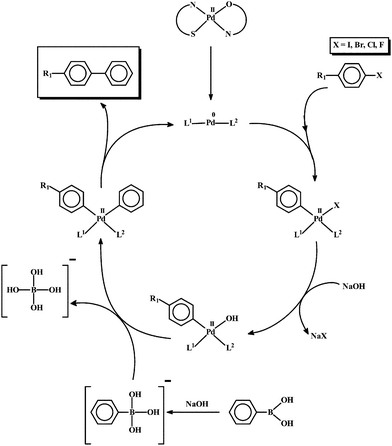 | ||
| Scheme 1 Probable mechanism for the observed C–C cross-coupling reactions. L1 and L2 represent the protonated thiosemicarbazone and N,O-donor ligands. | ||
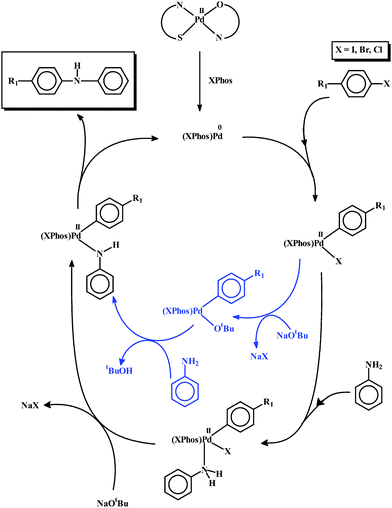 | ||
| Scheme 2 Probable mechanism for the observed C–N coupling reactions. | ||
Conclusions
The present study shows that mixed-bis complexes of palladium containing two different unsymmetric bidentate ligands, which is unprecedented, can be conveniently synthesized by following a rather simple method. This is demonstrated through the synthesis of two families of mixed-ligand complexes of palladium containing anions derived from 4-R-benzaldehyde thiosemicarbazone (HL–R) and a N,O-donor ancillary ligand, viz.1-nitroso-2-naphthol (Hnn) and quinolin-8-ol (Hq). Both the [Pd(L–R)(nn)] and [Pd(L–R)(q)] complexes are found to catalyze, with notable efficiency, Suzuki type C–C coupling and Buchwald type C–N coupling reactions.Experimental
General procedures
Palladium chloride was obtained from Arora Matthey, Kolkata, India. Na2[PdCl4] was prepared following a reported procedure.14 The para-substituted benzaldehydes and thiosemicarbazide were procured from E-Merck India Pvt. Ltd. The 4-R-benzaldehyde thiosemicarbazones were prepared by reacting equimolar amounts of thiosemicarbazide and the respective para-substituted benzaldehyde in a hot 1:1 ethanol–water mixture under stirring conditions. 1-Nitroso-2-naphthol was obtained from Loba Chemie, Mumbai, India, and quinolin-8-ol was purchased from E-Merck India Pvt. Ltd. All other chemicals and solvents were reagent grade commercial materials and were used as received. Microanalyses (C, H, N) were performed using a Heraeus Carlo Erba 1108 elemental analyzer. IR spectra were obtained on a Shimadzu FTIR-8300 spectrometer with samples prepared as KBr pellets. Electronic spectra were recorded on a JASCO V-570 spectrophotometer. 1H NMR spectra were recorded in CDCl3 solutions on a Bruker Avance DPX 300 NMR spectrometer using TMS as the internal standard. Molecular orbital calculations were carried out by a density functional theory (DFT) method using the Gaussian 03 (B3LYP/SDD-6-31G) package.15GC-MS analysis was performed using a Perkin Elmer CLARUS 680 instrument.
Synthesis of the complexes
[Pd(L–OCH3)(nn)]. To a solution of 4-methoxybenzaldehyde thiosemicarbazone (35 mg, 0.17 mmol) and 1-nitroso-2-naphthol (30 mg, 0.17 mmol) in hot ethanol (30 mL) was added triethylamine (34 mg, 0.34 mmol), followed by a solution of Na2[PdCl4] (50 mg, 0.17 mmol) in ethanol (10 mL). The solution was then heated at reflux for 6 h. The [Pd(L–OCH3)(nn)] complex started to precipitate as a red crystalline solid during reflux, which was allowed to settle by cooling of the mixture to room temperature and was then collected by filtration. The solid was washed thoroughly with water, followed by ethanol and then dried in air. Yield: 79%. Anal. Calc. for C19H16N4O3SPd: C, 46.87; H, 3.28; N, 11.51. Found: C, 47.03; H, 3.37; N, 11.42%. 1H NMR (300 MHz, CDCl3):16δ (ppm) = 3.88 (s, OCH3), 4.95 (s, NH2), 6.94 (d, J = 9.0 Hz, 2H), 6.99 (d, J = 9.0 Hz, 1H), 7.43 (t, J = 8.0 Hz, 1H), 7.57 (d + t, 2H)*, 7.77 (d, J = 9.5 Hz, 1H), 7.83 (s, azomethine), 8.19 (d, J = 9.0 Hz, 2H), 8.88 (d, J = 8.0 Hz, 1H). IR (wave number, cm−1): 446, 483, 661, 751, 824, 1092, 1158, 1177, 1205, 1260, 1319, 1398, 1505, 1590, 1603.
