Triphenylphosphine-mediated reaction of dialkyl azodicarboxylate with activated alkenes leading to pyrazolines†
Shoko
Yamazaki
*a,
Yuta
Maenaka
a,
Ken
Fujinami
a and
Yuji
Mikata
b
aDepartment of Chemistry, Nara University of Education, Takabatake-cho, Nara 630-8528, Japan. E-mail: yamazaks@nara-edu.ac.jp
bKYOUSEI Science Center, Nara Women's University, Nara 630-8506, Japan
First published on 12th July 2012
Abstract
Reaction of the Huisgen zwitterions, derived from triphenylphosphine and azodicarboxylates with activated alkenes has been studied. The reaction of various ethenetricarboxylates and diethyl azodicarboxylate with PPh3 gave pyrazolines efficiently. This pyrazoline synthesis is also applicable to 2-substituted acrylates such as trimethyl 2-phosphonoacrylate and methyl 2-(trifluoromethyl)acrylate. The possible reaction mechanism was discussed in comparison with the reaction of acetylenedicarboxylate and Huisgen zwitterions. The selective transformation of pyrazoline products have also been performed.
Introduction
Pyrazoline1 and its fully conjugated system pyrazole2 derivatives are an important class of organic compounds, because they possess a wide range of biological activities. They are also utilized as building blocks in complex molecule synthesis3 and as functional materials.4 Various synthetic methods for the construction of pyrazole rings have been reported. For example, the condensation reaction of 1,3-dicarbonyl or α,β-unsaturated carbonyl compounds with hydrazine derivatives has been widely used.5 The 1,3-dipolar cycloaddition reaction has also been effectively utilized.6 However, general methods for functionalized pyrazoline rings are relatively few. It is desirable to develop new synthetic methods for the construction of diversely substituted pyrazoline rings.The reaction of dialkyl azodicarboxylates 2 and triphenylphosphine leads to the formation of Huisgen zwitterions A (Scheme 1),7 which plays an important role in the Mitsunobu reaction.8 Brunn and Huisgen reported that the cycloaddition reaction of the zwitterion A with dimethyl acelylenedicarboxylate afforded pyrazoles.9 The reactions of the zwitterion and allenes, which led to pyrazolines and pyrazoles, have been reported.10
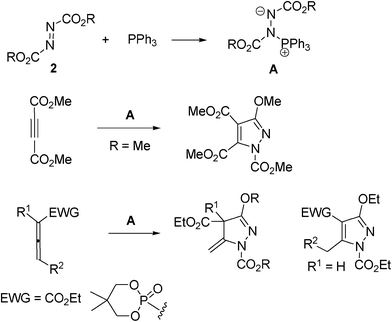
In addition, reactions of the zwitterion, with phenyl isocyanate, phenylisothiocyanate and ketone derivatives leading to various heterocycles and nitrogen-containing compounds have been studied.9a,11 However, few cycloaddition reactions of the zwitterion and alkenes leading to pyrazolines have been reported.12 In this work, cycloaddition reactions of the zwitterion and an activated alkene as a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C component has been investigated.
C component has been investigated.
Results and discussion
Ethenetricarboxylates 1 function as highly electrophilic Michael acceptors.13 It is interesting to develop cycloaddition reactions which lead to highly substituted pyrazoline derivatives. The reaction of ethenetricarboxylate 1a and diethyl azodicarboxylate 2a with 1 equivalent of PPh3 in ether at room temperature for 18 h gave pyrazoline 3a in 85% yield and quantitative triphenylphosphine oxide (eqn (1), Table 1). The structure of pyrazoline 3a was determined by spectroscopic data and confirmed on the basis of the X-ray analysis (Fig. S1 in the ESI†). The reaction of various ethenetricarboxylates 1b–f and 2a with PPh3 were also examined. The reaction gave pyrazolines in 58–89% yield. The additional alkene and alkyne groups on the 2-ester moiety in 1e,f did not participate in the cycloaddition reaction, and the products 3e,f were obtained efficiently (Table 1, entries 5–6).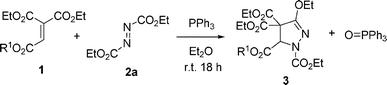 | (1) |
| Entry | 1 | R1 | 3 | Yield (%) |
|---|---|---|---|---|
| 1 | 1a | t Bu | 3a | 85 |
| 2 | 1b | Et | 3b | 86 |
| 3 | 1c | iPr | 3c | 76 |
| 4 | 1d | CH2Ph | 3d | 82 |
| 5 | 1e | CH2CHCH2 |
3e | 58 |
| 6 | 1f | CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH CH |
3f | 89 |
Ether was used as a standard solvent, based on the reported Mitsunobu reaction conditions.8a Various solvents were also examined. The reaction of 1a and 2a in benzene, CH2Cl2, THF and CH3CN for 18 h at room temperature gave the pyrazoline product 3a in 55, 62, 66, and 92% isolated yields, respectively. The polar aprotic solvent CH3CN gave the better yield.14
Next, various phosphines for the reaction of 1a and 2a were examined (Fig. 1). The reaction with phosphines 8a–e gave the pyrazoline product 3a in 63–93% yields, respectively. The reaction with phosphines 8f–h gave 3a in lower yields. The reaction of phosphines 8i–n did not give 3a. The byproducts, phosphine oxides, from 8e and 8f were removed by washing with 2 M HCl.15 Failure by using tri-tert-butylphosphine 8i, tri(o-tolyl)phosphine 8l, diphosphines 8h and 8n, and polymer-supported phosphine 8k16 could be understood by their steric hindrances. Although the reaction of phenylisocyanate, dimethyl azodicarboxylate and trimethyl phosphite 8g was reported to give a cyclized product, 1,2,4-triazolone in good yield,9b the reaction of 1a and 2a with 8g gave the product 3a in low yield. The reaction with a chiral phosphoramidite 8m17 did not give the product 3a. Use of phosphine 8j may lead to the decomposition of the starting materials, as alkyl phosphines are more reactive than aryl phosphines. The side reactions may be caused by the possible reactions between alkene 1a and some phosphines, as discussed later.
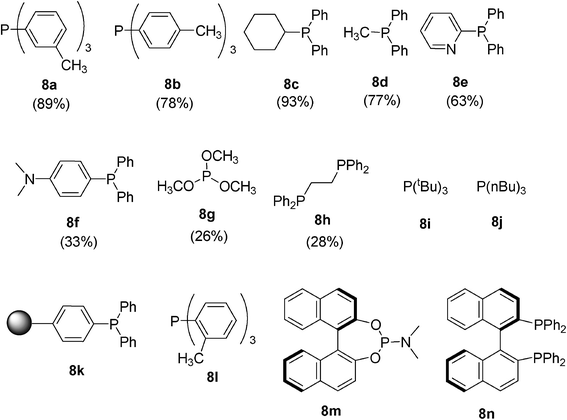 | ||
| Fig. 1 Phosphines examined in this study. | ||
The reaction of 1,1-diethyl 2-t-butyl ethenetricarboxylate (1a) and dimethyl and diisopropyl azodicarboxylates 2b–c in the presence of PPh3 was examined next. However, the reaction gave complex mixtures, possibly consisting of mixed esters 3x (eqn (2)–(3)). In the reaction of 1a and 2b, formation of a trace amount of 2-t-butyl ethenetricarboxylate 1,1-methyl/ethyl mixed ester 1ax was detected. Related ester migration was reported for the reaction of 3-substituted allenoates and dialkyl azodicarboxylate and triphenylphosphine.10a
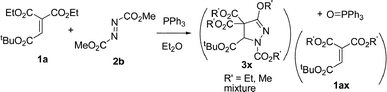 | (2) |
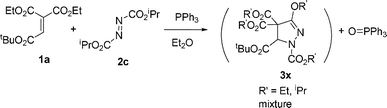 | (3) |
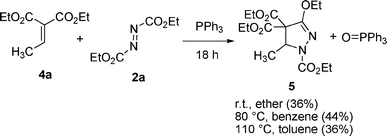
The reaction of less reactive diethyl ethylidenemalonate (4a) and 2a with PPh3 at room temperature in ether for 18 h gave the cycloadduct 5 in 36% yield, along with the remaining starting material 4a (eqn (4)). The reaction at 80 °C in benzene for 18 h gave 5 in 44% yield. Heating at 110 °C in toluene for 18 h decreased the yield of 5 to 36%. At all attempted temperatures, the starting material 4a remained. The reactions of diethyl benzylidenemalonate (4b) or diethyl/methyl maleates (4c), and 2a/2b with PPh3 gave inseparable mixtures, possibly containing cycloadducts with the starting alkenes at room temperature and higher temperatures (60 °C in THF, 80 °C in benzene). The reactions of methyl acrylate (4d), dimethyl fumarate (4e), maleic anhydride (4f), N-methyl maleimide (4g), diethyl p-nitrobenzylidenemalonate (4h), benzylidene Meldrum's acid (5-benzylidene-2,2-dimethyl-1,3-dioxane-4,6-dione) (4i),18 benzalmalononitrile (4j), tetracyanoethylene (4k), or tetraethyl ethenetetracarboxylate (4l) and 2a with PPh3 gave complex mixtures and/or starting materials (Fig. 2). The suitable reactivity of 1,1,2-trisubstituted activated alkenes 1 compared to these 1-, 1,2-, 1,1,2-, and 1,1,2,2-substituted activated alkenes was shown by the efficient cyclization reaction with 2a in the presence of PPh3.
