Green epoxidation of 1-hexene: O2 utilizing cis-MoO2 Schiff base complex
M. A.
Katkar
a,
S. N.
Rao
*a and
H. D.
Juneja
b
aDepartment of Chemistry, Priyadarshini Institute of Engineering and Technology, Hingna Road, Nagpur, 440019, India. E-mail: sumitarao2000@rediffmail.com; Fax: 91-07104-236458; Tel: 91-07104-236463
bDepartment of Chemistry, Rashtrasant Tukadoji Maharaj Nagpur University, Amaravati Road, Nagpur, 440033, India
First published on 6th July 2012
Abstract
The green epoxidation of 1-hexene using [hydrazine-N-salicylidene-N′-salicyloyl-] cis-dioxomolybdenum(VI) and its zeolite-Y composite as catalyst in DMF in the presence of molecular oxygen as oxidant and in the temperature range 333–363 K is reported. The ‘green’ factor of olefin oxidation catalysis using the Mo(VI) dioxo complex is its ability to use atmospheric oxygen which is non-hazardous and inexpensive and produces no harmful by-products. The effects of the initial concentration of the substrate and Mo complex and the temperature in the range 333–363 K on the rate of reaction are examined. The olefin conversion into different products is monitored using gas chromatography. The formation of 1,2-epoxyhexane, 1-hexanal and 2-hexanone is observed under both homogeneous and heterogeneous catalytic conditions; however, 2-hexanone did not form during the reactions using the zeolite composite. 1-Hexene conversion was found to be nearly complete and the product distribution into 1,2-epoxyhexane, 1-hexanal and 2-hexanone being 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:6 under homogeneous conditions for 6 h; while the final product is 1-hexanal under heterogeneous conditions for 6 h. The rate of 1-hexene oxidation showed first order dependence with respect to the catalyst and substrate concentration and it was an endothermic reaction (activation energy, 10.68 Kcal mol−1). A tentative mechanism for the oxidation of 1-hexene is proposed and the observed product formation was reasoned to involve epoxide isomerisation through participation of adventitious water in the reaction mixture and the resulting carbonium ions.
:20:6 under homogeneous conditions for 6 h; while the final product is 1-hexanal under heterogeneous conditions for 6 h. The rate of 1-hexene oxidation showed first order dependence with respect to the catalyst and substrate concentration and it was an endothermic reaction (activation energy, 10.68 Kcal mol−1). A tentative mechanism for the oxidation of 1-hexene is proposed and the observed product formation was reasoned to involve epoxide isomerisation through participation of adventitious water in the reaction mixture and the resulting carbonium ions.
Introduction
In keeping with growing environmental concerns as well as with the basic tenets of chemistry for sustainable development, the chemical processing industry (CPI) is eagerly awaiting the development of ‘green’ chemical processes. This has prompted serious efforts to improve chemical synthesis and manufacturing methods, as well as the development of new synthetic methodologies that minimise or completely eliminate by-products (pollutants). Hence, more attention has been focused on the use of safer and green raw materials/reagents, through the proper design of clean processes and products.Transition metal catalysed oxidation reactions are important both in biological and industrial processes. Metalloenzyme reactions are essential for energy transformation and storage and for the biosynthesis and metabolism of bio molecules, in biological processes, whereas, in industrial processes, transition metal based catalysts are important in the production of various oxygenated products.1
Epoxides are versatile and useful intermediates in organic synthesis and as a consequence the epoxidation of alkenes is a field of both academic and industrial importance. The catalysis of the oxidation of olefins using transition metal complexes into a variety of products such as epoxides, carbonyl compounds, diols and oxidative cleavage products of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C has received much attention, primarily due to interest in selective and partial oxidation. Various catalytic systems have been reported to be useful for the oxidation of olefins, viz., Mn(III)(salen) complexes in a homogeneous medium2,3, Mn(III)(salen) complexes encapsulated in zeolite-X,4 Mn(III) bipyridine complex cations immobilised on Al-MCM-41,5 titanium silicate molecular sieve TS-1,6,7 heteropoly tungstate,8 Mo- or Ti-containing mesoporous MCM-49 and Ru(II) complexes containing pyridine and picoline ligands.10
C has received much attention, primarily due to interest in selective and partial oxidation. Various catalytic systems have been reported to be useful for the oxidation of olefins, viz., Mn(III)(salen) complexes in a homogeneous medium2,3, Mn(III)(salen) complexes encapsulated in zeolite-X,4 Mn(III) bipyridine complex cations immobilised on Al-MCM-41,5 titanium silicate molecular sieve TS-1,6,7 heteropoly tungstate,8 Mo- or Ti-containing mesoporous MCM-49 and Ru(II) complexes containing pyridine and picoline ligands.10
While epoxides of 1-hexene, cyclohexene, styrene, cyclooctene, etc, are the targeted products using these transition metal catalysts, a number of problems are encountered during the actual course of the reaction, viz. the lower yield and selectivity for epoxides and the isomerisation of epoxides as in the case with the Mn(III)(salen) complex and TS-1 molecular sieves,4,6 the unwanted oxidative cleavage of double bonds promoted by Ru and Os catalysts10 and the overall low olefin conversion with the zeolite encapsulated catalysts due to diffusion resistance.4 Therefore, the search for selective epoxidation catalysts of transition metal origin is continuing and new metal complexes in different ligand environments that may strongly influence the catalytic behaviour are being prepared and examined for their catalytic activity.