[Pd(L–CH3)(nn)]. Yield: 77%. Anal. Calc. for C19H16N4O2SPd: C, 48.46; H, 3.40; N, 11.90. Found: C, 48.59; H, 3.45; N, 12.01%. 1H NMR (300 MHz, CDCl3): δ (ppm) = 2.44 (s, CH3), 4.97 (s, NH2), 6.58 (d, J = 9.3 Hz, 2H), 6.87 (d, J = 9.0 Hz, 1H), 7.45 (t, J = 8.0 Hz, 1H), 7.53 (d + t, 2H)*, 7.77 (d, J = 9.0 Hz, 1H), 7.86 (s, azomethine), 8.23 (d, J = 9.0 Hz, 2H), 8.34 (d, J = 9.0 Hz, 1H). IR (wave number, cm−1): 446, 484, 662, 752, 824, 1093, 1157, 1179, 1209, 1261, 1319, 1397, 1508, 1591, 1605.
[Pd(L–H)(nn)]. Yield: 75%. Anal. Calc. for C16H14N4O2SPd: C, 42.06; H, 3.06; N, 12.26. Found: C, 42.10; H, 3.09; N, 12.31%. 1H NMR (300 MHz, CDCl3): δ (ppm) = 4.45 (s, NH2), 6.58 (t, J = 9.6 Hz, 1H), 6.92 (t, J = 9.0 Hz, 2H), 7.02 (d, J = 9.2 Hz, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.59 (d + t, 2H)*, 7.78 (d, J = 9.5 Hz, 1H), 7.86 (s, azomethine), 8.16 (d, J = 9.4 Hz, 2H), 9.07 (d, J = 9.0 Hz, 1H). IR (wave number, cm−1): 444, 483, 660, 752, 824, 1095, 1157, 1178, 1207, 1261, 1320, 1396, 1507, 1592, 1605.
[Pd(L–Cl)(nn)]. Yield: 76%. Anal. Calc. for C18H13N4O2SClPd: C, 44.00; H, 2.84; N, 11.40. Found: C, 44.09; H, 2.89; N, 11.45%. 1H NMR (300 MHz, CDCl3): δ (ppm) = 5.77 (s, NH2), 6.58 (d, J = 9.2 Hz, 2H), 7.03 (d + d, 2H)*, 7.28 (s, azomethine), 8.11 (d, J = 9.0 Hz, 2H), 8.33 (t, J = 9.0 Hz, 1H), 8.37 (d, J = 9.3 Hz, 1H), 8.43 (t, J = 9.1 Hz, 1H), 9.07 (d, J = 9.0 Hz, 1H). IR (wave number, cm−1): 445, 482, 661, 751, 823, 1093, 1158, 1176, 1205, 1260, 1318, 1398, 1509, 1590, 1606.
[Pd(L–NO2)(nn)]. Yield: 72%. Anal. Calc. for C18H13N5O4SPd: C, 43.07; H, 2.84; N, 13.96. Found: C, 43.24; H, 2.89; N, 13.91%. 1H NMR (300 MHz, CDCl3): δ (ppm) = 5.87 (s, NH2), 6.78 (d, J = 9.2 Hz, 2H), 7.21 (d + d, 2H)*, 7.40 (s, azomethine), 8.20 (d, J = 8.8 Hz, 2H), 8.39 (t, J = 9.0 Hz, 1H), 8.41 (d, J = 9.0 Hz, 1H), 9.01 (t, J = 9.2 Hz, 1H), 9.16 (d, J = 9.0 Hz, 1H). IR (wave number, cm−1): 444, 485, 661, 751, 823, 1094, 1157, 1180, 1208, 1260, 1320, 1356, 1398, 1507, 1527, 1590, 1605.
[Pd(L–OCH3)(q)]. To a solution of 4-methoxybenzaldehyde thiosemicarbazone (35 mg, 0.17 mmol) and quinolin-8-ol (25 mg, 0.17 mmol) in hot ethanol (30 mL) was added triethylamine (34 mg, 0.34 mmol), followed by a solution of Na2[PdCl4] (50 mg, 0.17 mmol) in ethanol (10 mL). The mixture was heated at reflux for 6 h to yield an orange solution. Evaporation of this solution gave an orangish-yellow solid, which was subjected to purification by thin layer chromatography on a silica plate. With benzene as the eluant, a yellow band separated, which was extracted with acetonitrile. Upon evaporation of the acetonitrile extract [Pd(L–OCH3)(q)] was obtained as a crystalline yellow solid. Yield: 65%. Anal. Calc. for C18H16N4O2SPd: C, 47.12; H, 3.73; N, 12.21. Found: C, 47.31; H, 3.75; N, 12.15%. 1H NMR (300 MHz, CDCl3): δ (ppm) = 3.87 (s, OCH3), 4.98 (s, NH2), 6.88 (d + t, 3H)*, 7.04 (d, J = 9.0 Hz, 1H), 7.22 (t, J = 8.7 Hz, 1H), 7.36 (s, azomethine), 7.45 (d, J = 8.8 Hz, 1H), 8.12 (d, J = 8.7 Hz, 1H), 8.19 (d, J = 8.1 Hz, 2H), 8.22 (d, J = 8.4 Hz, 1H). IR (wave number, cm−1): 484, 514, 662, 744, 816, 1110, 1180, 1286, 1313, 1464, 1504, 1572, 1610.