 | (4) |
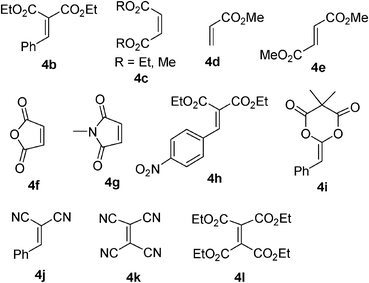 | ||
| Fig. 2 Other activated alkenes examined in this study. | ||
1,1-Disubstituted activated alkenes with two electron-withdrawing groups, acrylate derivatives 6, have been examined next (eqn (5), Table 2). Phosphonate19 and CF320 moieties work as electron-withdrawing groups, and they are biologically attractive and also synthetically useful. The reaction of trimethyl 2-phosphonoacrylate (6a) and dimethyl azodicarboxylate (2b) with 1 equivalent of PPh3 in ether at room temperature for 20 h gave pyrazoline 7a in 80% yield. The reaction of methyl 2-(trifluoromethyl)acrylate (6b) gave pyrazoline 7b in 61% yield. Then, the reaction of tert-butyl 2-(trifluoromethyl)acrylate (6c) and di-tert-butyl azodicarboxylate (2d) with PPh3 was examined, in order to avoid the mixed ester formation with consideration of the result in eqn (2)–(3). However, the reaction did not give the expected cycloadduct and only gave the complex mixture. On the other hand, the reaction of 6c and 2a with PPh3 gave pyrazoline 7c as a major product in 71% yield.
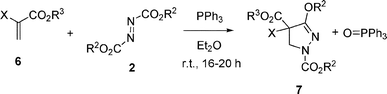 | (5) |
| Entry | 6 | X | R3 | 2 | R2 | 7 | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 6a | PO(OCH3)2 | CH3 | 2b | CH3 | 7a | 80 |
| 2 | 6b | CF3 | CH3 | 2b | CH3 | 7b | 61 |
| 3 | 6c | CF3 | tBu | 2a | CH2CH3 | 7c | 71 |
| 4 | 6d | CN | CH2CH3 | 2a | CH2CH3 | 7d | 44 |
| 5 | 6e | CO2tBu | tBu | 2a | CH2CH3 | 7e | 72 |
| 6 | 6e | CO2tBu | tBu | 2b | CH3 | 7f | 72 |
The reaction of 2-cyanoacrylate 6d and 2a with PPh3 gave pyrazoline 7d in lower yield (Table 2, entry 4). Highly reactive di-tert-butyl methylenemalonate (6e) and 2a with PPh3 was also examined and the reaction gave pyrazoline 7e as a major product in 72% yield. As well, the reaction of 6e and 2b with PPh3 gave 7f in 72% yield. The reaction of 6e and di-tert-butyl azodicarboxylate (2d) with PPh3 also gave a complex mixture.
In order to compare the selectivity of dialkyl acetylenedicarboxylate, the reaction of the dimethyl acetylenedicarboxylate and diethyl azodicarboxylate reported by Cookson and Locke was re-examined (eqn (6)).9b The reaction in ether at room temperature gave the reported pyrazole in 42% yield as a major isolable product, similar to the reported yield (in dioxane, 49% yield). Although unidentified by-product mixture was formed, the formation of mixed ester products could not be confirmed. The reaction of 1a (1 equiv.), 9 (1 equiv.), 2a (1 equiv.), and PPh3 (1 equiv.) was also attempted. The reaction gave pyrazole 10 as a major isolable product (31%). Formation of 3a was not detected, although possible mixed pyrazoles and starting materials of 1a and 9 might be formed.
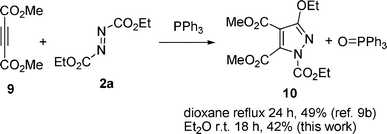 | (6) |
The formation of the pyrazoline ring may undergo a similar mechanism to the reaction of zwitterions A and dimethyl acetylenedicarboxylate or allenic esters (Scheme 1).9,10 To clarify the mechanisms for pyrazoline formation, we carried out density functional theory calculations for the cyclization reactions of the model compounds, trimethyl ethenetricarboxylate (1m), dimethyl azodicarboxylate (2b), and trimethylphosphine (Scheme 2). The structures of intermediates and transition states (TSs) were optimized by B3LYP/6-31G* calculations (Fig.S2 in the ESI†).21
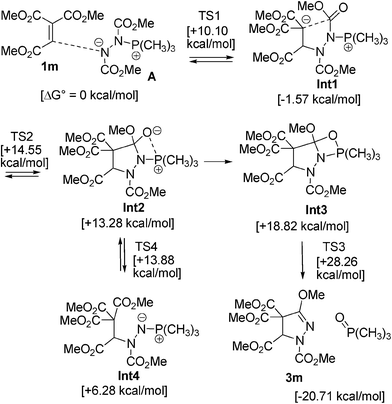 | ||
| Scheme 2 Proposed mechanism for the reaction of model compounds 1m, 2b and trimethylphosphine and B3LYP/6-31G* calculated Gibbs free energies (T = 298.15 K and P = 1 atm). | ||
Conjugate addition of nitrogen of the zwitterion A (generated from 2b and trimethylphosphine) to ethenetricaboxylate 1m gives the stable intermediate Int1 (ΔG° = −1.57 kcal mol−1). The use of highly electrophilic ethenetricarboxylates 1 or 1,1-disubstituted alkenes 6 may facilitate this addition step. The ring closure by nucleophilic attack of the generated malonate anion to the ester group of the azoester gives betaine intermediate Int2. The intermediate Int2 transforms to the oxaphosphetane intermediate Int4.22 Elimination of the phosphine oxide from Int3via a process similar to the Wittig reaction gives the cyclized product 3m.
The betaine intermediate Int2 is in equilibrium with the ylide intermediate Int4 by C–N bond cleavage. The small energy differences of 1m + A, Int1, Int2, Int4 and TS1,2,4) show the process is reversible. This is probably because of the stability of the malonate ester anion Int1. The proposed mechanism may explain the formation of the mixtures in eqn (2)–(3) (Scheme 3). The same ester groups in malonate and azodicaboxylate should be used for synthetic purposes, in order to obtain a single product. Meanwhile, reactions between t-butyl 1,1-disubstituted acrylate esters 6c and 6e and di-t-butyl azodicarboxylate did not give the cyclized products effectively, and the reactions of 6c and 6e with diethyl azodicarboxylate (2a) or dimethyl azodicarboxylate (2b) gave 3-ethoxy pyrazolines 7c and 7e, and 3-methoxy pyrazoline 7f as major products, respectively. Probably the selective reactions arise from steric reasons.