Mo(VI) complexes are well known as potent catalysts for the epoxidation of alkenes by hydroperoxides such as tert-butylhydroperoxide. Several authors20–23 have reported on the oxo-transfer activity of several dioxomolybdenum complexes in different Schiff base environments. We have been interested in five-coordinate octahedral cis-MoO2 complexes with non-symmetrical Schiff bases having mixed sets of donor atoms in which the sixth coordination site is occupied by a solvent molecule.11–19 The envisaged advantage of such complexes is that an olefin molecule may displace a solvent molecule in the sixth coordination site and undergo oxidation under suitable experimental conditions. The formal oxidation state of molybdenum cycles between 4+ and 6+ in reactions with olefin and oxidant (e.g., O2). Amongst all the complexes we made, [hydrazine-N-salicylidene-N′-salicyloyl-] cis-dioxomolybdenum(VI), formulated as cis-[MoO2(L)(solv)], has been found to show higher catalytic reactivity. We have reported the synthesis, characterization and crystal structure of the same including its catalytic activity.11
In this paper, we report the kinetics of the catalytic oxidation of 1-hexene in DMF using [hydrazine-N-salicylidene-N′-salicyloyl-] cis-dioxomolybdenum(VI) and its zeolite-Y composite as catalyst, in the presence of molecular oxygen as oxidant and in the temperature range 333–363 K. Zeolites with their cage like structures provide micro reactor cavities in which catalytic reactions occur with low energies of activation towards selective product formation. It is found that 1,2-epoxyhexane forms initially and undergoes isomerisation into 1-hexanal and 2-hexanone probably through the carbonium ion route. A tentative mechanism for 1-hexene oxidation is proposed and discussed.
Experimental
2.1 Materials
Ammonium molybdate, acetylacetonate, salicylaldehyde, salicylic acid hydrazide and 1-hexene were procured from Sisco Chem., Mumbai, India. Chromatography grade acetone and dimethyl formaldehyde were used. Partially dealuminated NaY zeolite (Si/Al = 4.18) was used. cis-MoO2(acac)2 was prepared and purified according to the reported method.24 The synthesis and characterisation of the salicylidene salicyloyl hydrazine Schiff base (L), the corresponding dioxomolybdenum complex [hydrazine-N-salicylidene-N′-salicyloyl-] cis-dioxomolybdenum(VI) and the encapsulation of this complex in zeolite-Y super cages have been reported elsewhere.11,12 The dioxomolybdenum complex [hydrazine-N-salicylidene-N′-salicyloyl-] cis-dioxomolybdenum(VI) is abbreviated as cis-MoO2(L)(H2O) in the remaining text for convenience, where L denotes [hydrazine-N-salicylidene-N′-salicyloyl-] or sal-SAH.2.2 Oxidation of 1-hexene