[Pd(L–CH3)(q)]. Yield: 68%. Anal. Calc. for C18H16N4OSPd: C, 48.82; H, 3.61; N, 12.65. Found: C, 48.47; H, 3.52; N, 12.58%. 1H NMR (300 MHz, CDCl3): δ (ppm) = 2.41 (s, CH3), 5.02 (s, NH2), 6.84 (d + t, 3H)*, 7.09 (d, J = 9.0 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 7.31 (s, azomethine), 7.42 (d, J = 8.3 Hz, 1H), 8.11 (d, J = 7.9 Hz, 1H), 8.24 (d, J = 8.1 Hz, 2H), 8.33 (d, J = 9.0 Hz, 1H). IR (wave number, cm−1): 482, 515, 665, 745, 816, 1111, 1180, 1287, 1314, 1464, 1503, 1572, 1611.
[Pd(L–H)(q)]. Yield: 70%. Anal. Calc. for C17H14N4OSPd: C, 47.62; H, 3.26; N, 13.07. Found: C, 47.83; H, 3.18; N, 13.11%. 1H NMR (300 MHz, CDCl3): δ (ppm) = 5.08 (s, NH2), 6.95 (d, J = 9.0 Hz, 2H), 7.04 (d, J = 9.0 Hz, 1H), 7.18 (t, J = 9.2 Hz, 2H), 7.25 (t, J = 8.3 Hz, 1H), 7.37 (t, J = 8.5 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.86 (s, azomethine), 8.01 (t, J = 9.0 Hz, 1H), 8.20 (d, J = 8.1 Hz, 1H), 8.29 (d, J = 8.0 Hz, 1H). IR (wave number, cm−1): 483, 515, 666, 746, 817, 1109, 1181, 1285, 1311, 1462, 1500, 1571, 1611.
[Pd(L–Cl)(q)]. Yield: 67%. Anal. Calc. for C17H13N4OSClPd: C, 44.06; H, 3.03; N, 12.09. Found: C, 44.12; H. 3.07; N, 11.91%. 1H NMR (300 MHz, CDCl3): δ (ppm) = 5.01 (s, NH2), 6.89 (d, J = 9.0 Hz, 1H), 6.96 (d, J = 9.0 Hz, 2H), 7.33 (d + t, 3H)*, 8.03 (s, azomethine), 8.08 (t, J = 9.1 Hz, 1H), 8.13 (d, J = 9.3 Hz, 1H), 8.23 (d, J = 9.2 Hz, 1H), 8.38 (d, J = 8.8 Hz, 1H). IR (wave number, cm−1): 482, 516, 662, 747, 817, 1110, 1180, 1288, 1311, 1465, 1504, 1570, 1612.
[Pd(L–NO2)(q)]. Yield: 58%. Anal. Calc. for C17H13N5O3SPd: C, 43.09; H, 2.74; N, 14.78. Found: C, 43.33; H, 2.81; N, 14.85%. 1H NMR (300 MHz, CDCl3): δ (ppm) = 5.29 (s, NH2), 6.93 (d, J = 9.0 Hz, 1H), 6.98 (d, J = 9.2 Hz, 2H), 7.37 (d + t, 3H)*, 8.07 (s, azomethine), 8.13 (t, J = 9.3 Hz, 1H), 8.17 (d, J = 9.0 Hz, 1H), 8.31 (d, J = 9.1 Hz, 1H), 8.47 (d, J = 8.4 Hz, 1H). IR (wave number, cm−1): 483, 513, 667, 745, 815, 1108, 1182, 1285, 1310, 1362, 1465, 1503, 1534, 1571, 1610.
Crystallography
Single crystals of [Pd(L–Cl)(nn)] were obtained by slow evaporation of the solvents from a solution of the complex in an acetonitrile–dichloromethane (3:1) mixture. Single crystals of [Pd(L–CH3)(q)] and [Pd(L–Cl)(q)] were grown by slow evaporation of the solvent from acetonitrile solutions of the respective complexes. Selected crystal data and data collection parameters are given in Table 7. Data on all the crystals were collected on a Bruker SMART CCD diffractometer. X-ray data reduction, structure solution and refinement were done using the SHELXS-97 and SHELXL-97 packages.17 The structures were solved by the direct methods.
| [Pd(L–Cl)(nn)] | [Pd(L–CH3)(q)] | |
|---|---|---|
| a R 1 = ∑ ||Fo| − |Fc||/∑|Fo|. b wR 2 = [∑ [w(Fo2 − Fc2)2]/∑ [w(Fo2)2]]1/2. c GOF = [∑ [w(Fo2 − Fc2)2]/(M − N)]1/2, where M is the number of reflections and N is the number of parameters refined. | ||
| Empirical formula | C18H13N4O2SClPd | C18H16N4OSPd |
| Formula weight | 491.26 | 442.84 |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | P21/c | Fdd2 |
| a/Å | 15.5491(14) | 27.9696(19) |
| b/Å | 7.6562(7) | 35.857(3) |
| c/Å | 15.7781(14) | 7.0330(5) |
| α/° | 90 | 90 |
| β/° | 105.304(7) | 90 |
| γ/° | 90 | 90 |
| V/Å3 | 1811.7(3) | 7053.4(9) |
| Z | 4 | 16 |
| D calcd/mg m−3 | 1.801 | 1.668 |
| F(000) | 976 | 3552 |
| λ | 0.71073 | 0.71073 |
| Crystal size/mm3 | 0.24 × 0.24 × 0.25 | 0.14 × 0.17 × 0.21 |
| Temp./K | 273 | 273 |
| μ/mm−1 | 1.309 | 1.184 |
| Collected reflections | 26837 |
25241 |
| R int | 0.132 | 0.066 |
| Independent reflections | 4193 | 4785 |
| R 1 a | 0.0597 | 0.0385 |
| wR 2 b | 0.2265 | 0.0835 |
| GOFc | 0.72 | 0.93 |
General procedure for Suzuki coupling reactions. In a typical run, an oven-dried 10 mL round bottom flask was charged with a known mole percent of catalyst and base, phenylboronic acid (1.2 mmol) and aryl halide (1 mmol) with the appropriate solvents (4 mL). The flask was placed in a preheated oil bath at the required temperature. After the specified amount of time, the flask was removed from the oil bath and water (20 mL) added, followed by extraction with ether (4 × 10 mL). The combined organic layers were washed with water (3 × 10 mL), dried over anhydrous Na2SO4, and filtered. Solvent was removed under vacuum. The residue was dissolved in hexane and analyzed by GC-MS.
General procedure for C–N coupling reactions. In a typical run, an oven-dried 10 mL round bottom flask was charged with a known mole percent of catalyst, NaOtBu (1.3 mmol), amine (1.2 mmol) and aryl halide (1 mmol) with the appropriate solvent(s) (4 mL). The flask was placed in a preheated oil bath at required temperature. After the specified amount of time the flask was removed from the oil bath, water (20 mL) was added, followed by extraction with ether (4 × 10 mL). The combined organic layers were washed with water (3 × 10 mL), dried over anhydrous Na2SO4, and filtered. Solvent was removed under vacuum. The residue was dissolved in acetonitrile and analyzed by GC-MS.
Acknowledgements
We thank the referees for their constructive comments, which have been helpful in preparing the revised manuscript. Financial assistance received from the Department of Science and Technology, New Delhi [Grant No. SR/S1/IC-29/2009 and SR/S1/RFIC-01/2009] is gratefully acknowledged. Jayita Dutta, Sayanti Datta and Dipravath Kumar Seth thank the Council of Scientific and Industrial Research, New Delhi, for their fellowship [Grant No. 09/096(0702)/2011-EMR-I, 09/096(0563)/2008-EMR-I and 9/096(0511)/2006-EMR-I respectively].References
- (a) J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS; (b) V. Guerchais and J. L. Fillaut, Coord. Chem. Rev., 2011, 255, 2448–2457 CrossRef CAS; (c) A. Sivaramakrishna, H. S. Clayton, M. M. Mogorosi and J. R. Moss, Coord. Chem. Rev., 2010, 254, 2904–2932 CrossRef CAS; (d) V. K. Jain and L. Jain, Coord. Chem. Rev., 2010, 254, 2848–2903 CrossRef CAS; (e) J. R. Berenguer, E. Lalinde and M. T. Moreno, Coord. Chem. Rev., 2010, 254, 832–875 CrossRef CAS; (f) D. Belli, D. Amico, L. Labella, F. Marchetti and S. Samaritani, Coord. Chem. Rev., 2010, 254, 635–645 CrossRef; (g) T. S. Lobana, R. Sharma, G. Bawa and S. Khanna, Coord. Chem. Rev., 2009, 253, 977–1055 CrossRef CAS; (h) S. D. Cummings, Coord. Chem. Rev., 2009, 253, 449–478 CrossRef CAS; (i) U. K. Lis, J. Ochocki and K. M. Wasowska, Coord. Chem. Rev., 2008, 252, 1328–1345 CrossRef; (j) J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef; (k) K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753–2766 CrossRef CAS; (l) K. M. C. Wong and V. W. W. Yam, Coord. Chem. Rev., 2007, 251, 2477–2488 CrossRef CAS; (m) H. Torrens, Coord. Chem. Rev., 2005, 249, 1957–1985 CrossRef CAS; (n) A. G. Quiroga and C. N. Ranninge, Coord. Chem. Rev., 2004, 248, 119–133 CrossRef CAS; (o) J. Slageren, A. Klein and S. Záliš, Coord. Chem. Rev., 2002, 230, 193–211 CrossRef; (p) K. Matsumoto and M. Ochiai, Coord. Chem. Rev., 2002, 231, 229–238 CrossRef CAS; (q) J. R. Dilwarth, P. Amold, D. Morales, Y. L. Wong and Y. Zheng, Mod. Coord. Chem., 2002, 217–230 Search PubMed; (r) D. X. West, A. E. Liberta, S. B. Padhye, R. C. Chikate, P. B. Sonawane, A. S. Kumbhar and R. G. Yerande, Coord. Chem. Rev., 1993, 123, 49–71 CrossRef CAS; (s) D. X. West, S. B. Padhye and P. B. Sonawane, Struct. Bonding, 1992, 76, 1–50 CrossRef; (t) I. Haiduc and C. Silvestru, Coord. Chem. Rev., 1990, 99, 253–296 CrossRef CAS; (u) S. B. Padhye and G. B. Kaffman, Coord. Chem. Rev., 1985, 63, 127–160 CrossRef CAS; (v) P. B. Critchlow and S. D. Robinson, Coord. Chem. Rev., 1978, 25, 69–102 CrossRef CAS; (w) M. J. M. Campbell, Coord. Chem. Rev., 1975, 15, 279–319 CrossRef CAS.
- (a) M. O'Connor, A. Kellett, M. McCann, G. Rosair, M. McNamara, O. Howe, B. S. Creaven, S. McClean, A. F. A. Kia, D. O'Shea and M. Devereux, J. Med. Chem., 2012, 55, 1957–1968 CrossRef CAS; (b) M. Skander, P. Retailleau, B. Bourrié, L. Schio, P. Mailliet and A. Marinetti, J. Med. Chem., 2010, 53, 2146–2154 CrossRef CAS; (c) S. H. van Rijt, A. Mukherjee, A. M. Pizarro and P. J. Sadler, J. Med. Chem., 2010, 53, 840–849 CrossRef CAS; (d) J. Hamberger, M. Liebeke, M. Kaiser, K. Bracht, U. Olszewski, R. Zeillinger, G. Hamilton, D. Braun and P. J. Bednarski, Anti-Cancer Drugs, 2009, 20, 559–572 CrossRef CAS; (e) R. Hrstka, D. J. Powell, V. Kvardova, E. Roubalova, K. Bourougaa, M. M. Candeias, P. Sova, F. Zak, R. Fåhraeus and B. Vojtesek, Anti-Cancer Drugs, 2008, 19, 369–379 CrossRef CAS; (f) B. Ma, P. I. Djurovich and M. E. Thompson, Coord. Chem. Rev., 2005, 249, 1501–1510 CrossRef CAS; (g) P. Sova, A. Mistr, A. Kroutil, F. Zak, P. Pouckova and M. Zadinova, Anti-Cancer Drugs, 2005, 16, 653–657 CrossRef CAS; (h) J. Turánek, A. Kašná, D. Záluská, J. Neca, V. Kvardová, P. Knötigová, V. Horváth, L. Šindlerová, A. Kozubík, P. Sova, A. Kroutil, F. Žák and A. Mistr, Anti-Cancer Drugs, 2004, 15, 537–543 CrossRef; (i) E. M. Jouad, X. D. Thanh, G. Bouet, S. M. Bonneau and A. Khan, Anticancer Res., 2002, 22, 1713–1716 CAS; (j) M. B. Ferrari, F. Bisceglie, G. Pelosi, M. Sassi, P. Tarasconi, M. Cornia, S. Capacchi, R. Albertini and S. Pinelli, J. Inorg. Biochem., 2002, 90, 113–126 CrossRef; (k) A. R. Cowly, J. R. Dilworth, P. S. Donnely, E. Labisbal and A. Sousa, J. Am. Chem. Soc., 2002, 124, 5270–5271 CrossRef; (l) R. I. Maurer, P. J. Blower, J. R. Dilworth, C. A. Reynolds, Y. Zheng and G. E. D. Mullen, J. Med. Chem., 2002, 45, 1420–1431 CrossRef CAS; (m) J. Patole, S. Dutta, S. Padhye and E. Sinn, Inorg. Chim. Acta, 2001, 318, 207–211 CrossRef CAS; (n) Z. Iakovidou, A. Papageorgiou, M. A. Demertzis, E. Mioglou, D. Mourelatos, A. Kotsis, P. N. Yadav and D. Kovala-Demertzi, Anti-Cancer Drugs, 2001, 12, 65–70 CrossRef CAS; (o) D. Kovala-Demertzi, J. R. Miller, N. Kourkoumelis, S. K. Hadjikakou and M. A. Demertzis, Polyhedron, 1999, 18, 1005–1013 CrossRef CAS; (p) M. C. Miller III, C. N. Stineman, J. R. Vance, D. X. West and I. H. Hall, Anticancer Res., 1998, 18, 4131–4139 Search PubMed; (q) A. Papageorgiou, Z. Iakovidou, D. Mourelatos, E. Mioglou, L. Boutis, A. Kotsis, D. Kovala-Demertzi, A. Domopoulou, D. X. West and M. A. Demertzis, Anticancer Res., 1997, 17, 247–252 CAS; (r) J. R. Dimmock, R. N. Puthucode, J. M. Smith, M. Hetherington, J. W. Quail, U. Pugazhenthi, J. Lechler and J. P. Stables, J. Med. Chem., 1996, 39, 3984–3997 CrossRef CAS.
- (a) D. K. Seth and S. Bhattacharya, J. Organomet. Chem., 2011, 696, 3779–3784 CrossRef CAS; (b) S. Datta, M. G. B. Drew and S. Bhattacharya, Indian J. Chem. Sec. A, 2011, 50, 1403–1409 Search PubMed; (c) S. Datta, D. K. Seth, R. J. Butcher and S. Bhattacharya, Inorg. Chim. Acta, 2011, 377, 120–128 CrossRef CAS; (d) P. Paul, S. Datta, S. Halder, R. Acharyya, F. Basuli, R. J. Butcher, S. M. Peng, G. H. Lee, A. Castineiras, M. G. B. Drew and S. Bhattacharya, J. Mol. Catal. A: Chem., 2011, 344, 62–73 CrossRef CAS; (e) N. S. Chowdhury, D. K. Seth, M. G. B. Drew and S. Bhattacharya, Inorg. Chim. Acta, 2011, 372, 183–190 CrossRef CAS; (f) S. Basu, R. Acharyya, F. Basuli, S. M. Peng, G. H. Lee, G. Mostafa and S. Bhattacharya, Inorg. Chim. Acta, 2010, 363, 2848–2856 CrossRef CAS; (g) S. Dutta, F. Basuli, A. Castineiras, S. M. Peng, G. H. Lee and S. Bhattacharya, Eur. J. Inorg. Chem., 2008, 4538–4546 CrossRef CAS; (h) S. Halder, S. M. Peng, G. H. Lee, T. Chatterjee, A. Mukherjee, S. Dutta, U. Sanyal and S. Bhattacharya, New J. Chem., 2008, 32, 105–114 RSC; (i) S. Halder, R. J. Butcher and S. Bhattacharya, Polyhedron, 2007, 26, 2741–2748 CrossRef CAS; (j) S. Basu, R. Acharyya, W. S. Sheldrick, H. Mayer-Figge and S. Bhattacharya, Struct. Chem., 2007, 18, 209–215 CrossRef CAS; (k) R. Acharyya, S. Dutta, F. Basuli, S. M. Peng, G. H. Lee, L. R. Falvello and S. Bhattacharya, Inorg. Chem., 2006, 45, 1252–1259 CrossRef CAS; (l) S. Dutta, F. Basuli, S. M. Peng, G. H. Lee and S. Bhattacharya, New J. Chem., 2002, 26, 1607–1612 RSC; (m) I. Pal, F. Basuli, T. C. W. Mak and S. Bhattacharya, Angew. Chem., Int. Ed., 2001, 40, 2923–2925 CrossRef CAS; (n) F. Basuli, S. M. Peng and S. Bhattacharya, Inorg. Chem., 2000, 39, 1120–1127 CrossRef CAS; (o) F. Basuli, M. Ruf, C. G. Pierpont and S. Bhattacharya, Inorg. Chem., 1998, 37, 6113–6116 CrossRef CAS; (p) F. Basuli, S. M. Peng and S. Bhattacharya, Inorg. Chem., 1997, 36, 5645–5647 CrossRef CAS.
- (a) H. Saarinen and J. Karvenranta, Acta Chem. Scand., Ser. A, 1975, 29a, 409–413 CrossRef; (b) A. K. Das, A. Rueda, L. R. Falvello, S. M. Peng and S. Bhattacharya, Inorg. Chem., 1999, 38, 4365–4368 CrossRef CAS.
- (a) A. Gerli, J. Reedijk, M. T. Lakin and A. L. Spec, Inorg. Chem., 1995, 34, 1836–1843 CrossRef CAS; (b) B. J. Coe, D. W. Thompson, C. T. Culberston, J. R. Schoonover and T. J. Meyer, Inorg. Chem., 1995, 34, 3385–3395 CrossRef CAS.
- (a) J. M. Antelo, L. Adrio, M. Teresa Pereira, J. M. Ortigueira, J. J. Fernandez and J. M. Vila, Cryst. Growth Des., 2010, 10, 700–708 CrossRef CAS; (b) S. Singh, N. Bharti and P. P. Mohapatra, Chem. Rev., 2009, 109, 1900–1947 CrossRef CAS; (c) D. Kovala-Demertzi, A. Alexandratos, A. Papageorgiou, P. N. Yadav, P. Dalezis and M. A. Demertzis, Polyhedron, 2008, 27, 2731–2738 CrossRef CAS; (d) K. Husain, A. R. Bhat and A. Azam, Eur. J. Med. Chem., 2008, 43, 2016–2028 CrossRef CAS; (e) L. Otero, C. Folch, G. Barriga, C. Rigol, L. Opazo, M. Vieites, D. Gambino, H. Cerecetto, E. Norambuena and C. Olea-Azar, Spectrochim. Acta, 2008, 70A, 519–523 CrossRef CAS; (f) R. V. Singh, M. K. Biyala, K. Sharma, M. Swami and N. Fahmi, Transition Met. Chem., 2008, 33, 377–381 CrossRef; (g) A. G. Quiroga, L. Cubo, P. J. Sanz Miguel, V. Moneo, A. Carnero and C. Navarro-Ranninger, Eur. J. Inorg. Chem., 2008, 1183–1187 CrossRef CAS; (h) R. Prabhakaran, S. V. Renukadevi, R. Karvembu, R. Huang, J. Mautz, G. Huttner, R. Subashkumar and K. Natarajan, Eur. J. Med. Chem., 2008, 43, 268–273 CrossRef CAS; (i) K. Husain, M. Abid and A. Azam, Eur. J. Med. Chem., 2008, 43, 393–403 CrossRef CAS; (j) J. S. Casas, E. E. Castellano, J. Ellena, M. S. García-Tasende, M. L. Pérez-Parallé, A. Sánchez, Á. Sánchez-González, J. Sordo and Á. Touceda, J. Inorg. Biochem., 2008, 102, 33–45 CrossRef CAS; (k) V. Kaur, J. S. Aulakh and A. K. Malik, Anal. Chim. Acta, 2007, 603, 44–50 CrossRef CAS; (l) D. Gambino, L. Otero, M. Vieites, M. Boiani, M. González, E. J. Baran and H. Cerecetto, Spectrochim. Acta, 2007, 68A, 341–348 CrossRef CAS; (m) A. I. Matesanz and P. Souza, J. Inorg. Biochem., 2007, 101, 1354–1361 CrossRef CAS; (n) P. Parameshwara, J. Karthikeyan, A. N. Shetty and P. Shetty, Ann. Chim., 2007, 97, 1097–1106 CrossRef CAS; (o) K. Husain, M. Abid and A. Azam, Eur. J. Med. Chem., 2007, 42, 1300–1308 CrossRef CAS; (p) T. S. Lobana, G. Bawa, A. Castineiras and R. J. Butcher, Inorg. Chem. Commun., 2007, 10, 506–509 CrossRef CAS; (q) D. Kovala-Demertzi, A. Boccarelli, M. A. Demertzis and M. Coluccia, Chemotherapy, 2007, 53, 148–152 CrossRef CAS; (r) A. I. Matesanz and P. Souza, J. Inorg. Biochem., 2007, 101, 245–253 CrossRef CAS; (s) A. I. Matesanz, C. Pastor and P. Souza, Inorg. Chem. Commun., 2007, 10, 97–100 CrossRef CAS.
- (a) M. B. Ferrari, S. Capacchi, F. Bisceglie, G. Pelosi and P. Tarasconi, Inorg. Chim. Acta, 2001, 312, 81–87 CrossRef CAS; (b) M. B. Ferrari, S. Capacchi, G. Reffo, G. Pelosi, P. Tarasconi, R. Albertini, S. Pinelli and P. Lunghi, J. Inorg. Biochem., 2000, 81, 89–97 CrossRef CAS; (c) N. T. Akinchan and U. Abram, Acta Crystallogr., Sect.C: Cryst. Struct. Commun., 2000, 56, 549–550 Search PubMed; (d) C. Y. Duan, X. Z. You and T. C. W. Mak, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 31–33 Search PubMed; (e) K. Ortner and U. Abram, Inorg. Chem. Commun., 1998, 1, 251–253 CrossRef CAS; (f) Y. P. Tion, C. Y. Duan, Z. L. Lu, X. Z. You, H. K. Fun and S. Kandasamy, Polyhedron, 1996, 15, 2263–2271 CrossRef; (g) N. T. Akinchan, R. Akinchan, U. J. Ibok, L. P. Battaglia, A. B. Corradi and P. Drozdzewski, J. Crystallogr. Spectrosc. Res., 1992, 22, 741–753 CrossRef CAS.
- A. S. A. Zidan and A. I. El-Said, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 1293–1305 CrossRef CAS.
- (a) V. Polshettiwar, C. Len and A. Fihri, Coord. Chem. Rev., 2009, 253, 2599–2626 CrossRef CAS; (b) C. Barnard, Platinum Met. Rev., 2008, 52, 38–45 CrossRef CAS; (c) A. Fihri, P. Meunier and J. C. Hierso, Coord. Chem. Rev., 2007, 251, 2017–2055 CrossRef CAS; (d) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318–5365 CrossRef CAS; (e) R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283–2321 CrossRef CAS; (f) C. J. Elsevier, Coord. Chem. Rev., 1999, 185, 809–822 CrossRef.
- (a) S. M. Islam, P. Mondal, K. Tuhina, A. S. Roy, S. Mondal and D. Hossain, J. Inorg. Organomet. Polym. Mater., 2010, 20, 264–277 CrossRef CAS; (b) M. Guo, Z. Zhu, H. Huang and Q. Zhang, Catal. Commun., 2009, 10, 865–867 CrossRef CAS; (c) C. Pan, M. Liu, L. Zhang, H. Wu, J. Ding and J. Cheng, Catal. Commun., 2007, 9, 508–510 CrossRef.
- (a) F. Godoy, C. Segarra, M. Poyatos and E. Peris, Organometallics, 2011, 30, 684–688 CrossRef CAS; (b) R. K. Das, B. Saha, S. M. W. Rahaman and J. K. Bera, Chem.–Eur. J., 2010, 16, 14459–14468 CrossRef CAS; (c) H. Firouzabadi, N. Iranpoor and M. Gholinejad, J. Mol. Catal. A: Chem., 2010, 321, 110–116 CrossRef CAS; (d) C. F. Fu, C. C. Lee, Y. H. Liu, S. M. Peng, S. Warsink, C. J. Elsevier, J. T. Chen and S. T. Liu, Inorg. Chem., 2010, 49, 3011–3018 CrossRef CAS; (e) M. C. Lipke, R. A. Woloszynek, L. Ma and J. D. Protasiewicz, Organometallics, 2009, 28, 188–196 CrossRef CAS.
- (a) G. K. Rao, A. Kumar, B. Kumar, D. Kumar and A. K. Singh, Dalton Trans., 2012, 41, 1931–1937 RSC; (b) I. B1aszczyk, A. Gniewek and A. M. Trzeciak, J. Organomet. Chem., 2012, 710, 44–52 CrossRef; (c) C. Xu, H. M. Li, H. Liu, Z. Q. Zhang, Z. Q. Wanga, W. J. Fu and Y. Q. Zhang, Inorg. Chim. Acta, 2012, 386, 22–26 CrossRef CAS; (d) D. Pandiarajan and R. Ramesh, J. Organomet. Chem., 2012, 708–709, 18–24 CrossRef CAS; (e) P. Mao, L. Yang, Y. Xiao, J. Yuan, X. Liu and M. Song, J. Organomet. Chem., 2012, 705, 39–43 CrossRef CAS; (f) T. Liu, X. Zhao, Q. Shen and L. Lu, Tetrahedron, 2012, 68, 6535–6547 CrossRef CAS; (g) J. Qiu, L. Wang, M. Liu, Q. Shen and J. Tang, Tetrahedron Lett., 2011, 52, 6489–6491 CrossRef CAS; (h) V. A. Kozlov, D. V. Aleksanyan, Y. V. Nelyubina, K. A. Lyssenko, P. V. Petrovskii, A. A. Vasil'ev and I. L. Odinets, Organometallics, 2011, 30, 2920–2932 CrossRef CAS; (i) Y. Q. Tang, H. Lv, J. M. Lu and L. X. Shao, J. Organomet. Chem., 2011, 696, 2576–2579 CrossRef CAS.
- (a) D. Maiti, B. P. Fors, J. L. Henderson, Y. Nakamura and S. L. Buchwald, Chem. Sci., 2011, 2, 57–68 RSC; (b) L. Chen, G. A. Yu, F. Li, X. Zhu, B. Zhang, R. Guo, X. Li, Q. Yang, S. Jin, C. Liu and S. H. Liu, J. Organomet. Chem., 2010, 695, 1768–1775 CrossRef CAS; (c) A. A. Sorrosa, F. E. Negrete, S. H. Ortega, R. A. Toscano and D. M. Morales, Inorg. Chim. Acta, 2010, 363, 1262–1268 CrossRef.
- L. Malatesta and M. Angoletta, J. Chem. Soc., 1957, 1186–1188 RSC.
- M. J. Frisch, G. W. Tracks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman Jr, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalamani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, I. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskroz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W Wong, C. Gonzalez and J. A. Pople, Gaussian 03, revision D01; Gaussian Inc.: Pittsburgh, PA, 2003 Search PubMed.
- Chemical shifts are given in ppm, and the multiplicity of the signals, along with the associated coupling constants, are given in parentheses. Overlapping signals are marked with an asterisk (*).
- G. M. Sheldrick, SHELXS-97 and SHELXL-97, Fortran programs for crystal structure solution and refinement, University of Gottingen: Gottingen, Germany 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 875871, 875872 and 875873 contain the supplementary crystallographic data for this paper. Crystallographic data for [Pd(L–Cl)(q)] (Table S1), structure of [Pd(L–Cl)(q)] (Fig. S1), selected bond lengths (Å) and angles (°) for [Pd(L–Cl)(q)] (Table S2) and contour plots of HOMO and LUMO of [Pd(L–Cl)(q)] (Fig. S2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21078a |
| This journal is © The Royal Society of Chemistry 2012 |