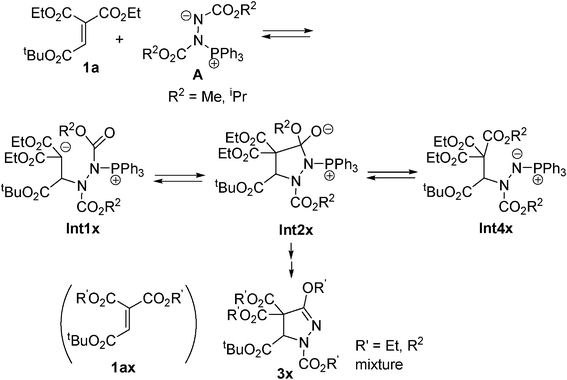 | ||
| Scheme 3 Proposed mechanism for the formation of the mixture in eqn (2)–(3). | ||
To explain the difference of the reactivity and selectivity between the alkynes and the alkenes, the mechanism for alkynes was also examined using density functional calculations. The calculations for dimethyl acetylenedicarboxylate (9), dimethyl azodicarboxylate (2b), and trimethylphosphine were carried out (Scheme 4). The structures of intermediates and TSs were obtained (Fig. S3 in the ESI†).
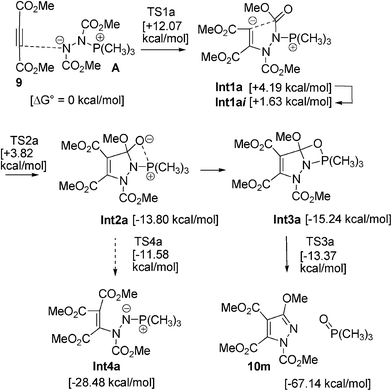 | ||
| Scheme 4 Proposed mechanism for the reaction of 9, 2b and trimethylphosphine and B3LYP/6-31G* calculated Gibbs free energies. | ||
Conjugate addition of the nitrogen of the zwitterion A to dimethyl acetylenedicarboxylate (9) gives the zwitterion intermediate Int1a. Int1a is transformed to the slightly more stable conformational isomer Int1aii, the precursor for cyclization. The ring closure gives the betaine intermediate Int2a. The intermediate Int2a changes to the oxaphosphetane intermediate Int3a.20 Elimination of the phosphine oxide from Int3a gives the cyclized product 10.
For the reaction of ethenetricarboxylate 1m, the phosphine oxide elimination step (Int3 → 3m + Me3PO) is rate-determining (TS3, ΔG‡ = +28.26 kcal mol−1). On the other hand, for the reaction of acetylenedicarboxylate 9, the addition step (9 + A → Int1a) is rate-determining (TS1a, ΔG‡ = +12.07 kcal mol−1). The activation energy of the phosphine oxide elimination step (Int3a → 10m + Me3PO, TS3a, ΔG‡ = −13.37 kcal mol−1) is much lower than that for ethenetricarboxylate 1m. Therefore, the formation of another intermediate Int4a and the equilibrium between 9 + A, Int1a, Int1aii, Int2a, and Int4a may not be facile. The small activation energy of the phosphine oxide elimination step and the stability of 10 + Me3PO indicate the formation of the aromatic pyrazole ring.
The reaction of the mixture of 1a, 9, 2a, and PPh3 leading to pyrazole 10 as a major product above described, can be understood by equilibrium for ethenetricarboxylate intermediates and the faster pyrazole ring formation.
It has been reported that phosphines react with dimethyl acetylenedicarboxylate (9) or electrophilic olefins.23 The relative stability of the phosphine-azodicarboxylate intermediate A compared to phosphine-acetylenedicarboxylate B and phosphine-ethenetricarboxylate C intermediates has also been calculated (Scheme 5). Their optimized structures are shown in the ESI† (Fig. S4).
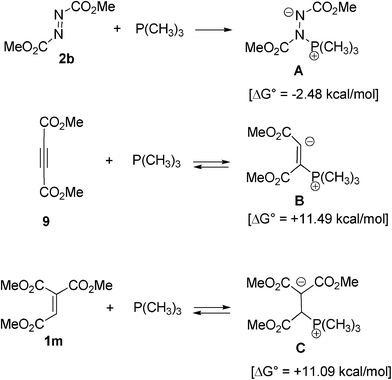 | ||
| Scheme 5 B3LYP/6-31G* calculated relative energies ΔG° for intermediates A, B, and C. ΔG° is the difference of Gibbs free energies (T = 298.15 K, P = 1 atom) relative to those of 2b, 9, 1m and trimethylphosphine (the reactants separated), respectively. | ||
A is 2.48 kcal mol−1 more stable than the reactants, 2b and trimethylphosphine. As the energy difference shows, facile formation of A may occur and the result agrees with the reported irreversibility of the process.24 On the other hand, intermediates B and C are less stable than their reactants. Although intermediates B and C may form reversibly, the stability of A leads to the selectivity of formation of the cyclized products, pyrazoline 3 and pyrazole 10. However, the possible formation of intermediate B causes the side reactions and lowers the yield of the product 10. The formation of intermediate C might also cause the side reactions for ethenetricarboxylate 1m and phosphines in Fig. 1. The stability of intermediate A may arise from the larger bond energy of P–N (ca. 290 kJ mol−1) than that of P–C (264 kJ mol−1).25
Next, in order to demonstrate the utility of the pyrazolines, transformation of 3a was performed. Alkaline hydrolysis of 3a gave trans-monocarboxylic acid 11 stereoselectively. Treatment of 11 with Me3SiCHN2 led to methyl ester 12 in 85% yield (Scheme 6). The trans structures of 11 and 12 were deduced by the coupling constants (5.1–5.3 Hz) between ring protons (C(4)H and C(5)H).26 Interestingly, monocarboxylic acid 11 is unstable at room temperature and gradually becomes decarboxylated to give 13 in CDCl3 or CHCl3 in about 3 days quantitatively (from 3a). Monocarboxylic acid 11 was also transformed by heating in ClCH2CH2Cl at 80 °C for 18 h to give 13 in 62% isolated yield (from 3a). Facile decarboxylation of 11 occurs most probably because of the existence of a –CN– moiety in the ring. Thus, selective transformation of pyrazoline 3a to pyrazoline 12 and the unusually ready transformation of the malonate group, C(CO2Et)2, to CH2 have been found. The detailed reaction mechanism for the transformations is under investigation.
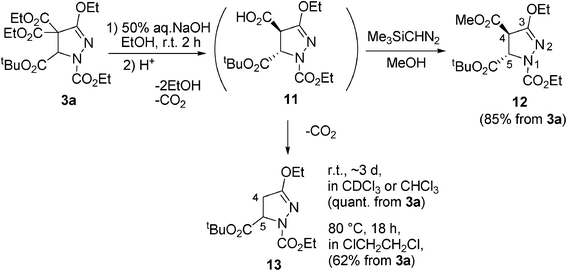 | ||
| Scheme 6 Transformation of pyrazoline 3a. | ||
In summary, the reaction of the Huisgen zwitterions, derived from triphenylphosphine and diethyl azodicarboxylate with activated alkenes afforded highly functionalized pyrazolines. This reaction demonstrates an efficient way to construct potentially useful pyrazolines. The selective transformation of pyrazoline products have been performed. Further study on the transformation of the products to biologically interesting compounds is under investigation.
Experimental section
General methods
1H chemical shifts are reported in ppm relative to Me4Si. 13C chemical shifts are reported in ppm relative to CDCl3 (77.1 ppm). 31P NMR chemical shifts are reported in ppm relative to 85% H3PO4. 19F chemical shifts are reported in ppm relative to CFCl3. 13C mutiplicities were determined by DEPT and HSQC. Peak assignments are made by 2D COSY, HSQC, NOESY, and HMBC spectra.Ethenetricarboxylates (1a,27a1b,c,f,27b1d,27c1e27d) were prepared according to the literature. All procedures using benzene should be carried out in a ventilated hood.
Typical experimental procedure for the preparation of 3, 7 and 10 (Table 1, entry 1)
To a solution of 1a (272.3 mg, 1 mmol) in ether (1 mL) was added diethyl azodicarboxylate 2a (40% in toluene, 0.45 mL, 1 mmol) and PPh3 (262 mg, 1 mmol). The mixture was stirred for 18 h at room temperature. After removal of the solvent under reduced pressure, the residue was purified by column chromatography over silica gel with hexane–ether as eluent to give 3a (368 mg, 85%) and then with CH2Cl2–ether as eluent to give triphenylphosphine oxide (Rf 0.4, CH2Cl2–ether = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, colorless crystals) quantitatively.
:1, colorless crystals) quantitatively.
3a: Rf 0.5 (CH2Cl2–ether = 4:1); colorless crystals; mp 98–100 °C (EtOAc); 1H NMR (400 MHz, CDCl3) δ (ppm) 1.29 (t, J = 7.1 Hz, 3H), 1.30 (t, J = 7.1 Hz, 3H), 1.31 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.1 Hz, 3H), 1.45 (s, 9H), 4.21–4.43 (m, 8H), 5.42 (s, 1H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 13.85 (CH3), 13.94 (CH3), 14.16 (CH3), 14.69 (CH3), 27.86 (CH3), 62.38 (CH2), 63.01 (CH2), 63.38 (CH2), 67.32 (CH2), 67.70 (CH), 68.39 (C), 83.18 (C), 152.67 (C), 156.88 (C), 163.81 (C), 164.57 (C), 165.60 (C). Selected HMBC correlations are between δ 5.42 (C(5)H) and δ 68.39 (C(4)), 156.88 (C(3)), between δ 4.21–4.43 (OCH2) and δ 156.88 (C(3)), and between δ 4.21–4.43 (OCH2) and δ 152.67 (NCO2); IR (KBr) 2980, 1739,1636, 1424, 1369, 1347, 1277, 1234, 1152, 1009 cm−1; MS (FAB) m/z 453 ((M + Na)+); HRMS M+ 430.1948 (calcd for C19H30N2O9 430.1951); Anal. Calcd for C19H30N2O9: C, 53.02; H, 7.02; N, 6.51. Found: C, 52.85; H, 6.88; N, 6.43.
3b (86%): Rf 0.6 (CH2Cl2–ether = 2:1); colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.27 (t, J = 7.1 Hz, 3H), 1.28 (t, J = 7.1 Hz, 3H), 1.27–1.31 (m, 3H), 1.32 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.1 Hz, 3H), 4.12–4.42 (m, 10H), 5.52 (s, 1H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 13.82 (CH3), 13.94 (CH3), 14.04 (CH3), 14.16 (CH3), 14.64 (CH3), 62.18 (CH2), 62.54 (CH2), 63.17 (CH2), 63.54 (CH2), 67.20 (CH), 67.43 (CH2), 68.42 (C), 152.68 (C), 156.85 (C), 163.86 (C), 164.47 (C), 166.82 (C). Selected HMBC correlations are between δ 5.52 (C(5)H) and δ 68.42 (C(4)) and between δ 4.12–4.42 (OCH2) and δ 156.85 (C(3)).; IR (neat) 2985, 1747, 1704, 1645, 1469, 1445, 1257, 1028 cm−1; MS (EI) m/z 402 (M+, 21), 211 (25), 85 (94), 83 (100%); HRMS M+ 402.1638 (calcd for C17H26N2O9 402.1638).
3c (76%): Rf 0.2 (hexane–ether = 1:1); colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.24 (d, J = 6.2 Hz, 3H), 1.28 (d, J = 6.2 Hz, 3H), 1.28 (t, J = 7.1 Hz, 3H), 1.30 (t, J = 7.3 Hz, 3H), 1.32 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.1 Hz, 3H), 4.20–4.44 (m, 8H), 5.02 (septet, J = 6.2 Hz, 1H), 5.48 (s, 1H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 13.76 (CH3), 13.88 (CH3), 14.10 (CH3), 14.58 (CH3), 21.50 (CH3), 21.65 (CH3), 62.41 (CH2), 63.01 (CH2), 63.43 (CH2), 67.23 (CH), 67.32 (CH2), 68.41 (C), 70.10 (CH), 152.66 (C), 156.79 (C), 163.69 (C), 164.44 (C), 166.26 (C). Selected HMBC correlations are between δ 5.48 (C(5)H) and δ 68.41 (C(4)), between δ 4.20–4.44 (OCH2) and δ 156.79 (C(3)), and between δ 5.48 (C(5)H) and δ 152.66 (NCO2).; IR (neat) 2984, 1746, 1703, 1645, 1469, 1445, 1383, 1330, 1256, 1227, 1106, 1019 cm−1; MS (EI) m/z 416 (M+, 41), 329 (21), 257 (94), 211 (80), 139 (100%); HRMS M+ 416.1792 (calcd for C18H28N2O9 416.1795); Anal. Calcd for C18H28N2O9: C, 51.92; H, 6.78; N, 6.73. Found: C, 51.52; H, 6.69; N, 6.75.
3d (82%): Rf 0.2 (hexane–ether = 1:1); colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.17 (t, J = 7.1 Hz, 3H), 1.21 (m, 3H), 1.30 (t, J = 7.1 Hz, 3H), 1.35 (t, J = 7.1 Hz, 3H), 4.00–4.43 (m, 8H), 5.15 (d, J = 12.4 Hz, 1H), 5.18 (d, J = 12.4 Hz, 1H), 5.58 (s, 1H), 7.31–7.36 (m, 5H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 13.79 (CH3), 13.94 (CH3), 14.18 (CH3), 14.55 (CH3), 62.59 (CH2), 63.17 (CH2), 63.57 (CH2), 67.24 (CH), 67.49 (CH2), 67.74 (CH2), 68.47 (C), 128.41 (CH), 128.50 (CH), 128.60 (CH), 134.98 (C), 152.25 (C), 156.91 (C), 163.78 (C), 164.43 (C), 166.69 (C). Selected HMBC correlations are between δ 5.58 (C(5)H) and δ 68.47 (C(4)), 156.91 (C3).; IR (neat) 2984, 1743, 1645, 1445, 1383, 1331, 1259, 1228, 1176, 1016 cm−1; MS (EI) m/z 464 (M+, 13), 273 (23), 160 (41), 91 (100%); HRMS M+ 464.1797 (calcd for C22H28N2O9 464.1795).
3e (58%): Rf 0.7 (CH2Cl2–ether = 1:1); colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.26 (t, J = 7.1 Hz, 3H), 1.29 (t, J = 7.1 Hz, 3H), 1.32 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.1 Hz, 3H), 4.17–4.42 (m, 8H), 4.60 (dddd, J = 13.2, 5.8, 1.4, 1.4 Hz, 1H), 4.66 (dddd, J = 13.2, 5.7, 1.4, 1.4 Hz, 1H), 5.25 (dddd, J = 10.5, 1.3, 1.3, 1.3 Hz, 1H), 5.35 (dddd, J = 17.2, 1.5, 1.5, 1.5 Hz, 1H), 5.56 (s, 1H), 5.90 (dddd, J = 17.2, 1.5, 1.5, 1.5 Hz, 1H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 13.67 (CH3), 13.79 (CH3), 14.02 (CH3), 14.48 (CH3), 62.44 (CH2), 63.13 (CH2), 63.43 (CH2), 66.49 (CH2), 67.03 (CH), 67.31 (CH), 68.32 (C), 118.73 (CH2), 131.12 (CH), 152.51 (C), 156.73 (C), 163.68 (C), 164.27 (C), 166.35 (C). Selected HMBC correlations are between δ 5.56 (C(5)H) and δ 68.32 (C(4)), 156.73 (C(3)).; IR (neat) 2985, 1742, 1704, 1645, 1446, 1383, 1331, 1255, 1186, 1018 cm−1; MS (EI) m/z 414 (M+, 64), 257 (66), 211 (84), 139 (100%); HRMS M+ 414.1634 (calcd for C18H26N2O9 414.1638); Anal. Calcd for C18H26N2O9: C, 52.17; H, 6.32; N, 6.76. Found: C, 51.88; H, 6.45; N, 6.90.
3f (89%): Rf 0.7 (CH2Cl2–ether = 1:1); colorless crystals; mp 98–100 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.28 (t, J = 7.1 Hz, 3H), 1.30 (t, J = 7.3 Hz, 3H), 1.32 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.1 Hz, 3H), 2.54 (t, J = 2.5 Hz, 1H), 4.21–4.42 (m, 8H), 4.73–4.74 (m, 2H), 5.56 (s, 1H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 13.69 (CH3), 13.79 (CH3), 14.02 (CH3), 14.47 (CH3), 53.31 (CH2), 62.54 (CH2), 63.29 (CH2), 63.51 (CH2), 66.84 (CH), 67.39 (CH2), 68.32 (C), 75.78 (CH), 76.52 (C), 152.42 (C), 156.70 (C), 163.58 (C), 164.18 (C), 166.03 (C). Selected HMBC correlations are between δ 5.56 (C(5)H) and δ 68.32 (C(4)), 156.70 (C(3)).; IR (neat) 3231, 2990, 2125, 1776, 1735, 1640, 1429, 1384, 1350, 1283, 1255, 1225, 1176, 1027, 1008 cm−1; MS (EI) m/z 412 (M+, 60), 339 (39), 293 (68), 257 (74), 221 (79), 211 (100%); HRMS M+ 412.1481 (calcd for C18H24N2O9 424.1482); Anal. Calcd for C18H24N2O9: C, 52.42; H, 5.87; N, 6.79. Found: C, 52.37; H, 5.73; N, 6.78.
5 (44%; reaction conditions, 80 °C/18 h/benzene): Rf 0.7 (CH2Cl2–ether = 1:1); colorless crystals; mp 48–50 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.288 (t, J = 7.1 Hz, 3H), 1.293 (t, J = 7.3 Hz, 3H), 1.32 (t, J = 7.1 Hz, 3H), 1.365 (t, J = 7.1 Hz, 3H), 1.374 (d, J = 6.7 Hz, 3H), 4.19–4.41 (m, 8H), 5.16 (d, J = 6.7 Hz, 1H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 13.94 (CH3), 14.03 (CH3), 14.21 (CH3), 14.70 (CH3), 15.67 (CH3), 61.10 (CH), 61.97 (CH2), 62.47 (CH2), 62.91 (CH2), 66.87 (CH2), 69.08 (C), 152.82 (C), 157.26 (C), 164.50 (C), 164.94 (C). Selected HMBC correlations are between δ 5.16 (C(5)H) and δ 69.08 (C(4)), 157.26 (C(3)), between δ 4.19–4.41 (OCH2) and δ 157.26 (C(3)), and between δ 4.19–4.41 (CO2CH2) and δ 152.82 (NCO2).; IR (KBr) 2984, 1740, 1636, 1444, 1331, 1265, 1056 cm−1; MS (EI) m/z 344 (M+, 100), 271 (83), 141 (63%); HRMS M+ 344.1584 (calcd for C15H24N2O7 344.1584); Anal. Calcd for C15H24N2O7: C, 52.32; H, 7.02; N, 8.13. Found: C, 52.57; H, 7.04; N, 8.09.
7a (80%): Rf 0.4 (CH2Cl2–ether = 1:1); colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 3.82 (s, 3H), 3.848 (d, JPH = 4.9 Hz, 3H), 3.852 (s, 3H), 3.88 (d, JPH = 4.8 Hz, 3H), 4.01 (s, 3H), 4.43 (dd, JPH = 21.6, JHH = 12.1 Hz, 1H), 4.55 (dd, JPH = 14.5, JHH = 12.1 Hz, 1H). Selected NOEs are between δ 4.43 (C(5)HH) and δ 4.55 (C(5)HH).; 13C NMR (100.6 MHz, CDCl3) δ (ppm) 53.39 (CH3), 54.07 (CH3), 54.41 (d, JPC = 2 Hz, CH2), 54.60 (d, JPC = 8 Hz, CH3), 54.67 (d, JPC = 7 Hz, CH3), 58.45 (CH3), 59.93 (C), 153.27 (C), 158.74 (d, JPC = 8 Hz, C), 166.22 (d, JPC = 2 Hz). Selected HMBC correlations are between δ 4.55 (C(5)HH) and δ 59.93 (C(4)), between δ 4.01 (OCH3), 4.43, 4.55 (C(5)H2) and δ 158.74 (C(3)), and between δ 3.82 (CO2CH3) and δ 153.27 (NCO2).; 31P NMR (161.9 MHz, CDCl3) δ (ppm) 18.49; IR (neat) 2959, 1742, 1704, 1644, 1463, 1442, 1393, 1262, 1032 cm−1; MS (EI) m/z 324 (M+, 52), 265 (26), 215 (100%); HRMS M+ 324.0722 (calcd for C10H17N2O8P 324.0723).
7b (61%): Rf 0.4 (hexane–ether = 1:1); colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 3.85 (s, 3H), 3.88 (s, 3H), 4.01 (s, 3H), 4.34 (d, J = 12.5 Hz, 1H), 4.49 (dq, JHH = 12.5, JFH = 0.8 Hz, 1H). Selected NOEs are between δ 4.34 (C(5)HH) and δ 4.49 (C(5)HH).; 13C NMR (100.6 MHz, CDCl3) δ (ppm) 53.25 (q, JFC = 1.5 Hz, CH2), 53.57 (CH3), 54.24 (CH3), 58.66 (CH3), 62.48 (C), 122.60 (q, JFC = 283 Hz, C), 153.14 (C), 156.69 (C), 164.15 (C). Selected HMBC correlations are between δ 4.34, 4.49 (C(5)H2) and δ 62.48 (C(4)), between δ 4.01 (OCH3), 4.34, 4.49 (C(5)H2) and δ 156.69 (C(3)), and between δ 3.85 (CO2CH3) and δ 153.14 (NCO2).; 19F NMR (376 MHz, CDCl3) δ (ppm) −70.71; IR (neat) 2961, 1755, 1709, 1653, 1477, 1443, 1401, 1306, 1205, 1135, 1070 cm−1; MS (EI) m/z 284 (M+, 89), 225 (65), 181 (71), 161 (76), 123 (74), 59 (100%); HRMS M+ 284.0619 (calcd for C9H11F3N2O5 284.0620).
7c (71%; including a small amount of impurity): Rf 0.7 (hexane–ether = 1:2); colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.33 (t, J = 7.1 Hz, 3H), 1.37 (t, J = 7.1 Hz, 3H), 1.50 (s, 9H), 4.28 (q, J = 7.1 Hz, 2H), 4.28 (d, J = 12.3 Hz, 1H), 4.36 (q, J = 7.1 Hz, 2H), 4.40 (dq, JHH = 12.3, JFH = 0.7 Hz, 1H). Selected NOEs are between δ 4.28 (C(5)HH) and δ 4.40 (C(5)HH).; 13C NMR (100.6 MHz, CDCl3) δ (ppm) 14.10 (CH3), 14.68 (CH3), 27.70 (CH3), 52.83 (q, JFC = 1.5 Hz, CH2), 62.40 (CH2), 63.22 (C), 67.46 (CH2), 85.29 (C), 122.81 (q, JFC = 282 Hz, C), 152.74 (C), 156.39 (C), 162.58 (q, JFC = 1.5 Hz, C). Selected HMBC correlations are between δ 4.28, 4.40 (C(5)H2) and δ 63.22 (C(4)), between δ 4.36 (OCH2), 4.28, 4.40 (C(5)H2) and δ 156.39 (C(3)), and between δ 4.28 (CO2CH2) and δ 152.74 (NCO2).; 19F NMR (376 MHz, CDCl3) δ (ppm) −70.64; IR (neat) 2984, 1745, 1648, 1471, 1446, 1373, 1345, 1305, 1206, 1155, 1058 cm−1; MS (EI) m/z 354 (M+, 37), 298 (43), 253 (44), 57 (100%); HRMS M+ 354.1403 (calcd for C14H21F3N2O5 354.1403).
7d (44%): Rf 0.3 (hexane–ether = 1:1); colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.33 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.1 Hz, 3H), 1.39 (t, J = 7.1 Hz, 3H), 4.26–4.42 (m, 6H), 4.46 (d, J = 12.0 Hz, 1H), 4.56 (d, J = 12.0 Hz, 1H). Selected NOEs are between δ 4.46 (C(5)HH) and δ 4.56 (C(5)HH).; 13C NMR (100.6 MHz, CDCl3) δ (ppm) 14.00 (CH3), 14.10 (CH3), 14.73 (CH3), 52.29 (C), 56.44 (CH2), 62.75 (CH2), 64.82 (CH2), 68.56 (CH2), 113.70 (C), 152.66 (C), 154.68 (C), 163.00 (C). Selected HMBC correlations are between δ 4.46, 4.56 (C(5)H2) and δ 52.29 (C(4)), between δ 4.26–4.42 (OCH2), 4.46, 4.56 (C(5)H2) and δ 154.68 (C(3)), and between δ 4.26–4.42 (CO2CH2) and δ 152.66 (NCO2).; IR (neat) 2985, 2253, 1752, 1701, 1654, 1444, 1383, 1317, 1252, 1126, 1095, 1014 cm−1; MS (EI) m/z 283 (M+, 70), 211 (41), 183 (43), 138 (91), 110 (100%); HRMS M+ 283.1169 (calcd for C12H17N3O5 283.1168).
7e (72%; including a small amount of impurity): Rf 0.7 (CH2Cl2–ether = 1:1); colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.32 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.1 Hz, 3H), 1.48 (s, 18H), 4.26 (q, J = 7.1 Hz, 2H), 4.33 (q, J = 7.1 Hz, 2H), 4.40 (s, 2H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 14.29 (CH3), 14.80 (CH3), 27.83 (CH3), 54.78 (CH2), 62.12 (CH2), 66.20 (C), 66.78 (CH2), 83.75 (C), 153.23 (C), 158.90 (C), 164.93 (C). Selected HMBC correlations are between δ 4.40 (C(5)H2) and δ 66.20 (C(4)), between δ 4.33 (OCH2), 4.40 (C(5)H2) and δ 158.90 (C(3)), and between δ 4.26 (CO2CH2) and δ 153.23 (NCO2).; IR (neat) 2980, 1734, 1636, 1446, 1370, 1285, 1254, 1159 cm−1; MS (EI) m/z 387 (M+ + 1, 14), 386 (M+, 6), 275 (48), 231 (62), 57 (100%); HRMS M+ 386.2054 (calcd for C18H30N2O7 386.2053).
7f (72%; including a small amount of impurity): Rf 0.7 (CH2Cl2–ether = 1:1); colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.47 (s, 18H), 3.82 (s, 3H), 3.98 (s, 3H), 4.43 (s, 2H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 27.74 (CH3), 53.23 (CH3), 55.02 (CH2), 58.00 (CH3), 65.94 (C), 83.95 (C), 153.38 (C), 159.78 (C), 164.66 (C). Selected HMBC correlations are between δ 4.43 (C(5)H2) and δ 65.94 (C(4)), between δ 3.98 (OCH3), 4.43 (C(5)H2) and δ 159.78 (C(3)), and between δ 3.82 (CO2CH3) and δ 153.38 (NCO2).; IR (neat) 2980, 1731, 1641, 1475, 1456, 1394, 1370, 1287, 1257, 1159 cm−1; MS (EI) m/z 358 (M+, 9.8), 202 (76), 57 (100%); HRMS M+ 358.1730 (calcd for C16H26N2O7 358.1740).
10 (42%): Rf 0.6 (CH2Cl2–ether = 1:1); colorless crystals; mp 90–92 °C (lit.9b mp 92.5–93.5 °C); 1H NMR (400 MHz, CDCl3) δ (ppm) 1.43 (t, J = 7.1 Hz, 3H), 1.46 (t, J = 7.1 Hz, 3H), 3.83 (s, 3H), 3.99 (s, 3H), 4.44 (q, J = 7.1 Hz, 2H), 4.49 (q, J = 7.1 Hz, 2H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 14.11 (CH3), 14.45 (CH3), 52.16 (CH3), 53.62 (CH3), 65.62 (CH2), 66.25 (CH2), 104.51 (C), 141.35 (C), 148.26 (C), 160.81 (C), 160.99 (C), 161.68 (C). Selected HMBC correlations are between δ 4.44 (OCH2), and δ 161.68 (C(3)), and between δ 4.49 (CO2CH2) and δ 148.26 (NCO2).; IR (KBr) 2982, 1763, 1708, 1591, 1514, 1473, 1378, 1330, 1278, 1059, 1026 cm−1; MS (EI) m/z 300 (M+, 21), 269 (15), 213 (34), 196 (41), 181 (49), 168 (100%); HRMS M+ 300.0958 (calcd for C12H16N2O7 300.0958).
Preparation of 12
To a solution of 3a (215 mg, 0.5 mmol) in EtOH (0.47 mL) was added dropwise a 50% (wt%) aqueous NaOH solution (43.9 μL) at 0 °C. The resulting solution was stirred for 2 h at room temperature. The EtOH was evaporated, and the remaining solution was diluted with water (2 mL). The aqueous phase was acidified with KHSO4 and extracted with Et2O twice. The combined organic extracts were dried (MgSO4) and evaporated to give crude 11 (169 mg, 100%).
11: Rf 0.6 (CH2Cl2–ether = 1:1); pale yellow oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.30 (t, J = 7.0 Hz, 3H), 1.36 (t, J = 7.1 Hz, 3H), 1.48 (s, 9H), 3.90 (d, J = 5.1 Hz, 1H), 4.19–4.37 (m, 4H), 5.05 (d, J = 5.1 Hz, 1H), 7.84 (bs, 1H); 13C NMR (100.6 MHz, CDCl3) as a mixture with 13, δ (ppm) 14.20 (CH3), 14.66 (CH3), 27.91 (CH3), 53.27 (CH), 62.48 (CH2), 63.85 (CH), 67.22 (CH2), 83.34 (C), 153.02 (C), 158.33 (C), 167.77 (C), 169.48 (C). Selected HMBC correlations are between δ 3.90 (C(4)H), 5.05 (C(5)H), 4.19–4.37 (OCH2) and δ 158.33 (C(3)).; IR (neat) 3307, 2982, 1748, 1641, 1445, 1223, 1154, 1022 cm−1; MS (EI) m/z 330 (M+); HRMS M+ 330.1430 (calcd for C14H22N2O7 330.1427).
To a solution of crude 11 (prepared from 3a (0.5 mmol)) in methanol (0.2 mL)–benzene (0.8 mL) was added (CH3)3SiCHN2 (ca. 10% hexane solution, 1.5 mL) at room temperature. The mixture was stirred for 30 min at room temperature and concentrated. The residue was purified by column chromatography over silica gel with hexane–ether as eluent to give 12 (145 mg, 85% from 3a).
12: Rf 0.2 (hexane–ether = 1:1); colorless crystals; mp 74–76 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.31 (t, J = 7.1 HZ, 3H), 1.35 (t, J = 7.1 Hz, 3H), 1.48 (s, 9H), 3.82 (s, 3H), 3.87 (d, J = 5.3 Hz, 1H), 4.23–4.37 (m, 4H), 5.00 (d, J = 5.3 Hz, 1H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 14.26 (CH3), 14.72 (CH3), 27.95 (CH3), 53.36 (CH), 53.41 (CH3), 62.30 (CH2), 64.01 (CH), 67.12 (CH2), 83.18 (C), 152.83 (C), 158.24 (C), 167.26 (C), 167.82 (C). Selected HMBC correlations are between δ 3.87 (C(4)H), 5.05 (C(5)H) and δ 158.33 (C(3)), and between δ 4.23–4.37 (CO2CH2) and δ 152.83 (NCO2).; IR (KBr) 2993, 1742, 1649, 1427, 1376, 1345, 1250, 1237, 1150, 1025, 1013 cm−1; MS (EI) m/z 334 (M+, 12), 243 (59), 69 (100%); HRMS M+ 344.1578 (calcd for C15H24N2O7 344.1584); Anal. Calcd for C15H24N2O7: C, 52.32; H, 7.02; N, 8.13. Found: C, 52.55; H, 7.23; N, 8.12.
Preparation of 13
A solution of crude 11 (prepared from 3a (0.5 mmol)) in ClCH2CH2Cl (1 mL) was heated at 80 °C for 18 h. After cooling, the solvent was removed under reduced pressure. The residue was purified by column chromatography over silica gel with hexane–ether as eluent to give 13 (89 mg, 62% from 3a).
13: Rf 0.2 (hexane–ether = 1:1); Colorless oil; 1H NMR (400 MHz, CDCl3) δ (ppm) 1.28 (t, J = 7.1 Hz, 3H), 1.34 (t, J = 7.1 Hz, 3H), 1.48 (s, 9H), 2.82 (dd, J = 17.3, 5.4 Hz, 1H), 3.22 (dd, J = 17.3, 12.1 Hz, 1H), 4.18–4.36 (m, 4H), 4.69 (dd, J = 12.1, 5.4 Hz, 1H). Selected NOEs are between δ 3.22 (C(4)HH) and δ 2.82 (C(4)HH), 4.69 (C(5)H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 14.32 (CH3), 14.76 (CH3), 27.94 (CH3), 35.46 (CH2), 59.91 (CH), 62.03 (CH2), 66.26 (CH2), 82.46 (C), 153.10 (C), 162.39 (C), 169.24 (C). Selected HMBC correlations are between δ 3.22 (C(4)HH), 2.82 (C(4)HH), 4.69 (C(5)H), 4.18–4.36 (OCH2) and δ 158.10 (C(3)).; IR (neat) 2983, 1743, 1696, 1643, 1439, 1340, 1222, 1156, 1023 cm−1; MS (EI) m/z 286 (M+, 41), 230 (25), 185 (98), 113 (100%); HRMS M+ 286.1528 (calcd for C13H22N2O5 286.1529).
Acknowledgements
This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology of the Japanese Government. We thank the Nara Institute of Science and Technology (NAIST) and Prof. K. Kakiuchi (NAIST) for mass spectra. We thank Prof. S. Umetani (Kyoto University) for elemental analyses. We also thank Mr M. A. Kahn, Mr M. Takebayashi, and Mr Y. Maitoko (Nara University of Education) for experimental help.References
- (a) M. Johnson, B. Younglove, L. Lee, R. LeBlanc, H. Holt Jr, P. Hills, H. Mackay, T. Brown, S. L. Mooberry and M. Lee, Bioorg. Med. Chem. Lett., 2007, 17, 5897 Search PubMed; (b) Z. Oezdemir, H. B. Kandilci, B. Guemuesel, U. Calis and A. A. Bilgin, Eur. J. Med. Chem., 2007, 42, 373 CrossRef; (c) D. Nauduri and G. B. Reddy, Chem. Pharm. Bull., 1998, 46, 1254 CAS; (d) F. C. Copp, P. J. Islip and J. E. Tateson, Biochem. Pharmacol., 1984, 33, 339 CrossRef CAS; (e) S. Manyem, M. P. Sibi, G. H. Lushington, B. Neuenswander, F. Schoenen and J. Aube, J. Comb. Chem., 2007, 9, 20 CrossRef CAS; (f) M. E. Camacho, J. Leon, A. Entrena, G. Velasco, M. D. Carrion, G. Escames, A. Vivo, D. Acuna-Castroviejo, M. A. Gallo and A. Espinosa, J. Med. Chem., 2004, 47, 5641 Search PubMed.
- (a) M. J. Genin, C. Biles, B. J. Keiser, S. M. Poppe, S. M. Swaney, W. G. Tarpley, Y. Yagi and D. L. Romero, J. Med. Chem., 2000, 43, 1034 CrossRef CAS; (b) A. Guzman-Perez, R. T. Webster, M. C. Allen, J. A. Brown, A. R. Buchholz, E. R. Cook, W. W. Day, E. S. Hamanaka, S. P. Kennedy, D. R. Knight, P. J. Kowalczyk, R. B. Marala, C. J. Mularski, W. A. Novomisle, R. B. Ruggeri, W. R. Tracey and R. J. Hill, Bioorg. Med. Chem. Lett., 2001, 11, 803 CrossRef CAS; (c) T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347 CrossRef CAS; (d) N. K. Terrett, A. S. Bell, D. Brown and P. Ellis, Bioorg. Med. Chem. Lett., 1996, 6, 1819 CrossRef; (e) H. H. Seltzmann, F. I. Carroll, J. P. Burgess, C. D. Wyrick and D. F. Burch, J. Chem. Soc., Chem. Commun., 1995, 1549 Search PubMed.
- (a) F. M. Guerra, M. R. Mish and E. M. Carreira, Org. Lett., 2000, 2, 4265 CrossRef CAS; (b) J. L. G. Ruano, S. A. A. de Diego, M. R. Martín, E. Torrente and A. M. M. Castro, Org. Lett., 2004, 6, 4945 CrossRef CAS.
- (a) L. Kang, Y. Chen, D. Xiao, A. Peng, F. Shen, X. Kuang, H. Fu and J. Yao, Chem. Commun., 2007, 2695 RSC; (b) K. Rurack and U. Resch-Genger, Chem. Soc. Rev., 2002, 31, 116 RSC; (c) T. Ritter, L. Kværnø, M. Werder, H. Hauser and E. M. Carreira, Org. Biomol. Chem., 2005, 3, 3514 RSC.
- (a) J. Elguero, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky, C. W. Rees and K. T. Potts, Pergamon Press, Oxford, 1984, vol. 5, p. 167 Search PubMed; (b) G. Coispeau and J. Elguero, Bull. Soc. Chim. Fr., 1970, 2717 Search PubMed; (c) J. Safaei-Ghomi, A. H. Bamoniri and M. Soltanian-Telkabadi, Chem. Heterocycl. Compd., 2006, 42, 892 CrossRef CAS; (d) A. R. Katritzky, M. Wang, S. Zhang and M. V. Voronkov, J. Org. Chem., 2001, 66, 6787 CrossRef CAS; (e) Y. R. Huang and J. A. Katzenellenbogen, Org. Lett., 2000, 2, 2833 CrossRef.
- (a) T. Kano, T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2006, 128, 2174 CrossRef CAS; (b) Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 11279 CrossRef CAS; (c) Y. Chen, Y. Lam and Y.-H. Lai, Org. Lett., 2003, 5, 1067 CrossRef CAS; (d) D. Simovic, M. Di, V. Marks, D. C. Chatfield and K. S. Rein, J. Org. Chem., 2007, 72, 650 CrossRef CAS; (e) M. R. Mish, F. M. Guerra and E. M. Carreira, J. Am. Chem. Soc., 1997, 119, 8379 CrossRef CAS; (f) T. J. J. Műller, M. Ansorge and D. Aktah, Angew. Chem., Int. Ed., 2000, 39, 1253 CrossRef CAS; (g) M. P. Sibi, L. M. Stanley and T. Soeta, Org. Lett., 2007, 9, 1553 CrossRef CAS.
- (a) R. Huisgen, in The Adventure Playground of Mechanisms and Novel Reactions: Profiles, Pathways and Dreams, ed. J. I. Seeman, American Chemical Society, Washington, DC, 1994, p. 62 Search PubMed; (b) R. Huisgen, H. Blaschke and E. Brunn, Tetrahedron Lett., 1966, 7, 405 Search PubMed.
- (a) O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS; (b) D. L. Hughes, Org. React., 1992, 42, 335 CAS; (c) D. L. Hughes, Org. Prep. Proced. Int., 1996, 28, 127 CrossRef CAS.
- (a) E. Brunn and R. Huisgen, Angew. Chem., Int. Ed. Engl., 1969, 8, 513 CrossRef CAS; (b) C. Cookson and J. M. Locke, J. Chem. Soc., 1963, 6062 Search PubMed.
- (a) V. Nair, A. T. Biju, K. Mohanan and E. Suresh, Org. Lett., 2006, 8, 2213 CrossRef CAS; (b) M. Chakravarty, N. N. B. Kumar, K. V. Sajna and K. C. K. Swamy, Eur. J. Org. Chem., 2008, 4500 CrossRef CAS.
- (a) R. D. Otte, T. Sakata, I. A. Guzei and D. Lee, Org. Lett., 2005, 7, 495 CrossRef CAS; (b) S.-L. Cui, J. Wang and Y.-G. Wang, Org. Lett., 2008, 10, 13 CrossRef CAS; (c) X.-G. Liu, Y. Wei and M. Shi, Org. Biomol. Chem., 2009, 7, 4708 RSC.
- V. Nair, S. C. Mathew, A. T. Biju and E. Suresh, Angew. Chem., Int. Ed., 2007, 46, 2070 CrossRef CAS.
- For our recent studies using ethenecarboxylates, see: (a) S. Yamazaki, S. Morikawa, K. Miyazaki, M. Takebayashi, Y. Yamamoto, T. Morimoto, K. Kakiuchi and Y. Mikata, Org. Lett., 2009, 11, 2796 CrossRef CAS; (b) S. Yamazaki, Y. Yamamoto, Y. Fukushima, M. Takebayashi, T. Ukai and Y. Mikata, J. Org. Chem., 2010, 75, 5216 Search PubMed; (c) S. Morikawa, S. Yamazaki, M. Tsukada, S. Izuhara, T. Morimoto and K. Kakiuchi, J. Org. Chem., 2007, 72, 6459 CrossRef CAS; (d) S. Morikawa, S. Yamazaki, M. Tsukada, S. Izuhara, T. Morimoto and K. Kakiuchi, J. Org. Chem., 2006, 71, 3540 CrossRef CAS.
- The solvents were not fully optimized in all the reactions.
- (a) M. von Itzstein and M. Mocerino, Synth. Commun., 1990, 20, 2049 CAS; (b) D. Camp and I. D. Jenkins, Aust. J. Chem., 1988, 41, 1835 CrossRef CAS.
- (a) A. R. Tunoori, D. Dutta and G. I. Georg, Tetrahedron Lett., 1998, 39, 8751 CrossRef CAS; (b) R. A. Amos, R. W. Emblidge and N. Havens, J. Org. Chem., 1983, 48, 3598 CrossRef CAS.
- (a) Z. Li, Z. Zhou, L. Wang, Q. Zhou and C. Tang, Tetrahedron: Asymmetry, 2002, 13, 145 CrossRef CAS; (b) R. Hulst, A. van Basten, K. Fitzpatrick and R. M. Kellogg, J. Chem. Soc., Perkin Trans. 1, 1995, 2961 RSC.
- A. M. Dumas, A. Seed, A. K. Zorzitto and E. Fillion, Tetrahedron Lett., 2007, 48, 7072 Search PubMed.
- (a) D. V. Patel, K. Rielly-Gauvin and D. E. Ryono, Tetrahedron Lett., 1990, 31, 5587 CrossRef CAS; (b) J. Wang, T. L. Taylor, A. J. Mical, S. Spitz and T. M. Reilly, Tetrahedron Lett., 1992, 33, 7667 CrossRef CAS; (c) K. Moonen, I. Laureyn and C. V. Stevens, Chem. Rev., 2004, 104, 6177 CrossRef CAS; (d) K. A. Schug and W. Lindner, Chem. Rev., 2005, 105, 67 CrossRef CAS; (e) H. Busseb, Bioorg. Med. Chem., 2006, 14, 1047 CrossRef CAS; (f) C.-F. Chien, J.-D. Wu, T. W. Ly, K.-S. Shia and H.-J. Liu, Chem. Commun., 2002, 248 Search PubMed; (g) C.-C. Liao and J.-L. Zhu, J. Org. Chem., 2009, 74, 7873 CrossRef CAS.
- (a) J. T. Welch, Tetrahedron, 1987, 43, 3123 CrossRef CAS; (b) J. T. Welch and S. Eswarakrishnan, Fluorine in Bioorganic Chemistry, John Wiley & Sons, New York, NY, 1991 Search PubMed; (c) Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications, ed. R. Filler, Y. Kobayashi and L. M., Yagupolskii, Elsevier, Amsterdam, 1993 Search PubMed; (d) G. Resnati and V. A. Soloshnok, Fluoroorganic chemistry: synthetic challenges and biomedical rewards (Tetrahedron Symposium-in-Print Number 58), Tetrahedron, 1996, 52, 1 CrossRef CAS; (e) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef CAS; (f) M. A. Honey, R. Pasceri, W. Lewis and C. J. Moody, J. Org. Chem., 2012, 77, 1396 Search PubMed; (g) L. Wen, Q. Shen, X. Wan and L. Lu, J. Org. Chem., 2011, 76, 2282 CrossRef CAS.
- Some other conformational isomers for the intermediates and TSs regarding ester moieties were also calculated. The reaction pathway composed of the most stable isomers is shown here.
- TS for the formation of intermediates Int3, Int3a from Int2, Int2a could not be obtained by the calculation level.
- (a) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS; (b) N. N. B. Kumar, M. Chakravarty and K. C. K. Swamy, New J. Chem., 2006, 30, 1614 RSC; (c) K. C. K. Swamy, G. Gangadhararao, V. Srinivas, N. N. B. Kumar, E. Balaraman and M. Chakravarty, Inorg. Chim. Acta, 2011, 372, 374 Search PubMed.
- (a) D. Crich, H. Dyker and R. J. Harris, J. Org. Chem., 1989, 54, 257 CrossRef CAS; (b) R. D. Guthrie and I. D. Jenkins, Aust. J. Chem., 1982, 35, 767 CAS; (c) S. Schenk, J. Weston and E. Anders, J. Am. Chem. Soc., 2005, 127, 12566 CrossRef CAS.
- J. Emsley and D. Hall, The chemistry of phosphorus: environmental, organic, inorganic, biochemical and spectroscopic aspects, Harper and Row, London, 1976, p. 35 Search PubMed.
- The coupling constants Jcis and Jtrans for C(4)H and C(5)H in 13 are 5.4 Hz and 12.1 Hz, respectively. The trans assignment for 12 also agrees with the dihedral angles C(4)H-C(5)H, 126° (trans) and 23° (cis) in the B3LYP/6-31G* calculated structures of trans-12 and cis-12 (Fig. S5 in the ESI†).
- (a) S. Yamazaki, H. Kumagai, T. Takada and S. Yamabe, J. Org. Chem., 1997, 62, 2968 CrossRef CAS; (b) S. Yamazaki, K. Ohmitsu, K. Ohi, T. Otsubo and K. Moriyama, Org. Lett., 2005, 7, 759 CrossRef CAS; (c) S. Yamazaki, S. Morikawa, Y. Iwata, M. Yamamoto and K. Kuramoto, Org. Biomol. Chem., 2004, 2, 3134 RSC; (d) S. Yamazaki and M. Takebayashi, J. Org. Chem., 2011, 76, 6432 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data, the optimized geometries, and 1H and 13C NMR spectral data. CCDC reference number 877668. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21249h |
| This journal is © The Royal Society of Chemistry 2012 |