The oxidation of 1-hexene was carried out in a water-jacketed glass reactor fitted with a condenser, thermometer and sampling port. Water from a constant temperature water bath was re-circulated through the water-jacketed glass reactor to attain a desired temperature in the range 333–363 K. The complex (0.04 g, 0.1 mmol) was dissolved in DMF (10 ml) and when the desired temperature was attained, a DMF solution of 1-hexene (0.03 ml, 0.3 mmol) was added. Throughout the experiment that lasted for 6 h, O2 (99.99%) was bubbled while the reaction mixture was stirred magnetically and the liquid samples (0.1 ml) were collected at chosen time intervals for analysis. The progress of the reaction was monitored using gas chromatography (Autosystem ASXL, Shimadzu) using 4% Dexsil 400 on a Chromosorb column (FID, 250 °C; column temperature programmed between 60–180 °C, 10 °C min−1; injector temperature, 200 °C; N2 carrier gas, 10 mL min−1). The oxidation runs using the zeolite-Y encapsulated complex were also conducted in a similar manner except that 0.1 g of the zeolite-composite was used instead of the complex. The retention times of the solvent, 1-hexene, 1,2-epoxyhexane, 1-hexanal and 2-hexanone were confirmed by comparing them with their authentic samples.Results and discussion
3.1 Oxidation of 1-hexene using cis-[MoO2(sal-SAH)(H2O)]
The oxidation of 1-hexene in DMF using cis-[MoO2(sal-SAH)(H2O)] at 333 K in the presence of molecular oxygen was confirmed to yield 1,2-epoxyhexane, 1-hexanal and 2-hexanone. The stack chromatograms shown in Fig. 1 illustrate the GC peaks corresponding to 1-hexene, 1,2-epoxyhexane, 1-hexanal and 2-hexanone. DMF eluted at 3.41 min; however, only sections of the chromatograms displaying peaks for 1-hexene (RT = 0.49 min), 1,2-epoxyhexane (0.48), 1-hexanal (0.60–0.66) and 2-hexanone (0.88) are shown for clarity. The time course variation in the percentage product distribution during the catalyzed oxidation of 1-hexene is presented in Table 1. The percentage of product (%) is calculated as the peak area due to the product divided by the sum total of the peak areas of all the products multiplied by 100. It can be seen that 1-hexene gradually converts into 1,2-epoxyhexane which further undergoes transformation into 1-hexanal and 2-hexanone. 1-Hexene conversion was found to be nearly complete, the product distribution into 1,2-epoxyhexane, 1-hexanal and 2-hexanone being 1:20:6 under homogeneous conditions for 6 h; while the final product was 1-hexanal under heterogeneous conditions for 6 h.
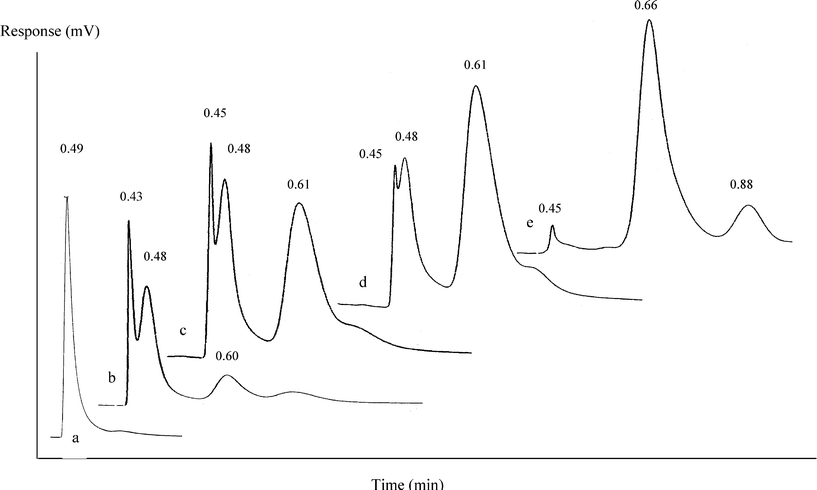 | ||
| Fig. 1 Stack gas chromatograms corresponding to 1-hexene; 1,2-epoxyhexane; 1-hexanal and 2-hexanone (a, 0 h; b, 1 h; c, 2 h; d, 3 h; e, 6 h). | ||
| Reaction time (h) | 1-Hexene (%) | 1,2-Epoxy hexane (%) | 1-Hexanal (%) | 2-Hexanone (%) |
|---|---|---|---|---|
| cis-MoO2(L)(solv) | ||||
| 1 | 33.88 | 46.49 | 19.63 | — |
| 2 | 10.31 | 31.43 | 58.26 | — |
| 3 | 7.46 | 28.26 | 64.28 | — |
| 6 | — | 3.77 | 74.14 | 22.09 |
| Complex–Zeolite composite | ||||
| 1 | 1.22 | 13.62 | 85.16 | — |
| 2 | — | 10.74 | 89.26 | — |
| 3 | — | 6.38 | 93.62 | — |
| 4 | — | — | 100.00 | — |
Subsequently, the effects of the substrate and catalyst concentrations and temperature on the initial rate of oxidation of 1-hexene using cis-[MoO2(sal-SAH)(H2O)] were determined.
