Formation of graphene oxide gel via the π-stacked supramolecular self-assembly
Wei
Ai
ab,
Zhu-Zhu
Du
a,
Ju-Qing
Liu
a,
Fei
Zhao
a,
Ming-Dong
Yi
a,
Ling-Hai
Xie
*a,
Nai-En
Shi
a,
Yan-Wen
Ma
a,
Yan
Qian
a,
Qu-Li
Fan
a,
Ting
Yu
*b and
Wei
Huang
*a
aKey Laboratory for Organic Electronics & Information Displays (KLOEID) and Institute of Advanced Materials (IAM), Nanjing University of Posts and Telecommunications, 9 Wenyuan Road, Nanjing, 210046, P. R. China. E-mail: iamlhxie@njupt.edu.cn; wei-huang@njupt.edu.cn; Fax: +86 25 8349 2333; Tel: +86 25 8349 2333
bDivision of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, 637371, Singapore. E-mail: yuting@ntu.edu.sg; Fax: +65 6316 7899; Tel: +65 6316 7899
First published on 4th October 2012
Abstract
Graphene oxide gel (GOG) possesses intrinsic three-dimensional (3D) networking architecture with large surface area and high porosity. Here, we report a novel method of fabrication of GOG by self-assembling a ferrocene-decorated graphene oxide sheets (GOS) at room temperature. Our systematic investigations reveal that Fc plays a critical role in the formation of such unique 3D architecture, as it functions as an effective interlayer cross-linker through the π–π interaction. The morphology, crystal structure, chemical bonding, porosity and thermal stability of the as-prepared GOG have been studied. This work successfully provides a facile and efficient way to form GOG and will extend the potentials of GOG as a promising electro-active material in carbon-based electronics or catalytic reactors.
Introduction
Two-dimensional (2D) graphene and its derivatives have been regarded as some of the most promising materials because of their outstanding mechanical, thermal and electrical properties.1–3 Previous work has demonstrated that ‘paper-like’ materials, thin films and free-standing membranes of graphene and chemically modified graphene can serve as electrodes for flexible electronics, as well as energy storage/conversion materials.4–8 With respect to 2D monolayers, 3D graphene-based materials exhibit several advantages, such as large surface area, high porosity and different physical and electronic properties. They offer more potential in a wide range of applications, such as catalysis, drug delivery, tissue engineering, energy storage, super-capacitors, sensors and actuators.9–12 To meet the demands of such applications, the main challenge is to effectively assemble the 2D graphene-based sheets into a 3D networking architecture like gels, especially with controllable pore structure or interlayer spacing.13 It has been reported that 3D graphene hydrogels can be formed through the π–π interactions between the sheets or by introducing a linker of noble-metal nanocrystals or a divalent ion after the hydrothermal process.14–16 Recently, graphene oxide hydrogel has also been achieved by adjusting the supramolecular interactions of graphene oxide sheets (GOS), such as hydrogen bonding or electrostatic interaction, through the introduction of a hydrophilic polymer or metal ion as the cross-linker.17–20 However, there is still no report of the self-assembly of 2D GOS into a 3D graphene oxide gel (GOG) by employing a hydrophobic interlayer cross-linker via supramolecular π–π interaction at room temperature.Ferrocene (Fc) is a typical sandwich-like metallocene, consisting of two cyclopentadienyl rings bound to the opposite sides of a central iron atom.21,22 Therefore, it is expected that by using Fc as a two-functional linker, the supramolecular interactions of GOS could be significantly adjusted, facilitating the formation of GOG. Here, we report the self-assembly of 2D GOS into 3D GOG at room temperature by using hydrophobic Fc as the interlayer cross-linker via the π–π interactions between the aromatic sheets of GO and cyclopentadienyl rings of Fc. The as-prepared 3D network structures of GOG have been characterized by scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), Raman spectroscopy and thermogravimetric analysis (TGA). The formation mechanism of GOG is proposed and the pore structure of GOG is investigated by the Brunauer–Emmett–Teller (BET) method.
Experimental
Graphite powder (325 mesh) was purchased from Baichuan Graphite Co., Ltd. (Qingdao, China). Sulfuric acid (H2SO4), potassium permanganate (KMnO4), hydrogen peroxide (H2O2) and Fc were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).Preparation of graphite oxide
GO was prepared by a modified Hummers' method.16 In a typical procedure, graphite (1.0 g) was added into 50 ml of concentrated H2SO4 (98%) under stirring in an ice bath. Then, 6.0 g of KMnO4 was added slowly to the suspension to prevent a rapid rise in temperature. The mixture was then stirred at 30 °C for 1 h. Subsequently, 80 ml of deionized water was added dropwise under vigorous stirring with the temperature increasing to 90 °C, and this was further stirred for 30 min. Afterwards, 200 ml of deionized water and 6 ml of H2O2 (30%) was added with the color of the solution turning from dark brown to yellow. The diluted suspension was stirred for an additional 10 min, then centrifuged and washed with deionized water repeatedly, and finally dried under vacuum condition.Preparation of GOG
100 mg GO was suspended in a 2.5 ml:2.5 ml mixture of ethanol and deionized water to form a homogeneous suspension (20 mg ml−1) by sonication for 1 h, after which 5 ml of Fc in ethanol solution (5 mg ml−1) was added quickly and shaken violently. The mixture was kept at rest for 30 min and GOG was formed. The ethanol was removed by exchanging with water and then freeze-dried in a freeze drying machine. It was noticed that GOG could also be formed with the GO suspension of the concentration of 10 mg ml−1. However, the as-prepared GOG will be broken during the removal of ethanol by exchanging with water, due to its weak mechanical strength.Characterization
Scanning electron microscope (SEM) images were taken by a S-3400N II scanning electron microscope. X-ray photoelectron spectroscopy (XPS) analysis was carried out on a PHI5000 Versa Probe X-ray photoelectron spectrometer. The crystal structure of the samples were examined by a Bruker D/MAX 2500 X-ray diffractometer (XRD) with Cu-K radiation (λ = 1.54056 Å). Raman spectra were collected using a WITEC CRM200 Raman system with 532 nm excitation laser. The Brunauer–Emmett–Teller (BET) method was used to determine the surface area by measuring the adsorption of N2 using an ASAP2020 volumetric adsorption analyzer (Micromeritics, USA). TGA was recorded by a Shimadzu DTG-60H under air condition (heating rate: 5 °C min−1).Results and discussion
GOG was prepared through a self-assembly process, as schematically illustrated in Fig. 1. The Fc firstly anchors on the GOS through the π–π interactions between the aromatic carbon system of GOS and the cyclopentadienyl rings of Fc. When the amount of the anchored Fc is sufficiently high and the induced π–π interaction is strong enough, the Fc-decorated GOS start linking with each other and eventually GOG is formed. | ||
| Fig. 1 The proposed mechanism for the formation of GOG. The major driving force for the formation of GOG is the π–π interactions between the GOS and the cyclopentadienyl rings of Fc. | ||
Fig. 2 shows the corresponding optical graphs of the fabrication of GOG. As normal, the high quality and concentrated GO suspension is stable and a brown–black color (Fig. 2a). The Fc in ethanol appears as a transparent yellow suspension (Fig. 2b). Fig. 2c displays the optical graph of GOG after adding the Fc into the GO suspension for 30 min. After removing the ethanol by exchanging with water, GOG was freeze-dried and the xerogel with 3D structure was obtained.
 | ||
| Fig. 2 Photographs of the fabrication process of GOG. 20 mg ml−1 GO suspension (a), 5 mg ml−1 Fc suspension (b), GOG after GO mixed with Fc (c). The formation of GOG was confirmed by inversing. | ||
The microscopic morphology of the freeze-dried GO and GOG was investigated by SEM, as shown in Fig. 3. The freeze-dried GO has a porous and disordered network owing to a force balance among the GOS and the random aggregation (Fig. 3a,b).17–20 After adding the Fc as a two-functional linker with graphene sheets, GOG with a 3D porous structure could be formed. Fig. 3c,d illustrate that GOG presents an interconnected 3D porous network assembly of GOS. Furthermore, no isolated Fc clusters or particles were observed in the freeze-dried GOG, indicating that the Fc has effectively and uniformly anchored on the surfaces of GOS, which eventually connected to each other and formed the 3D GOG.
 | ||
| Fig. 3 SEM images of the freeze-dried GO (a, b) and GOG (c, d) with the concentration of Fc suspension at 5 mg ml−1. | ||
In order to probe the components of the synthesized GOG, the GO samples and GOG were analyzed by XPS (see Fig. 4). The binding energy of 284.6, 286.7, 288.0 and 289.0 eV in the C 1s XPS spectrum of the GO correspond to the C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O, CO and O–CO bonds, respectively. The C1s/O1s ratio is 2.00. Differently, for GOG, the binding energy of the C–C/CC peak is downshifted to 284.5 eV, which suggests the existence of the π–π interaction between the GOS and Fc,23 and that is also clearly reflected in its Raman spectrum (see Fig. 5). The intensity of the C–C/CC groups of GOG is much stronger than that of GO, which could be attributed to the C–C/CC groups from the cyclopentadienyl rings of Fc. This also explains the increase of the C1s/O1s ratio of GOG to 2.54. Compared with GO, the additional two peaks in the XPS spectrum of GOG (Fig. 4c) at 708.3 eV and 721.1 eV are attributed to the Fe 2p3/2 and Fe 2p1/2 of Fe(II) from the Fc, respectively, which indicates that the chemical structure of Fc remains unchanged after the formation of GOG.24 Thus, the self-assembly of GOG reported in this work is a physical adsorption process.
C, C–O, CO and O–CO bonds, respectively. The C1s/O1s ratio is 2.00. Differently, for GOG, the binding energy of the C–C/CC peak is downshifted to 284.5 eV, which suggests the existence of the π–π interaction between the GOS and Fc,23 and that is also clearly reflected in its Raman spectrum (see Fig. 5). The intensity of the C–C/CC groups of GOG is much stronger than that of GO, which could be attributed to the C–C/CC groups from the cyclopentadienyl rings of Fc. This also explains the increase of the C1s/O1s ratio of GOG to 2.54. Compared with GO, the additional two peaks in the XPS spectrum of GOG (Fig. 4c) at 708.3 eV and 721.1 eV are attributed to the Fe 2p3/2 and Fe 2p1/2 of Fe(II) from the Fc, respectively, which indicates that the chemical structure of Fc remains unchanged after the formation of GOG.24 Thus, the self-assembly of GOG reported in this work is a physical adsorption process.
 | ||
| Fig. 4 The C 1s XPS spectra of GO (a), GOG (b) and the Fe 2p XPS spectra (c) of GOG with the concentration of Fc suspension at 5 mg ml−1. | ||
 | ||
| Fig. 5 Raman spectra of GO and GOG with the concentration of Fc suspension at 5 mg ml−1. | ||
The atomic concentration of GO and GOG with the concentration of Fc suspension at 5 mg ml−1 from the XPS data are shown in Table 1. Every Fe atom in GOG corresponds to one Fc molecule and 10 carbon atoms from Fc. From the atomic ratio of C to Fe determined by XPS in GOG, we can calculate the distribution of Fc in GOG, that is: NC = (70.98 − 1.02 × 10)/1.02 =59.59 where Nc represents the number of carbon atoms in GO. The result means that ∼60 atoms in GO share one Fc cross-linker.
| Sample | C1s(%) | C1s(%) | Fe2p(%) |
|---|---|---|---|
| GO | 66.63 | 33.37 | 0 |
| GOG | 70.98 | 28 | 1.02 |
Raman spectroscopy was performed to study the structural and electronic characterization of GO and GOG, and the coupling between the Fc and GOS. As can be seen in Fig. 5, the typical G band of GO is at 1603 cm−1, with a disordered-induced D band at 1356 cm−1. While in the Raman spectrum of GOG, the D and G band shift to 1352 and 1588 cm−1, respectively. The obvious redshift of the G band for GOG is solid evidence of the strong coupling and charge transfer between the Fc and GOS.25,26
The crystal structure of the Fc-linked GOG was studied by XRD. As shown in Fig. 6, the strong characteristic 2θ peak for GO (Fig. 6a) appears at 9.98°, corresponding to a layer-to-layer distance of 8.85 Å. After adding the Fc, the XRD peak of GOG shows a shift towards a smaller angle. Such a downshift becomes more remarkable by increasing the concentration of Fc suspension. Fig. 6b–d shows that the interlayer distances of GOGs could be increased to 9.02 Å, 9.33 Å and 9.57 Å when the concentrations of Fc suspension were controlled to be 3, 4, and 5 mg ml−1, respectively. It's obvious that the interlayer spacing of GOG strongly depends on the concentration of Fc suspension. This offers us an effective way to control fairly well the interlayer distance and the porosity of GOG.
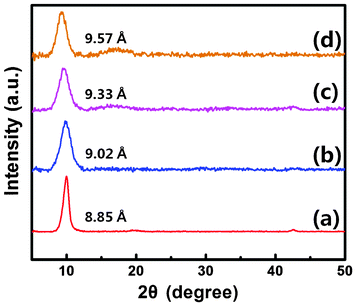 | ||
| Fig. 6 X-ray diffraction of GO (a) and GOGs (b–d) with the concentration of Fc suspension at 3, 4, 5 mg ml−1, respectively. | ||
To prove our hypothesis that the Fc could effectively trigger the formation of gel as the interlayer cross-linker through π–π interaction, we studied the effect of the Fc concentration on the dynamic formation process of GOG. As shown in Fig. 7, the duration needed to form GOG decreases with increasing concentrations of Fc and become stable when the concentration of Fc reaches a certain level. The major driving force for the formation of GOG is the π–π interactions between the GOS and the cyclopentadienyl rings of Fc. So, if the content of Fc is low in the suspension, that will result in a weak π–π interaction and it will need more time to link the GOS together. The higher content of the Fc is, the stronger the π–π interactions will be and the quicker the formation of GOG can be achieved. However, when the concentration of the GOS was low, i.e. less than 5 mg ml−1, it would be difficult for the Fc to link them together due to the weak mechanical strength of GOS skeleton. Consequently, only some flocculus can be achieved.
 | ||
| Fig. 7 The time required to form GOG as a function of the concentrations of Fc suspensions. | ||
Isolated graphene sheets have a theoretical specific surface area of 2600 m2 g−1. However, the GO powder usually has a much lower specific surface area than the theoretical value of an individual graphene sheet due to the agglomeration among the GOS.27 The N2 adsorption–desorption isotherms of freeze-dried GO and GOG in this case are shown in Fig. 8a. The isotherm for GO is straight, while that for GOG is type IV belonging to mesoporous structures. BET surface areas of GO and GOG are 76 m2 g−1 and 165 m2 g−1, respectively. In comparison with GO, GOG possesses an enhanced surface area because the GOS existing in the gel were twisted and curved by the connection of Fc molecules. The pore size distribution of GOG calculated from the desorption branch by the Barrett–Joyner–Halenda (BJH) method is given in Fig. 8b. The average pore size of GOG is 6.61 nm and the total pore volume is 0.48 cm3 g−1, indicating its potential applications as an electro-active material in carbon-based electronics or catalytic reactors.
 | ||
| Fig. 8 (a) Nitrogen adsorption–desorption isotherms at 77 K for powdery GO and GOG with the concentration of Fc suspension at 5 mg ml−1. (b) A pore-size distribution plot of GOG. | ||
Fig. 9 shows the thermogravimetric curves of GO and GOG. The mass loss for GO is about 18% with increasing the temperature to 276 °C, which could be attributed to the evaporation of trapped water and the removal of the functional groups from the GO.13 For GOG, however, there is no significant mass loss till about 312 °C, and only 9% mass loss occurs at that temperature. Compared to GO, the mass loss rate with temperature for GOG is significantly lower and more residues are observed, which indicates that the thermal stability of GOG is significantly improved.
 | ||
| Fig. 9 Thermogravimetric curves of GO and GOG with the concentration of Fc suspension at 5 mg ml−1 under nitrogen atmosphere. | ||
Conclusion
In summary, we have successfully fabricated a 3D networking GOG by using Fc as an interlayer cross-linker. This method provides a convenient and environmental friendly way to prepare GOG with a porous structure. In consideration of Fc being capable of engaging in electron transfer processes, GOG is expected to be interesting as an electro-active material in carbon-based electronics or catalytic reactors.Acknowledgements
This work was supported by the “973” project (No. 2009CB930600, No. 2012CB933301) NNSFC (Grants No. 21274064, No 21144004, No. 20974046, No. 21101095, No. 21003076, No. 50428303), The Program for New Century Excellent Talents in University (NCET-11-0992), the Ministry of Education of China (No. IRT1148), the NSF of Jiangsu Province (Grants No. SBK201122680, No. 11KJB510017, No. BK2008053, No. 11KJB510017 and No. BZ2010043) and NUPT (No. NY210030 and NY211022). The authors acknowledge Prof. Hui Xu and Tai-Xing Tan from the Key Laboratory of Functional Inorganic Material Chemistry (Heilongjiang University) and the Ministry of Education for help with the XRD characterization. L. H. Xie thanks the Jiangsu Overseas Research & Training Program for University Prominent Young & Middle-aged Teachers and Presidents.References
- C. X. Guo, M. Wang, T. Chen, X. W. Lou and C. M. Li, Adv. Energy Mater., 2011, 1, 736–741 CrossRef CAS.
- C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014–3017 CrossRef CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS.
- D. Li and R. B. Kaner, Science, 2008, 320, 1170–1171 CrossRef CAS.
- J. Q. Liu, Z. Y. Yin, X. H. Cao, F. Zhao, A. P. Lin, L. H. Xie, Q. L. Fan, F. Boey, H. Zhang and W. Huang, ACS Nano, 2010, 4, 3987–3992 CrossRef CAS.
- C. X. Guo, G. H. Guai and C. M. Li, Adv. Energy Mater., 2011, 1, 448–452 CrossRef CAS.
- C. M. Chen, Q. H. Yang, Y. G. Yang, W. Lv, Y. F. Wen, P. X. Hou, M. Z. Wang and H. M. Cheng, Adv. Mater., 2009, 21, 3541–3541 CAS.
- W. W. Zhou, J. X. Zhu, C. W. Cheng, J. P. Liu, H. P. Yang, C. X. Cong, C. Guan, X. T. Jia, H. J. Fan, Q. Y. Yan, C. M. Li and T. Yu, Energy Environ. Sci., 2011, 4, 4954–4961 CAS.
- C. X. Guo, X. T. Zheng, Z. S. Lu, X. W. Lou and C. M. Li, Adv. Mater., 2010, 22, 5164–5167 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2009, 48, 1525–1525 CrossRef CAS.
- P. Gupta, K. Vermani and S. Garg, Drug Discovery Today, 2002, 7, 569–579 CrossRef CAS.
- S. J. Hollister, Nat. Mater., 2005, 4, 518–524 CrossRef CAS.
- J. W. Burress, S. Gadipelli, J. Ford, J. M. Simmons, W. Zhou and T. Yildirim, Angew. Chem., Int. Ed., 2010, 49, 8902–8904 CrossRef CAS.
- Y. X. Xu, K. X. Sheng, C. Li and G. Q. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS.
- Z. H. Tang, S. L. Shen, J. Zhuang and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 4603–4607 CrossRef CAS.
- X. Jiang, Y. W. Ma, J. J. Li, Q. L. Fan and W. Huang, J. Phys. Chem. C, 2010, 114, 22462–22465 CAS.
- H. Bai, C. Li, X. L. Wang and G. Q. Shi, Chem. Commun., 2010, 46, 2376–2378 RSC.
- Y. X. Xu, Q. O. Wu, Y. Q. Sun, H. Bai and G. Q. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS.
- H. Bai, C. Li, X. L. Wang and G. Q. Shi, J. Phys. Chem. C, 2011, 115, 5545–5551 CAS.
- C. C. Huang, H. Bai, C. Li and G. Q. Shi, Chem. Commun., 2011, 47, 4962–4964 RSC.
- W. Kaminsky, A. Funck and H. Hahnsen, Dalton Trans., 2009, 8803–8810 RSC.
- H. G. Alt, E. H. Licht, A. I. Licht and K. J. Schneider, Coord. Chem. Rev., 2006, 250, 2–17 CrossRef CAS.
- Y. J. Gao, G. Hu, W. Zhang, D. Ma and X. H. Bao, Dalton Trans., 2011, 40, 4542–4547 RSC.
- C. M. Woodbridge, D. L. Pugmire, R. C. Johnson, N. M. Boag and M. A. Langell, J. Phys. Chem. B, 2000, 104, 3085–3093 CrossRef CAS.
- X. C. Dong, D. L. Fu, W. J. Fang, Y. M. Shi, P. Chen and L. J. Li, Small, 2009, 5, 1422–1426 CrossRef CAS.
- B. Das, R. Voggu, C. S. Rout and C. N. R. Rao, Chem. Commun., 2008, 5155–5157 RSC.
- G. D. Jiang, Z. F. Lin, C. Chen, L. H. Zhu, Q. Chang, N. Wang, W. Wei and H. Q. Tang, Carbon, 2011, 49, 2693–2701 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |