Modulated morphologies and tunable thiol-responsive shedding of aqueous block copolymer aggregates†
Qian
Zhang
,
Na
Re Ko
and
Jung
Kwon Oh
*
Department of Chemistry and Biochemistry, Concordia University and Center for Nanoscience Research (CENR), 7141 Sherbrooke St. W., Montreal, Quebec, Canada H4B 1R6. E-mail: joh@alcor.concordia.ca
First published on 2nd August 2012
Abstract
Controlled morphologies of aqueous sheddable block copolymer aggregates having tunable thiol-responsive degradation are reported. New thiol-responsive degradable block copolymers consisting of a hydrophilic poly(ethylene oxide) (PEO) block and a hydrophobic polystyrene (PSt) block having disulfide linkages at block junctions were synthesized by atom transfer radical polymerization in the presence of a newly-synthesized PEO-based bromine macroinitiator labeled with disulfides. These well-controlled PEO–ss–PSt block copolymers self-assembled to form aqueous aggregates having various morphologies, including spheres, vesicles, and rod-like aggregates, which was controlled not only by structural parameters, such as PSt block length and disulfides, but also by using the aqueous micellization method. Such structural and morphological variation of aqueous aggregates led to the control of their thiol-responsive degradation rate.
Introduction
Well-defined block copolymers (BCPs) have offered a broad choice of materials for the construction of multifunctional and nanostructured materials used as templates, coatings, elastomers, and nanomedicine.1–6 Diverse hydrophilic and hydrophobic building blocks where structures and chain lengths can easily be controlled have made possible the self-assembly of aggregates with various morphologies, including spheres, cylinders, and lamellas in solid states, as well as micellar aggregates with various structures in selective solvents.7 More desirable is the incorporation of degradable linkages into BCP-based micelles that can be cleaved in response to external stimuli such as light, low pH, or ultrasound as well as reductive, oxidative, or enzymatic reactions.8,9 These stimuli-responsive degradable nano-aggregates can be disassembled in a controlled fashion upon external stimuli, but otherwise are stable under physiological conditions.10,11Recently, sheddable BCPs having cleavable linkages at junctions of hydrophobic and hydrophilic blocks (shBCPs) have been developed for various applications as controlled delivery nanocarriers,12–17 nanocapsules,18,19 and nanoporous films.20,21 Several approaches including coupling reaction,22–25 immobilization,26,27 and combined polymerization28,29 have been reported; they allow for the synthesis of a variety of shBCPs with photocleavable, acid-labile, and disulfide linkages at block junctions. They self-assemble to form sheddable aggregates that can shed their hydrophilic coronas from hydrophobic cores in water; however, they typically result in mostly aqueous aggregates having spherical morphology. For bio-related applications in complex environments, a variation of morphologies exhibiting tunable degradation rates of shBCP-based micellar aggregates in aqueous solution can be required. Besides, the value-added strategy that introduces shedding groups to BCPs is greatly beneficial in exploiting their morphology19 as well as morphology-dependent stimuli-responsive degradation.
Herein, we report on controlled morphologies of sheddable aggregates with thiol-responsive degradation in aqueous solution (Scheme 1). This important advance centers on the design and synthesis of new shBCPs consisting of a well-established hydrophilic poly(ethylene oxide) (PEO) block and a hydrophobic polystyrene (PSt) block, having junction disulfide linkages (PEO–ss–PSt, ssBCPs) as a proof-of-concept degradable protocol. A disulfide is cleaved into corresponding thiols in a reducing environment or through a disulfide–thiol exchange.30,31 In biological systems, glutathione (GSH, a tripeptide containing cysteine) as a water-soluble thiol is found at different levels between intracellular and extracellular environments,32,33 attaining biodegradation. Further, the use of disulfide chemistry features the generation of thiol groups that can be further used for functionalization and fabrication of polymer-based nanomaterials.34–37 Due to the amphiphilic nature, the PEO–ss–PSt ssBCPs self-assembled in aqueous solution; the final aggregate morphology is controlled not only by structural parameters, such as PSt block length and disulfide, but also by using the aqueous micellization method. Furthermore, with such structural and morphological variation, the degradation rate of aqueous aggregates by cleavage of disulfides in reducing environments through a disulfide-thiol exchange was tuned.
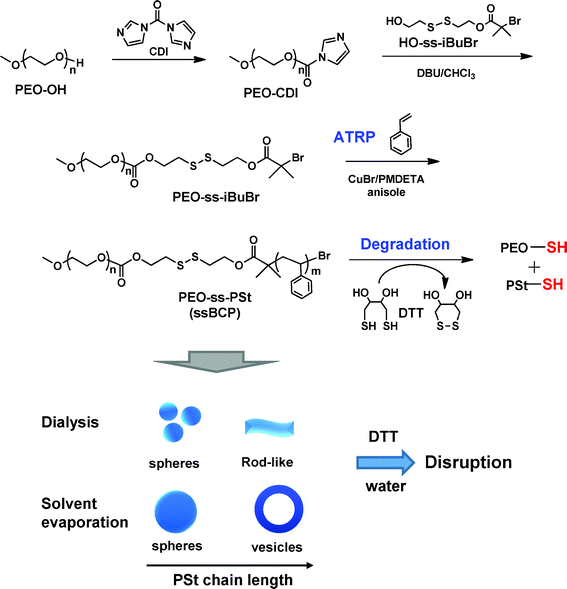 | ||
| Scheme 1 Our approach to synthesis, self-assembly, and degradation of new PEO–ss–PSt (ssBCPs). | ||
Experimental
Instrumentation and materials
1H-NMR spectra were recorded using a 500 MHz Varian spectrometer. The CDCl3 singlet at 7.27 ppm was selected as the reference standard. Spectral features are tabulated in the following order: chemical shift (ppm); multiplicity (s – singlet, d – doublet, t – triplet, m – complex multiplet); number of protons; position of protons. Molecular weight and molecular weight distribution were determined by gel permeation chromatography (GPC) with a Viscotek VE1122 pump and a refractive index (RI) detector. Two PolyAnalytik columns (PAS-103L, 106L, designed to determine molecular weight up to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 g mol−1) were used with THF as an eluent at 30 °C at a flow rate of 1 mL min−1. Linear polystyrene (PS) standards were used for calibration. Aliquots of polymer samples were dissolved in THF and the clear solutions were filtered using a 0.25 μm PTFE filter to remove any THF-insoluble species. A drop of anisole was added as a flow rate marker. The size of micelles in hydrodynamic diameter by volume was measured by dynamic light scattering (DLS) at a fixed scattering angle of 173° at 25 °C with a Malvern Instruments Nano S ZEN1600 equipped with a 633 nm He–Ne gas laser.
000000 g mol−1) were used with THF as an eluent at 30 °C at a flow rate of 1 mL min−1. Linear polystyrene (PS) standards were used for calibration. Aliquots of polymer samples were dissolved in THF and the clear solutions were filtered using a 0.25 μm PTFE filter to remove any THF-insoluble species. A drop of anisole was added as a flow rate marker. The size of micelles in hydrodynamic diameter by volume was measured by dynamic light scattering (DLS) at a fixed scattering angle of 173° at 25 °C with a Malvern Instruments Nano S ZEN1600 equipped with a 633 nm He–Ne gas laser.
Poly(ethylene oxide)monomethyl ether (PEO–OH with MW = 5000 g mol−1 and EO unit# = 113), 2-hydroxylethyl disulfide (SS–DOH), α-bromoisobutyl bromide (Br–iBuBr), 1,1′-carbonyldiimidazole (CDI, reagent grade), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 98%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, >98%), triethylamine (Et3N), copper(I) bromide (CuBr, >99.99%), anisole (anhydrous, >99%), and chloroform (ACS, >99%) purchased from Aldrich as well as DL-dithiothritol (DTT, 99%) purchased from Acros Organics were used as received unless specified. PEO–OH was dried in a vacuum oven at 100 °C overnight to remove residual moisture before use. Chloroform was extracted with water three times to remove a trace of ethanol, and then dried over MgSO4. Styrene was purified by passing through a column of basic alumina to remove inhibitors.
Synthesis of 2-hydroxyethyl-2′-(bromoisobutyryl)ethyl disulfide (HO–ss–iBuBr)
Br–iBuBr (8.2 g, 35.7 mmol) dissolved in THF (50 mL) was added drop-wise at 0 °C over 20 min to a solution consisting of SS–DOH (5.0 g, 32.4 mol), Et3N (6.0 ml, 42.8 mmol), and THF (150 mL) purged with dry nitrogen for 15 min. The resulting solution was stirred for 14 hours at room temperature. White solids (Et3N–HCl adducts) formed during the reaction were removed by vacuum filtration. Filtrates were concentrated by rotary evaporation and washed with aqueous acidic and basic solutions three times. The residuals were extracted with ethyl acetate (50 ml) three times, and then dried over anhydrous MgSO4. Solvents were removed by rotary evaporation and the product was purified by silica column chromatography with a mixture of ethyl acetate–hexane (3/7 v/v). The product was collected as the second of the total two bands off a silica gel column. The product was isolated by evaporation of solvents and further dried in a vacuum oven at room temperature for 12 hours to form a yellow oily residue. Yield = 4.3 g (43%). Rf = 0.36 on silica (3/7 ethyl acetate/hexane). 1H-NMR (CDCl3, ppm) 4.45 (t, 2H, nCH2OC(O)–), 3.9 (t, 2H, HO–CH2–), 3.0 (t, 2H, –SS–CH2–CH2OC(O)–), 2.9 (t, 2H, HOCH2–CH2–SS–), 1.95 (s, 6H, –C(CH3)2Br). 13C-NMR (CDCl3, ppm) 30.4, 36.2, 41.0, 55.5, 59.9, 63.4, 171.2. Mass calculated for C8H19BrNaO3S2: 324.59382. Found: 324.95491.Synthesis of CDI-activated PEO–OH (PEO–CDI)
A clear solution of PEO–OH (18.9 g, 3.8 mmol) dissolved in chloroform (15 mL) was added drop-wise to a dispersion of CDI (3.7 g, 22.7 mmol) partially dissolved in chloroform (25 mL) for 1 h at room temperature. The resulting translucent mixture was stirred for 3 days at room temperature, washed with an aqueous saturated NaCl solution (75 mL) four times, and then dried over MgSO4. The products were isolated by removal of solvents using rotary evaporation and further dried in vacuum at room temperature for 18 h. Yield = 18.1 g (94%). 1H-NMR (CDCl3, ppm) 8.16 (s, 1H, –NCH=N–), 7.45 (s, 1H, –NCH=CH–), 7.07 (s, 1H, –NCH=CH–), 4.56 (t, 2H, –CH2OC(O)N–), 3.4–3.9 (m, EO protons –CH2–), 3.38 (s, 3H, H3CO–).Synthesis of PEO–ss–iBuBr
PEO–CDI (6.0 g, 1.18 mmol) dissolved in chloroform (20 mL) was added to a mixture of HO–ss–iBuBr (0.5 g, 1.47 mmol) and DBU (32.6 mg, 0.21 mmol) at room temperature. After 15 hours, the resulting mixture was washed with water twice, and then dried over MgSO4. The product was precipitated from hexane three times, and then isolated by vacuum filtration. Yield = 5.9 g (93%). 1H-NMR (CDCl3, ppm) 4.44 (t, 2H, –CH2OC(O)C(CH3)2Br), 4.39 (t, 2H, –CH2OC(O)O–), 4.30 (t, 2H, –OC(O)OCH2–), 3.4–3.9 (m, EO proton –CH2–), 3.38 (s, 3H, H3CO–), 2.97 (m, 4H, –CH2SSCH2–), 1.94 (s, 6H, –C(CH3)2Br).Synthesis of PEO–ss–PSt ss BCP block copolymers
A normal ATRP of St was conducted in the presence of the PEO–ss–iBuBr macroinitiator in anisole at 100 °C (or 120 °C). Typically, PEO–ss–iBuBr (0.3 g, 0.06 mmol), St (3.3 mL, 28.6 mmol), PMDETA (6.4 mg, 0.04 mmol), and anisole (5.3 mL) were added to a 25 mL Schlenk flask. The resulting mixture was deoxygenated by three freeze–pump–thaw cycles. The reaction flask was filled with nitrogen and then CuBr (4.4 mg, 0.03 mmol) was added to the frozen solution. The flask was closed, evacuated with vacuum, and backfilled with nitrogen, three times. The mixture was thawed and then the flask was immersed in an oil bath preheated at 100 or 120 °C to start the polymerization. Aliquots were withdrawn at different time intervals to measure molecular weight by GPC. The polymerization was stopped by cooling and exposing the reaction mixture to air.The resulting ssBCPs were purified as follows: as-synthesized polymer solutions passed through a basic alumina column to remove copper complex, and then solvents were removed by rotary evaporation. The products were precipitated from hexane three times, and then dried in a vacuum oven at room temperature for 18 hours.
Aqueous self-assembly of ssBCPs using different methods
For the solvent evaporation method,38 deionized water (20 mL) was added drop-wise into a clear polymer solution containing an aliquot of the purified, dried ssBCP (1 mg) dissolved in THF (1 mL). The resulting dispersion was stirred overnight to remove THF, allowing for colloidally stable micellar dispersion at 0.05 mg mL−1 concentration.For the dialysis method,39 deionized water (0.2 mL) was added drop-wise to a clear solution of ssBCP-1 (10 mg) dissolved in DMF (490 mg). The resulting cloudy dispersion was transferred into a dialysis tubing with MWCO = 3500 g mol−1, and then dialyzed over fresh deionized water for 5 days. Solids formed were removed by centrifugation (13000 rpm × 1 min at 25 °C). A similar procedure was used with water (10 mL), ssBCP-2 (1 mg), and DMF (1 mL).
Transmission electron microscopy (TEM)
TEM images were taken using a Philips CM200 HR-TEM, operated at 200 kV electrons and equipped with a thermionic LaB6 cathode filament (electron emission mode), anti-contamination cold finger, Genesis EDAX system, and AMT V600 2k X2K CCD camera. The point-to-point resolution and the line resolution of the machine are 0.24 nm and 0.17 nm, respectively. To prepare specimens, as-prepared micellar dispersions were dropped onto TEM copper grids (400 mesh, carbon coated). The grids were dried in air.Construction of calibration curves using GPC
A series of mixtures with different wt ratios of PEO–Br/ssBCP was dissolved in THF. From their GPC chromatograms based on RI signal, the areas under the peaks were calculated to construct the calibration curve for each ssBCP.Thiol-responsive degradation of ssBCPs in DMF
GPC was used to examine thiol-responsive cleavage of disulfide linkages of ssBCPs in DMF at room temperature. ssBCP (50 mg) was dissolved in DMF (10 mL) and mixed with a stock solution of DTT in DMF (0.03 mg mL−1, 58 μL for ssBCP-1 and 40 μL for ssBCP-2; 5 mole equivalents to disulfides). Aliquots of polymer solutions were injected into GPC at given time intervals.Thiol-responsive degradation of micellar aggregates in aqueous solution
Aliquots of micellar dispersions (5 mL, 0.1 mg mL−1) were mixed with a stock solution of DTT in water (10 mg mL−1, 138.7 μL for ssBCP-1 and 95.2 μL for ssBCP-2, 400 mole equivalents to disulfides). Samples taken at 4 and 24 hours respectively were dissolved in THF for GPC measurements after the removal of water by rotary evaporation.Results and discussion
Synthesis of PEO–ss–PSt block copolymers
Our approach to the synthesis of well-controlled ssBCPs employs atom transfer radical polymerization (ATRP),40,41 a successful controlled radical polymerization method that enables the synthesis of (co)polymers with pre-determined molecular weight and narrow molecular weight distribution.42 For the approach, a new PEO-based ATRP macroinitiator labeled with a disulfide (PEO–ss–iBuBr) was first synthesized by the reaction of 2-hydroxyethyl-2′-(bromoisobutyryl)ethyl disulfide (HO–ss–iBuBr) with a CDI-activated PEO (PEO–CDI). PEO–CDI was synthesized by reacting PEO–OH with CDI. Their structures were confirmed by 1H-NMR (Fig. 1). The synthesis of HO–ss–iBuBr using a carbodiimide coupling reaction of SS–DOH with 2-bromoisobutyric acid in the presence of N,N′-dicyclohexylcarbodiimide (DCC) was described.28 Here, the synthetic procedure was modified with the reaction of SS–DOH with Br-iBuBr in the presence of Et3N for higher yield.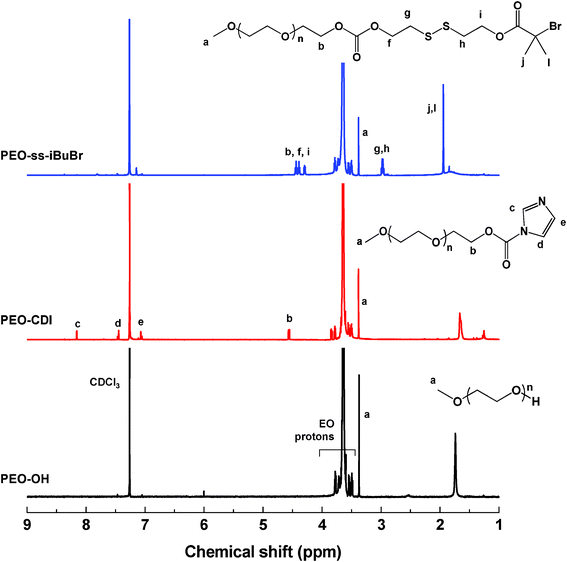
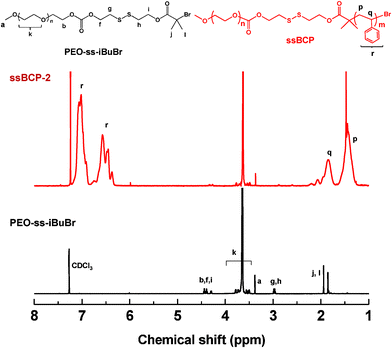
In the presence of the purified, dried PEO–ss–iBuBr macroinitiator, a series of ATRP of St was carried out in anisole mediated with CuBr/PMDETA active catalysts. Table 1 summarizes polymerization conditions and molecular weights. GPC results suggest the evolution of GPC traces of ssBCPs to the high molecular weight region with no significant traces of macroinitiators, suggesting the continuous growth of PSt chains from PEO–ss–iBuBr macroinitiators (Fig. 2). The molecular weight distribution was narrow, with Mw/Mn < 1.15. Furthermore, 1H-NMR results indicate the formation of PEO113–ss–PSt163 (ssBCP-1) in Fig. S1 in the ESI† and PEO113–ss–PSt260 (ssBCP-2) in Fig. 3. The DP of PSt block was determined by the integral ratio of peaks (r) corresponding to phenyl protons in the PSt block to a peak (a) corresponding to methoxy protons in PEO block. These GPC and 1H-NMR results confirmed the successful synthesis of well-controlled ssBCPs with different PSt chain lengths, which allowed for the investigation of aqueous micellization and morphology-driven thiol-responsive degradation.
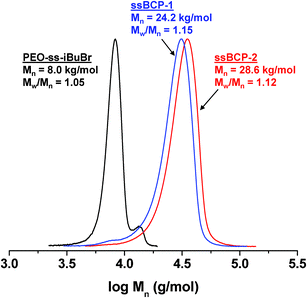 | ||
| Fig. 2 GPC traces of two ssBCPs having different PSt chain lengths, compared with PEO–ss–iBuBr macroinitiator, after purification. | ||
| ssBCPsa | Temp (°C) | Polym (hours) | DPc | M n d (g mol−1) | M w/Mnd |
|---|---|---|---|---|---|
| a Conditions for ATRP: [St]0/[PEO–ss–iBuBr]0/[CuBr/PMDETA]0 = 500/1/0.5; St/anisole = 0.7/1 v/v. b ATRP macroinitiator. c Degree of polymerization determined by 1H-NMR based on DP = 113 of PEO block. d Determined by GPC calibrated with PSt standards. | |||||
| PEO–ss–iBuBrb | — | — | 113 | 7900 | 1.05 |
| ssBCP-1 | 120 | 4.5 | 163 | 24200 | 1.15 |
| ssBCP-2 | 100 | 12.5 | 260 | 28600 | 1.12 |
Aqueous micellization and morphology studies
Aqeuous solution self-assembly of ssBCPs having different PSt chain lengths using different micellization methods was investigated. A dialysis method in DMF using dialysis tubing (MWCO = 3500 g mol−1) over water was first examined to prepare aqueous micellar aggregates. TEM images show that ssBCP-1 containing 41 mol% PEO (based on DP) formed primarily spheres with an average diameter of 20.4 ± 1.8 nm and ssBCP-2 containing 30 mol% PEO formed a mixture of rod-like aggregate and spheres (Fig. 4). DLS results suggest that aqueous ssBCP-1 micellar aggregates had monomodal distribution with a hydrodynamic diameter = 48 nm. However, ssBCP-2 micellar aggregates had three populations; one with the hydrodynamic diameter = 140 nm and the other two with a hydrodynamic diameter = 1.5 and 5.2 μm (Fig. S2 in the ESI†). In addition, small amount of precipitates were observed in the dialysis tubing during aqueous micellization, suggesting that ssBCP-2 with larger PSt chain length has a high tendency to aggregate. These results suggest that the morphology can be controlled from spheres to rod-like aggregates with varying PEO mol% from 41 to 30 mol% in PEO113–ss–PStx copolymers having disulfide linkages at block junctions. In addition, the presence of “coexistence” suggests that aqueous micellization by dialysis method is kinetic-controlled. Further experiments including the effect of the concentration of block copolymers in DMF on micellar morphologies are currently under investigation.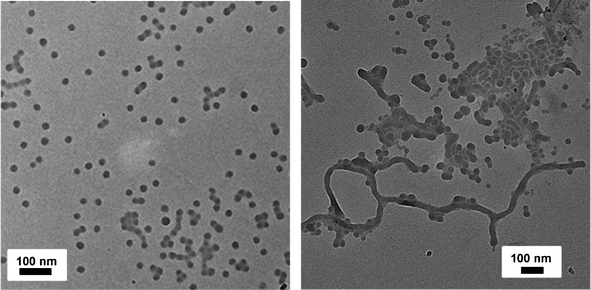 | ||
| Fig. 4 TEM images of micellar aggregates from aqueous solutions of ssBCP-1 showing primarily spheres with short rod-like aggregates (left) and ssBCP-2 showing primarily long rod-like aggregates (right) prepared by dialysis method. Scale bar = 100 nm. The copolymer concentration in the initial DMF solution was 2.0 wt%. | ||
Similar results were reported by Eisenberg and his coworkers who investigated aqueous micellization of PEOx-b-PSt240 copolymers with no disulfide linkages.43 In this case, PSt chain length was fixed as DP = 240, while DP of PEO (x) varied from 15 to 180. It was found that PEO180-b-PSt240 containing 43 mol% PEO primarily formed large micron-sized spheres with diameters = 0.5–2 μm, while PEO80-b-PSt240 containing 25 mol% PEO primarily formed rod-like aggregates having an average diameter of ca. 60 nm and small individual spheres having a diameter of ca. 35 nm. Although the variation is similar for the morphologies from spheres to rod-like aggregates consisting of spheres with PEO mol% varying from 43 to 25 mol% in PEOx-b-PSt240 copolymers, the sizes and diameters are different from our PEO113–ss–PStx systems with disulfide linkages at block junctions.
Further, a solvent evaporation method using THF was examined. Here, TEM images show that ssBCP-1 formed spheres having average diameter of 201.0 ± 50.1 nm (Fig. 5a), which is larger than those of small spheres formed in the dialysis method. Interestingly, ssBCP-2 formed primarily vesicles (Fig. 5b), although additional species such as spheres were also seen on some micrographs. The average diameter of vesicles was 168.2 ± 30.9 nm with 98.2 ± 28.8 nm inner voids and 35.0 ± 6.4 nm shell thickness. Dynamic light scattering (DLS) results indicate a hydrodynamic diameter of 316.3 ± 20.2 nm for ssBCP-1 and 324.8 ± 28.3 nm for ssBCP-2, suggesting no significant change in diameter with PSt chain lengths of DP = 163 and 260 in hydrated states (Fig. S3 in the ESI†). These results suggest that the morphologies of PEO–ss–PSt copolymers can be adjusted with PSt chain lengths and micellization methods.
 | ||
| Fig. 5 TEM images of micellar aggregates from aqueous solutions of ssBCP-1 showing spheres (left, scale bar = 500 nm) and ssBCP-2 showing primarily vesicles (right, scale bar = 100 nm) prepared by solvent evaporation. The inset in the image on the right clearly shows the vesicle with outer diameter = 124 nm and inner diameter = 65.3 nm. The final concentration of micellar dispersions after evaporation of THF was 0.05 mg mL-1. | ||
Thiol-responsive degradation
Degradation of new ssBCPs in response to thiols was first examined in DMF in the presence of DTT. GPC was used to determine the extent of degradation of ssBCPs with different PSt chain lengths. It is likely that the degraded products of PEO–ss–PSt include PSt–SH and PEO–SH (MW ≈ 5000 g mol−1). Fig. 6a–b show the evolution of GPC traces of ssBCPs in the presence and absence of DTT in DMF at different times of 20 min and 2 hours. A new peak corresponding to PEO–SH appeared due to the cleavage of disulfides in ssBCPs. Furthermore, the peak corresponding to ssBCP evolved to the low molecular weight region. A gradual increase in a new GPC peak corresponding to PEO–SH suggests the occurrence of the cleavage of disulfide linkages in the presence of DTT.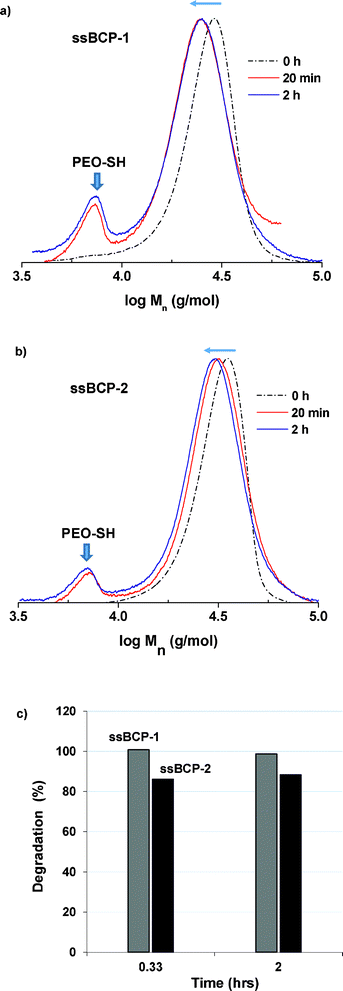 | ||
| Fig. 6 Overlaid GPC traces of ssBCP-1, ssBCP-2 (b), and thiol-responsive degradation (c) in the presence of DTT in DMF. Grey: ssBCP-1 and black: ssBCP-2 in (c). | ||
For the quantitative analysis of the degradation of ssBCPs, a series of mixtures with various wt ratios of PEO–ss–iBuBr (to mimic PEO–SH) to ssBCPs was prepared. From their GPC measurements, their calibration curves based on the ratio of GPC signal and wt ratio of PEO–Br to ssBCP were constructed (Fig. 7). They were then used to calculate wt% PEO in degraded products over time, which was normalized with a theoretically calculated wt% PEO in ssBCPs (15.5 wt% for ssBCP-1 and 23.3 wt% for ssBCP-2) to evaluate the degradation of ssBCPs in response to DTT. Fig. 6c shows the extent of degradation of both ssBCPs in DMF, suggesting a fast cleavage of disulfide linkages, thus leading to a rapid degradation of >99% for ssBCP-1 and >85% for ssBCP-2, within 20 min. A slightly faster degradation of ssBCP-1 could be attributable to a shorter PSt chain length.
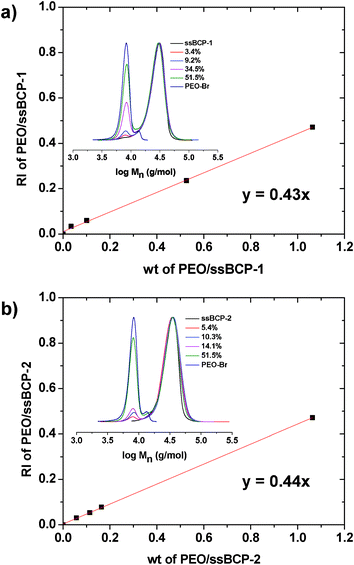 | ||
| Fig. 7 Calibration curve and GPC traces (inset) for various mixtures with different wt% of PEO–ss–iBuBr/ssBCP-1 (a) and ssBCP-2 (b). | ||
Further degradation of micellar aggregates with different morphologies was also investigated: spheres for ssBCP-1 and vesicles for ssBCP-2, both of which had similar sizes by DLS, when prepared by solvent evaporation method. In the presence of DTT, PEO coronas can be shed from PSt cores by the cleavage of disulfide linkages. From GPC results (Fig. S4 in the ESI†) with the constructed calibration curves, degradation was 56% for spheres, but ≈25% for vesicles, after 4 hours (Fig. 8). The lower degradation of vesicles, compared to spheres with similar diameters, is plausibly due to the location of disulfide linkages in both outer and inner shells and the limited access of DTT to disulfides in inner shells through hydrophobic shells. However, it further increased to 63% for ssBCP-1 and 52% for ssBCP-2, after 1 day. Such degradation resulted in destabilization of micellar aggregates, which was confirmed by DLS measurements (Fig. 9). These results suggest that degradation of polymers and aqueous aggregates by cleavage of disulfides in reducing environments can be modulated with structural and morphological variations.
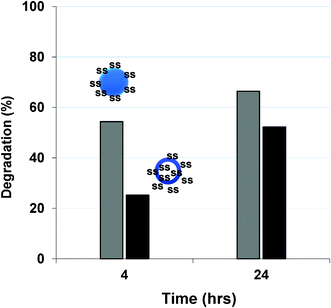 | ||
| Fig. 8 Thiol-responsive degradation of micellar aggregates in water in the presence of DTT, determined by GPC using calibration curves. Grey: ssBCP-1 and black: ssBCP-2. | ||
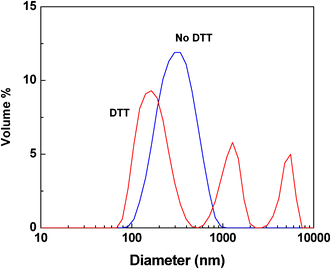 | ||
| Fig. 9 An example of particle size distribution of vesicles based on ssBCP-2 before and after the addition of DTT in water. | ||
Conclusions
We developed new thiol-responsive degradable ssBCPs consisting of PEO and PSt blocks with disulfides at block junctions by employing ATRP in the presence of newly-synthesized PEO–ss–iBuBr macroinitiators. GPC and 1H-NMR results suggest that the polymerizations proceeded in a living manner, yielding well-controlled PEO–ss–PSt with different PSt chain lengths and narrow molecular weight distributions. Through aqueous self-assembly, these block copolymers exhibited modulated morphologies of aggregates in aqueous solutions from spheres to vesicles and from small spheres to rod-like aggregates with different PSt chain lengths and aqueous micellization methods. Furthermore, with structural and morphological variation, thiol-responsive degradation of these micellar aggregates is controlled. These significant results of various morphologies exhibiting tunable degradation rates of aqueous micellar aggregates can be promising for bio-related applications in complex environments.Acknowledgements
Financial support from Concordia University, NSERC Canada, and CRC award are greatly appreciated. JKO is a Tier II Canada Research Chair in Nanobioscience as well as a member of Centre Québécois sur les Matériaux Fonctionnels (CQMF) funded by FQRNT as well as of Concordia Composite (CONCOM) Center at Concordia University. The authors thank Drs K. S. Sears and X. D. Liu at McGill University for training TEM facilities.References
- M. A. Hillmyer, Adv. Polym. Sci., 2005, 190, 137 CrossRef CAS.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647 CrossRef CAS.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and A. Winnik Mitchell, Science, 2007, 317, 644 CrossRef CAS.
- L. Zhang, K. Yu and A. Eisenberg, Science, 1996, 272, 1777 CAS.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707 CrossRef CAS.
- J. Hu, G. Liu and G. Nijkang, J. Am. Chem. Soc., 2008, 130, 3236 CrossRef CAS.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146 CrossRef CAS.
- C. J. F. Rijcken, O. Soga, W. E. Hennink and C. F. van Nostrum, J. Controlled Release, 2007, 120, 131 CrossRef CAS.
- Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849 CrossRef CAS.
- K. Loomis, K. McNeeley and R. V. Bellamkonda, Soft Matter, 2011, 7, 839 RSC.
- Q. Zhang, N. R. Ko and J. K. Oh, Chem. Commun., 2012, 48, 7542 RSC.
- J. Liu, Y. Pang, W. Huang, Z. Zhu, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 2407 CrossRef CAS.
- J. Liu, Y. Pang, W. Huang, X. Huang, L. Meng, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 1567 CrossRef CAS.
- T.-B. Ren, Y. Feng, Z.-H. Zhang, L. Li and Y.-Y. Li, Soft Matter, 2011, 7, 2329 RSC.
- H.-Y. Wen, H.-Q. Dong, W.-J. Xie, Y.-Y. Li, K. Wang, G. M. Pauletti and D.-L. Shi, Chem. Commun., 2011, 47, 3550 RSC.
- H. Sun, B. Guo, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2009, 30, 6358 CrossRef CAS.
- X.-J. Cai, H.-Q. Dong, W.-J. Xia, H.-Y. Wen, X.-Q. Li, J.-H. Yu, Y.-Y. Li and D.-L. Shi, J. Mater. Chem., 2011, 21, 14639 RSC.
- M. Guo and M. Jiang, Soft Matter, 2009, 5, 495 RSC.
- W.-F. Dong, A. Kishimura, Y. Anraku, S. Chuanoi and K. Kataoka, J. Am. Chem. Soc., 2009, 131, 3804 CrossRef CAS.
- M. Zhang, L. Yang, S. Yurt, M. J. Misner, J.-T. Chen, E. B. Coughlin, D. Venkataraman and T. P. Russell, Adv. Mater., 2007, 19, 1571 CrossRef CAS.
- H. Zhao, W. Gu, E. Sterner, T. P. Russell, E. B. Coughlin and P. Theato, Macromolecules, 2011, 44, 6433 CrossRef CAS.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830 CrossRef CAS.
- J. S. Katz, S. Zhong, B. G. Ricart, D. J. Pochan, D. A. Hammer and J. A. Burdick, J. Am. Chem. Soc., 2010, 132, 3654 CrossRef CAS.
- R. P. Brinkhuis, T. R. Visser, F. P. J. T. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 550 RSC.
- A. Klaikherd, S. Ghosh and S. Thayumanavan, Macromolecules, 2007, 40, 8518 CrossRef CAS.
- M. Kang and B. Moon, Macromolecules, 2009, 42, 455 CrossRef CAS.
- S. Yurt, U. K. Anyanwu, J. R. Scheintaub, E. B. Coughlin and D. Venkataraman, Macromolecules, 2006, 39, 1670 CrossRef CAS.
- B. Khorsand Sourkohi, A. Cunningham, Q. Zhang and J. K. Oh, Biomacromolecules, 2011, 3819 CrossRef CAS.
- J.-M. Schumers, J.-F. Gohy and C.-A. Fustin, Polym. Chem., 2010, 1, 161 RSC.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009 CrossRef CAS.
- C. Li, J. Madsen, S. P. Armes and A. L. Lewis, Angew. Chem., Int. Ed., 2006, 45, 3510 CrossRef CAS.
- S. Carelli, A. Ceriotti, A. Cabibbo, G. Fassina, M. Ruvo and R. Sitia, Science, 1997, 277, 1681 CrossRef CAS.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199 CrossRef CAS.
- W. Li, J. A. Yoon and K. Matyjaszewski, J. Am. Chem. Soc., 2010, 132, 7823 CrossRef CAS.
- W. Lv, S. Liu, W. Feng, J. Qi, G. Zhang, F. Zhang and X. Fan, Macromol. Rapid Commun., 2011, 32, 1101 CrossRef CAS.
- Z. Jia, L. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106 CrossRef CAS.
- D. J. Phillips and M. I. Gibson, Chem. Commun., 2012, 48, 1054 RSC.
- A. Nelson-Mendez, S. Aleksanian, M. Oh, H.-S. Lim and J. K. Oh, Soft Matter, 2011, 7, 7441 RSC.
- R. Zhu, Y. Wang and W. He, Eur. Polym. J., 2005, 41, 2088 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18 CrossRef CAS.
- K. Yu and A. Eisenberg, Macromolecules, 1996, 29, 6359 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra21209a |
| This journal is © The Royal Society of Chemistry 2012 |