3.2 Effect of substrate concentration
The substrate concentration was varied in the range 0.2 to 0.4 mmol at 353 K and 0.1 mmol catalyst concentrations. The time course variation in the fraction of 1-hexene converted at its different initial concentrations and the dependence of the initial rate of reaction on the initial concentration of 1-hexene are depicted in Fig. 2(a and b). From Fig. 2(a), it is found that 40–80% conversion takes place during the first hour at all the initial concentrations of 1-hexene; while it increases slowly there after until 6 h. The percentage conversion increased with the increase in 1-hexene concentration. The initial rates were deduced from concentration vs. time plots, considering the initial data points. The initial rate of reaction increased approximately proportionately with the increase in the initial concentration of 1-hexene indicating that the reaction follows pseudo-first order kinetics with 1-hexene concentration (Fig. 2(b)).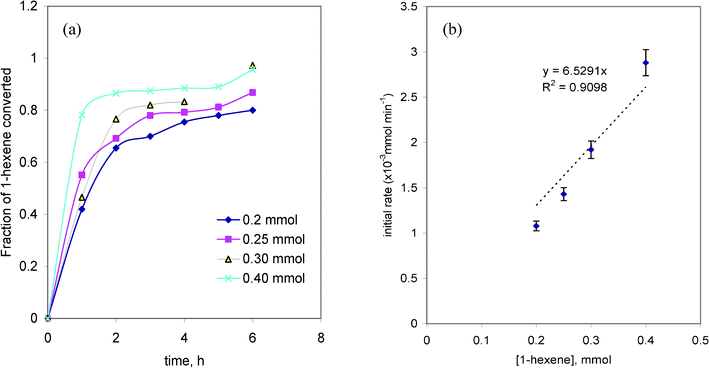 | ||
| Fig. 2 (a) The time course variation in the fraction of 1-hexene converted at its different initial concentrations and (b) the dependence of the initial rate of reaction on the initial concentration of 1-hexene. | ||
3.3 Effect of catalyst concentration
The effect of the catalyst concentration is evaluated by varying it from 0.05 to 0.2 mmol at 0.3 mmol 1-hexene concentration and at 353 K. The time course variation in the fraction of 1-hexene converted at different catalyst concentrations and the dependence of the initial rate of reaction on the catalyst concentration are depicted in Fig. 3(a and b). From Fig. 3(a), it is found that the conversion was <50% at 0.05 mmol catalyst concentration; however more than 80% conversion takes place in 5–6 h. The initial rates were deduced from concentration vs. time plots, considering the initial data points. The initial rate of reaction increased approximately proportionately with the increase in the initial concentration of the catalyst at the low concentration end, but the rate was practically independent at higher catalyst concentrations (Fig. 3(b)).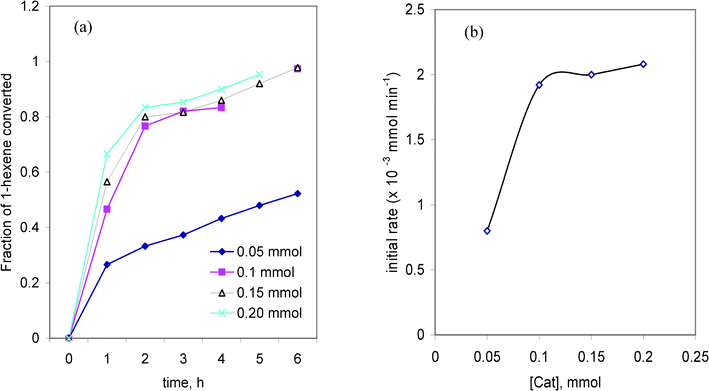 | ||
| Fig. 3 (a) The time course variation in the fraction of 1-hexene converted at different catalyst concentrations and (b) the dependence of the initial rate of reaction on the catalyst concentration. | ||
3.4 Effect of temperature
The oxidation of 1-hexene was studied by varying the temperature in the range 333–363 K at a fixed substrate concentration (0.3 mmol) and catalyst concentration (0.1 mmol). The fraction of 1-hexene converted as well as the initial rate of reaction increased gradually with the increase in temperature as depicted in Fig. 4(a). Since the rate of conversion of 1-hexene increased at higher temperatures, the reaction may be recognized as endothermic. A plot of −ln rate vs. 1/T (as shown in Fig. 4(b)) was used to determine the activation energy for the 1-hexene oxidation reaction. From the slope of the plot and using the relation −Ea = R × slope, the activation energy was deduced as 10.68 Kcal mol−1. This is also supported by the observation that percentage conversions were higher at higher temperatures.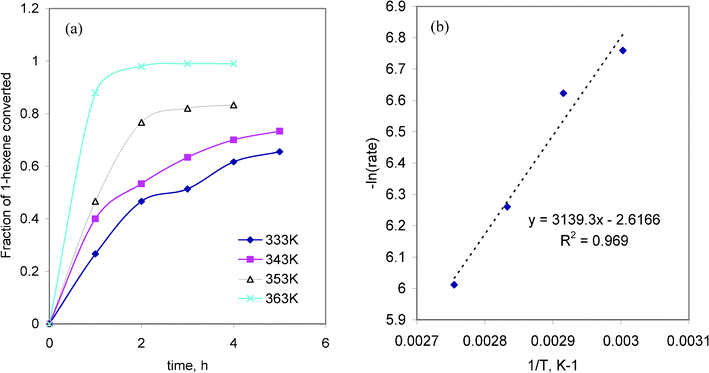 | ||
| Fig. 4 (a) The effect of temperature on the conversion of 1-hexene and (b) the Arrhenius plot of −ln rate vs. 1/T for the 1-hexene oxidation reaction. | ||
3.5 Oxidation of 1-hexene using [MoO2(sal-SAH)(H2O)]–NaY composite
In this experiment, 0.1 g MoO2(L)–NaY composite was reacted with 0.3 mmol 1-hexene in 10 ml DMF at 353 K and the reaction was continued for 6 h under oxygen bubbling. The corresponding time course variation in the product evolution and distribution are mentioned previously in Table 1. About 13.62% 1,2-epoxyhexane and 85.16% 1-hexanal were formed during the first hour leaving behind very low concentrations of 1-hexene in the reaction mixture. The epoxide gradually converted into aldehyde with time leading to 1-hexanal with 100% selectivity at 6 h.3.6 Rate constants (kobs)
The oberserved rate constant (kobs) for the oxidation of 1-hexene, under homogenous and heterogeneous conditions, at different concentrations of substrate and catalyst and at different temperatures are summarised in Table 2. The −logCt/Covs. time plots (not shown) were constructed using normalised concentration data, which were found to be linear with a satisfactory regression coefficient (>0.9). The slopes of these linear plots represented pseudo first order rate constants, kobs. The kobs for 0.20–0.4 mmol substrate concentrations were in the range 1.80–4.00 × 10−3 min−1; for 0.05–0.20 mmol catalyst concentrations kobs varied from 0.07–4.01 × 10−3 min−1. A five-fold increase in kobs was observed when the reaction temperature was increased to 363 K from 333 K. When the complex–zeolite composite was used as catalyst, the kobs was found to be 5.60 × 10−3 mmol min−1.| Serial No. | Experimental Variables | Concentration/Values | Rate constant (kobs × 10−3 min−1) |
|---|---|---|---|
| 1 | Substrate (mmol) | 0.2 | 1.80 |
| 0.25 | 2.00 | ||
| 0.3 | 4.00 | ||
| 0.4 | 3.60 | ||
| 2 | Catalyst (mmol) | 0.05 | 0.07 |
| 0.1 | 4.01 | ||
| 0.15 | 3.90 | ||
| 0.2 | 3.90 | ||
| 3 | T/K | 333 | 2.20 |
| 343 | 2.40 | ||
| 353 | 4.00 | ||
| 363 | 10.00 | ||
| 4 | Zeolite–Y complex composite | 1-hexene = 0.3 mmol | 5.60 |
| [Cat] = 0.1 g | |||
| T = 343 K |
3.7 Probable mechanism for 1-hexene oxidation
We have characterized the cis-[MoO2(sal-SAH)(H2O)]–NaY composite and reported it earlier.17 The Mo content of [MoO2(sal-SAH)]–NaY is 1.2 wt% which corresponds to about two Mo atoms per unit cell of zeolite-Y (i.e. one Mo atom per four super cages). Based on 1.2 wt% Mo loading, the turnover in 1-hexene conversion can be estimated as 242. Thus, an eight-fold increase in turnover was achieved by encapsulation of the complex in zeolite as compared to the corresponding homogeneous case with a turnover of 29 mol−1 of Mo in catalyst at 353 K. Similar observations were reported earlier.18,25,26The above results confirm the epoxidation of 1-hexene in DMF using the complex [MoO2(sal-SAH)H2O]. However, the epoxide is found to undergo isomerisation into 1-hexanal and 2-hexanone. Molybdenum complexes are well established catalysts for the epoxidation of olefins. The nature of the metal species responsible for the oxygen transfer has been extensively debated in the literature. In all cases, the active species is believed to be the Mo(VI) compound.27,28 It has been found that different complexes follow different mechanistic pathways. Although the selectivity of the Mo(VI) complexes in epoxidation reactions seemed to exclude the radical or homolytic pathways of oxidative reactions because they are believed to yield a mixture of products,29 the homolytic pathway could not be completely excluded due to the work of Kok and Skibida.30
A tentative scheme for the epoxidation reaction of 1-hexene to 1-hexanal is proposed (Scheme 1). It is proposed that the water molecule in 1 is displaced by a more powerful solvent molecule DMF. In a pre-equilibrium step, species 2 reacts with a molecule of 1-hexene to give an organometallic intermediate 3 followed by the [2+2] cycloaddition of the CC double bond to the oxo MoO bond leading to the formation of the molybdeno-oxetane intermediate 4. The metal oxetane type intermediate is frequently invoked for explaining epoxide formation catalysed by metal–oxo complexes.19,27,31 This transforms into 1,2-epoxyhexane and Mo(IV) complex 5. Complex 5 is oxidised to the MoO2(VI) complex by molecular O2. A similar mechanism was proposed earlier by us for the catalytic air oxidation of olefins using molybdenum dioxo complexes with dissymmetric tridentate O, N, S-donor Schiff base ligands derived from o-hydroxyacetophenone and S-benzyldithocarbazate or S-methyldithiocarabazate.19 Moreover, the favourable effect of temperature on the overall conversion of 1-hexene, found in the present study implies that the abstraction of the oxygen atom by the 1-hexene moiety via the molybdeno-oxetane intermediate to form 1,2- epoxyhexane is accelerated. This step can be endothermic with an activation energy of about 10.6 Kcal mol−1. The 1,2-epoxyhexane appears to undergo a proton catalysed isomerisation as shown in Scheme 1, to form two types of carbonium ions, A and B; A leading to 1-hexanal and B leading to 2-hexanone. Although, we have not deliberately added any acid to the reaction mixture, we predict that the carbonium intermediates may have formed with the aid of adventitious water in the reaction mixture on the basis of the observed products. Further, from the observation that only 1-hexanal and not 2-hexanone formed when the zeolite composite was used indicates that the allylic carbonium ion intermediate predominates in this case. This view is further strengthened by the observation that 1-hexene quickly converts into 85% 1-hexanal through the rapid isomerisation of 1,2-epoxyhexane. The isomerisation reaction is probably facilitated by the surface functionality of the zeolite. The selective isomerisation of epoxides catalysed by various heterogeneous catalysts has been reported elsewhere.32 Van Grieken et al.33 reported that Lewis and Brønsted acids and strong bases catalyse epoxide rearrangement homogeneously; and acid catalyzed rearrangement generally leads to ketones and aldehydes. Although water is assumed to act as a nucleophile,33 in view of the amphoteric nature of water and on the basis of the observed products, we propose that water participates as an electrophile (H3O+) under the experimental conditions in the present study and leads to the formation of 1-hexanal and 2-hexanone. The products formed by the rearrangement of epoxides are useful intermediates in organic synthesis.
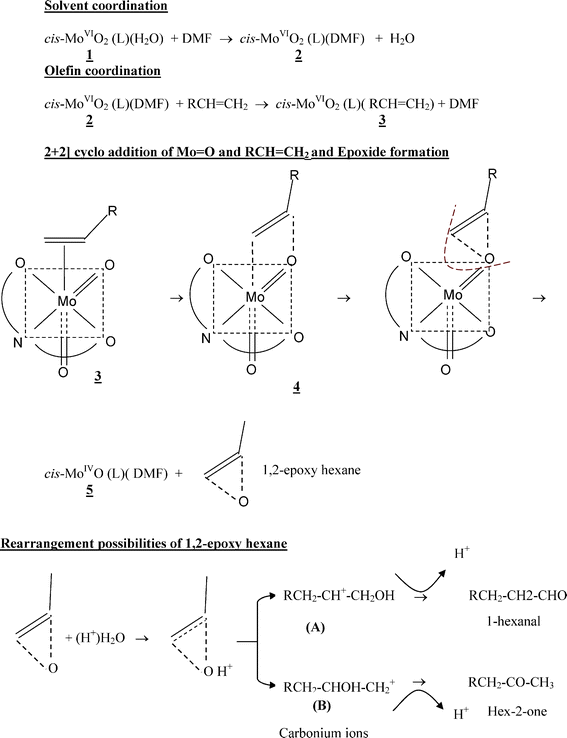 | ||
| Scheme 1 | ||
Conclusion
Both cis-[MoO2(sal-SAH)(H2O)] and its zeolite composite are useful catalysts for the oxidation of 1-hexene. The products of oxidation are 1,2-epoxyhexane, 1-hexanal and 2-hexanone which form in the ratio 1:20:6 under homogeneous conditions for 6 h. On the other hand, the final product is 1-hexanal under heterogeneous conditions for 6 h. The 1-hexene oxidation follows first order dependence with respect to the substrate and catalyst concentrations. The turn over number is eight-fold greater with the zeolite encapsulated complex. The reaction products, 1-hexanal and 2-hexanone, form through the rapid isomerisation of 1,2-epoxyhexane.
The ‘green’ factor of olefin oxidation catalysis using the Mo(VI) dioxo complex is its ability to use atmospheric oxygen which is non-hazardous and inexpensive and produces no harmful by-products. However, we experienced that this critically depends on the interplay of the coordination geometry around the Mo atom, the structural factors such as the zeolite type and the size of the channels and cages and chemical factors such as the Si–Al ratio and surface hydration. Spectroscopic techniques capable of selectively probing the active sites can provide detailed mechanistic insight and assist in the design and development of MoO2 based selective epoxidation catalysts.
Acknowledgements
MAK & SNR wish to thank Dr C. G. Dethe, Principal, PIET for support and Dr N. N. Rao (Senior Scientist, NEERI, Nagpur) for his useful discussions.References
- J. P. Caradonna, in Encyclopedia of Inorganic Chemistry Vol. 6, ed. R. B. King, John Wiley and Sons, Chichester, 1994, p. 2866 Search PubMed.
- E. N. Jacobsen, L. Deng, Y. Furukawa and L. E. Martinez, Enantioselective catalytic epoxidation of cinnamate esters, Tetrahedron, 1994, 50, 4323–4334 CrossRef CAS.
- T. Katasuki, Catalytic asymmetric oxidations using optically active (salen) manganese(III) complexes as catalysts Coord, Chem. Rev., 1995, 140, 189–214 Search PubMed.
- S. P. Varkey and C. R. Jacob, Selective oxidation of styrene using zeolite encapsulated manganese complexes, Indian J. Chem., 1998, 37A, 407–412 CAS.
- S. S. Kim, W. Zhang and T. J. Pinnavaia, Catalytic oxidation of styrene by manganese(II) bipyridine complex cations immobilized in mesoporous Al-MCM-41, Catal. Lett., 1997, 43, 149–154 CrossRef CAS.
- S. B. Kumar, S. P. Mirajkar, G. C. G. Pias, P. Kumar and R. Kumar, Epoxidation of styrene over a titanium silicate molecular sieve TS1 using dilute H2O2 as oxidizing agent, J. Catal., 1995, 156, 163–166 CrossRef CAS.
- J. S. Reddy, U. R. Khire, P. Ratnaswamy and R. B. Mitra, Cleavage of the carbon–carbon double bond over zeolites using hydrogen peroxide, J. Chem. Soc., Chem. Commun., 1992, 1234–1235 RSC.
- N. Mizuno, T. Hirose and R. Iwamoto, Highly selective epoxidation of olefins on mono-transition-metal-substituted Keggin-type heteropolytungstates by molecular oxygen in the presence of aldehyde, Stud. Surf. Sci. Catal., 1994, 82, 593–600 CrossRef CAS.
- R. K. Rana and B. Viswnathan, Oxidation of styrene on Mo-MCM 41, Bull. Catal. Soc. Ind., 1999, 9(1&2), 11–15 Search PubMed.
- A. S. Khanmani and S. Vancheesan, Oxidation of cyclohexene and styrene catalysed by ruthenium(II) complexes under homogeneous conditions, Stud. Surf. Sci. Catal., 1998, 113, 285–292 CrossRef.
- S. N. Rao, K. N. Munshi, N. N. Rao, M. M. Bhadbhade and E. Suresh, Synthesis, spectral and X-ray structural characterization of cis-[MoO2(L)(solv)] (L = salicylidene salicyloyl hydrazine) and its use as catalytic oxidant, Polyhedron, 1999, 18, 2491–2497 CrossRef CAS.
- S. N. Rao, K. N. Munshi and N. N. Rao, Catalytic air oxidation of cyclooctene using cis-[MoO2(sal-SH)(DMF)] (sal-SH = salicylidene salicyloyl hydrazinato-) and its zeolite-Y encapsulated composite, J. Mol. Catal. A: Chem., 1999, 145, 203–210 CrossRef CAS.
- S. N. Rao, M. N. Jaiswal, D. D. Mishra, R. C. Maurya and N. N. Rao, Synthesis and characterization of cis-dioxomolybdenum(VI) Schiff base complex derived from 1-phenyl-3-methyl-4-benzoyl-5-pyrazolone, Polyhedron, 1993, 12, 2045–2050 CrossRef.
- N. S. Rao, D. D. Mishra, R.C. Maurya and N. N. Rao, Synthesis and characterization of Ru(II) Schiff base complexes derived from sulpha drugs, Synth. React. Inorg. Met.-Org. Chem., 1995, 25, 437–449 CrossRef CAS.
- S. N. Rao, D. D. Mishra, R.C. Maurya and N. N. Rao, Novel 8-coordinate cis-MoO(VI) complexes with some 4-aminoantipyrine Schiff base derivatives, Bull. Chem. Soc. Jpn., 1995, 68, 1589–1592 CrossRef.
- R. C. Maurya, D. D. Mishra, S. N. Rao, R. Verma and N. N. Rao, Synthesis and characterization of cis-dioxomolybdenum Schiff base complexes derived from 2-benzoyl pyridine, Indian J. Chem., 1997, 36A, 599–601 CAS.
- N. Kathale, N. Sumita Rao, N. N. Rao and K. N. Munshi, Synthesis and characterisation of some dioxomolybdenum(VI) complexes with O, N, S-donor Schiff bases derived from S-methyldithiocarbazate, S-benzyldithiocarbazate, 2-aminoethanethiol and substituted salicylaldehydes, Synth. React. Inorg. Met.-Org. Chem., 2001, 31, 859–871 CrossRef CAS.
- S. N. Rao, K. N. Munshi and N. N. Rao, Catalytic oxidation of styrene using cis-[MoO2(L)(solv)] (L = salicylidene salicyloyl hydrazine) and its zeolite composite as catalysts in the presence of molecular oxygen, J. Mol. Catal. A: Chem., 2000, 156, 205–211 CrossRef CAS.
- S. N. Rao, N. Kathale, N. N. Rao and K. N. Munshi, Catalytic air oxidation of olefins using molybdenum dioxo complexes with dissymmetric tridentate O, N, S-donor Schiff base ligands derived from o-hydroxyacetophenone and S-benzyldithiocarbazate or S-methyldithiocarbazate, Inorg. Chim. Acta, 2007, 360, 4010–4016 CrossRef CAS.
- P. K. Bhattacharya, Homogeneous catalysis of epoxidation of olefins by mono- and binuclear Schiff base complexes, J. Chem. Sci., 2008, 102, 247–250 Search PubMed.
- R. Debel, A. Buchholz and W. Plass, Structural and catalytic aspects of dioxomolybdenum(VI) complexes with ω-hydroxy functionalized N-salicylidene hydrazides, Z. Anorg. Allg. Chem., 2008, 634, 2291–2298 CrossRef CAS.
- M. Bagherzadeh, R. Latifi, L. Tahasini, V. Amani and A. E. L. Keith Woo, Synthesis, characterization and crystal structure of a dioxomolybdenum(VI) complex with a N,O type bidentate Schiff base ligand as a catalyst for homogeneous oxidation of olefins, Polyhedron, 2009, 28, 2517–2521 CrossRef CAS.
- M. Salavati -Niasari and M. Bazarganipour, Synthesis, characterization and catalytic epoxidation of cyclohexene using dimeric cis-dioxomolybdenum(VI) bis-bidentate Schiff base complexes supported on single wall carbon nanotubes, Transition Met. Chem., 2008, 33, 751–757 CrossRef CAS.
- G. J. J. Chen, J. W. McDonald and W. E. Newton, Synthesis of molybdenum(IV) and molybdenum(V) complexes using oxo abstraction by phosphines. Mechanistic implications, Inorg. Chem., 1976, 15, 2612–2615 CrossRef CAS.
- C. Jin, W. Fan, Y. Jia, B. Fan, J. Maa and R. Li, Encapsulation of transition metal tetrahydro-Schiff base complexes in zeolite Y and their catalytic properties for the oxidation of cycloalkanes, J. Mol. Catal. A: Chem., 2006, 249, 23–30 CrossRef CAS.
- M. R. Maurya, A. K. Chandrakar and S. I. Chand, Zeolite-Y encapsulated metal complexes of oxovanadium(VI), copper(II) and nickel(II) as catalyst for the oxidation of styrene, cyclohexane and methyl phenyl sulfide, J. Mol. Catal. A: Chem., 2007, 274, 192–201 CrossRef CAS.
- K. A. Jorgensen, Transition-metal-catalysed epoxidations, Chem. Rev., 1989, 89, 431–458 CrossRef.
- M. K. Trost and R. G. Bergman, Cp*MoO2Cl-catalyzed epoxidation of olefins by alkyl hydroperoxides, Organometallics, 1991, 10, 1172–1178 CrossRef CAS.
- C.-C. Su, J. W. Reed and E. S. Gould, Metal ion catalysis of oxygen-transfer reactions. II. Vanadium and molybdenum chelates as catalysts in the epoxidation of cycloalkenes, Inorg. Chem., 1973, 12, 337–343 CrossRef CAS.
- P. Skibida and P. Kok, Mechanism of the catalytic epoxidation of olefins by hydroperoxides in the presence of complex compounds of Mo(VI), Russ. Chem. Bull., 1974, 23(12), 2599–2604 CrossRef.
- M. Santos, PhD thesis, Technische Universität München (Munich), 2000.
- H. Raptis and M. Garcia, Stratakis, Selective isomerization of epoxides to allylic alcohols catalyzed by TiO2-supported gold nanoparticles, Angew. Chem. Int. Ed., 2009, 48, 3133–3136 CrossRef.
- R. van Grieken, D. P. Serrano, J. A. Melero and A. Garcia, Effect of the solvent in the liquid phase rearrangement of 1,2-epoxyoctane over Al-MCM-41 and Al-TS-1 catalysts, J. Mol. Catal. A: Chem., 2004, 222, 167–174 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |