Nickel and cobalt complexes of benzoic acid (2-hydroxy-benzylidene)-hydrazide ligand: synthesis, structure and comparative in vitro evaluations of biological perspectives†
Paramasivam Krishnamoorthya, Palanisamy Sathyadevia, Packianathan Thomas Muthiahb and Nallasamy Dharmaraj*a
aDepartment of Chemistry, Bharathiar University, Coimbatore – 641 046, India. E-mail: dharmaraj@buc.edu.in; Fax: +91 422 2422387; Tel: +91 422 2428316
bSchool of Chemistry, Bharathidasan University, Tiruchirappalli – 620 024, India
First published on 29th October 2012
Abstract
Reactions of tridentate chelating hydrazone Schiff bases benzoic acid (2-hydroxyl-benzylidene)-hydrazide (H2L) (1) obtained from salicylaldehyde and benzhydrazide with [NiCl2(PPh3)2] and [CoCl2(PPh3)2] afforded respective metal hydrazone complexes of the composition [Ni(L)(PPh3)] (2) and [Co1(L)2]2[Co2(H2O)4(OPPh3)2] (3). The molecular structure of both complexes 2 and 3 determined by single crystal X-ray diffraction revealed that complex 2 is neutral in charge with distorted square planar geometry. However, complex 3 was found to have distorted octahedral geometry. All of the synthesised compounds 1–3 were studied by interaction with calf thymus DNA (CT DNA) and bovine serum albumin (BSA). In addition in vitro free radical scavenging and cytotoxic potential of all the synthesised compounds were also investigated.
Introduction
Transition metal ions with variable oxidation states have an important role in bioinorganic chemistry and redox enzyme systems and may provide the basis of models for active sites of biological systems. Artificial metallonucleases are in high demand as cellular regulators of DNA for therapeutic or biochemical purposes.1 Metal complexes containing site-specific substructures and multiple reactive sites constitute a group of promising candidates for nucleases because of their electronic and structural advantages.2 Moreover, these complexes generate highly cationic species that favor the electrostatic attraction to the anionic phosphate backbone of DNA. Incorporation of a DNA-targeting moiety into the ligands can improve the selectivity of metallonucleases.3 Therefore, there is an exigency to identify effective metal-based therapeutics, particularly those that overcome both inherent and acquired resistance to drug therapy and show improved therapeutic properties, stimulating the ongoing investigations of alternative molecular targeted metal-based drugs.4Anticancer drugs targeting biomolecules represent a rational advancement in modern drug discovery. It is a general fact that considerable achievement is realized in enhancing the targeting and efficiency of metal-chelators through ligand design that can significantly alter the biological properties by modifying reactivity or substitution-related inertness and play a major role in their binding to DNA. Large planar ligands promote intercalative binding of the metal complexes to DNA,5–10 whereas non-planar ligands or ligands that do not have extended planarity promote groove binding, particularly with octahedral metal complexes.11–13 Since nucleic acids are very important genetic substances, they provide different binding sites and binding modes for covalent and non-covalent interactions.14–19 Interest in the binding of metal complexes to DNA has been motivated not only by a desire to understand the basics of these interaction modes, but also for the development of metal complexes as anti-inflammatory, antifungal, antibacterial and anticancer agents. Hence, much attention has been devoted to the design of metal-based complexes that bind to DNA.20–23
Serum albumins, the most abundant proteins in blood plasma, have many physiological functions. Since most of the administered drugs bind extensively and reversibly to serum albumin to form protein–drug complexes, the biological properties of the drug, such as the overall distribution, metabolism and efficacy in the body, are correlated with the affinity towards serum albumin. Among serum albumins, bovine serum albumin (BSA) is an appropriate protein model for studying the interaction between serum albumins and drugs because of its medically important, unusual ligand-binding properties, ease of availability, low cost and structural homology with human serum albumin (HSA).24,25 Therefore, the interaction between serum albumins and drugs not only provide useful information on the structural features that determine the therapeutic effectiveness of drugs, but is also crucial to studying the pharmacological response of drugs and design of dosage forms. Hence, it has become an interesting research field that attracted the attention of biologists, chemists, pharmacists and therapists.26–29
It is well-known that many hydrazones and their corresponding metal complexes have displayed diverse spectra of biological and pharmaceutical activities, such as anticancer, antitumor and antioxidative activities, as well as the inhibition of lipid peroxidation etc.30–36 Hence, our present investigation focuses on the design and synthesis of Ni and Co complexes containing the hydrazone ligand obtained from the reaction of salicylaldehyde and benzhydrazide as an anchoring group that can provide a well-defined binding environment, as well as increase the stability of the resultant metal complexes. All of the synthesized compounds have been subjected to various experiments to assess their interacting ability with DNA/BSA and free radical scavenging and cytotoxic potentials.
Results and discussion
The reactions between [MCl2(PPh3)2] (where M = Ni(II) (or) Co(II)) and the hydrazone ligand benzoic acid-(2-hydroxy-benzylidene)-hydrazide (1) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio yielded complexes of the type [Ni(L)(PPh3)] (2) and [Co(L)2]2[Co(H2O)4(OPPh3)2] (3) (Scheme 2). The analytical data of the above said complexes are in good agreement with the proposed molecular formulae with 1:1 and 1:2 metal to ligand stoichiometries (presented under the experimental part), respectively. All of the three synthesised compounds are quite stable in air and light and soluble in most of the organic solvents, such as MeOH, EtOH, CH2Cl2, CHCl3, DMF and DMSO, and are well characterised using several physico-chemical techniques.
:1 molar ratio yielded complexes of the type [Ni(L)(PPh3)] (2) and [Co(L)2]2[Co(H2O)4(OPPh3)2] (3) (Scheme 2). The analytical data of the above said complexes are in good agreement with the proposed molecular formulae with 1:1 and 1:2 metal to ligand stoichiometries (presented under the experimental part), respectively. All of the three synthesised compounds are quite stable in air and light and soluble in most of the organic solvents, such as MeOH, EtOH, CH2Cl2, CHCl3, DMF and DMSO, and are well characterised using several physico-chemical techniques.Structural description of compounds 2 and 3
An attempt to synthesise 1:1 metal–ligand stoichiometric hydrazone complexes led to an interesting observation that the hydrazone showed a variable behaviour between nickel and cobalt systems, which encouraged us to carry out a systematic study on the structure of the complexes. It was found that the reaction of H2L with [NiCl2(PPh3)2] yielded the complex [Ni(L)(PPh3)] (2) with distorted square planar geometry, whereas a similar reaction with [CoCl2(PPh3)2] produced a novel complex of the composition [Co(H2O)4(OPPh3)2][Co(N2O2C14H10)2]2[OPPh3]2[(H2O)0.8]2 (3). The molecular structure of complexes 2 and 3 with the corresponding atom numbering scheme are shown in Fig. 1 and 2, respectively, with the relevant bond distances and angles collected in Tables 1 and 2.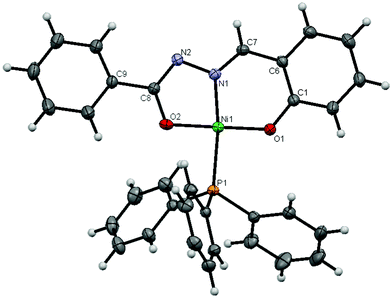 | ||
| Fig. 1 The molecular structure of complex 2, with displacement ellipsoids drawn at the 25% probability level. | ||
 | ||
| Fig. 2 The molecular structure of complex 3 with displacement ellipsoids drawn at the 25% probability level, including hydrogen bonding. Hydrogen atoms, water and phosphonium oxide molecules are omitted for clarity. Note: atoms generated by symmetry are not labelled. | ||
| Complex 2 | Complex 3 | |
|---|---|---|
| Formula | C32 H25 N2 Ni O2 P | C128 H111.20 Co3 N8 O17.60 P4 |
| Formula weight | 559.22 | 2343.72 |
| T/K | 293 | 293(2) |
| λ/Å | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/c | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Cell dimensions | ||
| a/Å | 14.6153(2) | 10.5181(5) |
| b/Å | 10.1839(1) | 14.2200(6) |
| c/Å | 17.6301(2) | 19.9005(9) |
| α (°) | 90 | 77.689(3) |
| β (°) | 93.013(1) | 84.636(3) |
| γ (°) | 90 | 79.795(3) |
| Z | 4 | 1 |
| hkl limits | −21 < = h = > 21 | −10 < = h = > 10 |
| −15 < = k = > 8 | −14 < = k = > 14 | |
| −23 < = l = > 27 | −20 < = l = > 20 | |
| Dc/Mg m−3 | 1.415 | 1.362 |
| F(000) | 1156 | 1217 |
| Crystal size/mm | 0.26 × 0.22 × 0.15 | 0.30 × 0.26 × 0.26 |
| Reflections collected | 35579 | 30411 |
| Data/restraints/parameters | 9769/0/343 | 6655/0/727 |
| Goodness–of–fit on F2 | 1.039 | 1.172 |
| Final R indices [I > 2σ(I)] | R1 = 0.0594, wR2 = 0.1909 | R1 = 0.0668, wR2 = 0.1451 |
| R indices (all data) | R1 = 0.1161, wR2 = 0.1606 | R1 = 0.1302, wR2 = 0.2063 |
| C32 H25 N2 Ni O2 P (2) | C128 H111.20 Co3 N8 O17.60 P4 (3) | ||||||
|---|---|---|---|---|---|---|---|
| Bond lengths | Bond angles | Bond lengths | Bond angles | ||||
| Ni1–N1 | 1.848(2) | N1–Ni1–O1 | 95.8(1) | Co1–O1 | 1.903(6) | O1–Co1–N2 | 83.3(3) |
| Ni1–O1 | 1.814(2) | N1–Ni1–O2 | 83.6(1) | Co1–N2 | 1.888(7) | N2–Co1–O2 | 96.0(3) |
| Ni1–O2 | 1.812(2) | O2–Ni1–P1 | 88.34(7) | Co1–O2 | 1.873(6) | O3–Co1–N4 | 83.1(3) |
| Ni1–P1 | 2.2199(7) | P1–Ni1–O1 | 92.27(7) | Co1–O3 | 1.837(7) | N4–Co1–O4 | 95.9(3) |
| N1–N2 | 1.405(3) | O1–Ni1–O2 | 176.3(1) | Co1–N4 | 1.880(7) | N2–Co1–N4 | 172.6(3) |
| N2–C8 | 1.299(4) | N1–Ni1–P1 | 171.86(7) | Co1–O4 | 1.880(6) | O1–Co1–O2 | 178.8(3) |
| C8–O2 | 1.318(4) | C6–C7–N1–N2 | 175.9(3) | C1–O1 | 1.308(9) | O3–Co1–O4 | 178.6(3) |
| C1–O1 | 1.316(4) | C9–C8–N2–N1 | −179.8(2) | C1–N1 | 1.313(10) | O1–Co1–O3 | 88.5(3) |
| C1–C6 | 1.400(4) | C9–C8–O2–Ni1 | 179.6(2) | N1–N2 | 1.385(9) | O2–Co1–O4 | 90.4(3) |
| C6–C7 | 1.435(4) | C2–C1–O1–Ni1 | 177.6(2) | C8–N2 | 1.280(9) | C1–N1–N2–C8 | 172.1(8) |
| C7–N1 | 1.285(4) | C8–C9 | 1.433(11) | N1–N2–C8–C9 | −177.8(8) | ||
| C9–C14 | 1.422(11) | C15–N3–N4–C22 | −175.8(8) | ||||
| C14–O2 | 1.295(10) | N3–N4–C22–C23 | 177.8(8) | ||||
| C15–O3 | 1.329(10) | O2W–Co2–O3W | 92.4(2) | ||||
| C15–N3 | 1.311(11) | O5–Co2–O3W | 93.1(2) | ||||
| N3–N4 | 1.377(9) | O5–Co2–O2W | 88.7(2) | ||||
| C22–N4 | 1.283(10) | P1–O5–Co2 | 144.8(4) | ||||
| C22–C23 | 1.422(12) | ||||||
| C23–C28 | 1.397(12) | ||||||
| C28–O4 | 1.357(10) | ||||||
| Co2–O2W | 2.102(6) | ||||||
| Co2–O3W | 2.126(6) | ||||||
| P1–O5 | 1.492(6) | ||||||
| Co2–O5 | 2.057(5) | ||||||
The crystals of complex 2 belong to the monoclinic crystal system with P21/c space group. The coordination geometry around nickel was distorted square planar in [Ni(L)(PPh3)], in which the dianionic ligand acted as a planar tridentate forming a five-membered and a six-membered metallocycle involving the nickel ion.
As expected, the hydrazone ligand is bonded to the nickel ion via phenolate oxygen, the enolate oxygen and the imine nitrogen atom; the corresponding bite angles being 95.8(1)° and 83.6(1)° for [O1–Ni1–N1] and [N1–Ni1–O2]. The ONO donor atoms of the ligand occupy three coordination sites of the square planar geometry, with the fourth coordination site utilised by the phosphorus atom of the triphenylphosphine. The unequal bond lengths of [Ni1–N1] 1.847(2) Å, [Ni1–O1] 1.814(2) Å, [Ni1–O2] 1.812(2) Å and [Ni–P1] 2.2200(7) Å, as well as unequal bond angles generated by these bonds at the Ni-acceptor center indicate a distortion from the perfect square planar geometry. These values are similar to those reported for a few metal(II) hydrazones chelates.37
The molecular structure of complex 3 shown in Fig. 2 is quite different from that of 2 with respect to unit cell parameters, composition and nature. Complex 3 consists of a pair of Co(III) anionic species [Co(L)2] and a cationic Co(II) entity in between them. The anionic complexes with the trivalent cobalt metal center (Co metal labeled as Co1) were coordinated by two units of binegative tridentate hydrazone ligand (H2L) that led to a distorted octahedral environment with 1:2 metal–ligand stoichiometry. To compensate the charge 3− on each of the [Co1(L)2], a tetra aqua Co(II) cationic complex [Co(H2O)4(OPPh3)2] in which Co is labeled as Co2 was formed and present in the unit cell. Furthermore, two molecules each of phosphonium oxide and water were also found in the same unit cell.
The four equatorial positions of cobalt atom in the anionic complex is occupied by phenolate –O and the enolate –O atoms and the two axial positions by imine –N of the deprotonated H2L ligand in its enol form. However, in the cationic complex part, four equatorial positions were occupied by water molecules and the two axial positions by phosphonium oxide through the oxygen atoms in a neutral manner form an octahedral geometry. In one of the above said anionic parts, [Co1(L)2], the coordination behaviour of the two equivalents of ligands are very similar and indicate that the ligand adopted the enol form and was deprotonated prior to the coordination with the metal ion. However, the crystallographic dimensions of the ligands were non-equivalent and hence they are only chemically equivalent. The two oxygen and nitrogen atoms exhibited ligand–metal–ligand bite angles of 83.3(3)° [O1–Co1–N2], 96.0(3)° [N2–Co1–O2], 83.1(3)° [O3–Co1–N4] and 95.9(3)° [N4–Co1–O4]. The bond length of [Co1–O1] 1.903(5) Å, [Co1–N2] 1.888(6) Å, [Co1–O2] 1.872(6) Å, [Co1–O3] 1.837(7) Å, [Co1–N4] 1.879(7) Å, [Co1–O4] 1.880(6) Å are comparable in length to those of the earlier reports.38 The trans angles, namely [N2–Co1–N4] 172.6(3)°, [O1–Co1–O2] 178.6(3)° and [O3–Co1–O4] 178.8(3)° are constrained from the ideal value of 180°. This showed that the complex [Co1(L)2] possesses distorted octahedral geometry. However, in the case of the cationic cobalt part, we observed a symmetrical equivalence between a set of water molecules coordinated to metal, as well as the triphenylphosphonium oxides to the cobalt ions with corresponding bond lengths of [Co2–O3W] 2.125(6) Å, [Co2–O5] 2.057(5) Å, [Co2–O2W] 2.102(7) Å, [P1–O5] 1.492(6) Å and bond angles of [O5–Co2–O2W] 88.7(2)°, [O5–Co2–O3W] 93.3(2)°, [O2W–Co2–O3W] 87.6(2)°, [P1–O5–Co2] 144.8(4)°. From the crystallographic study, it was found that Co2 lies on an inversion center. Thermal parameters of O1w indicated a possible partial occupancy; upon refinement the occupancy of O1w refined to 0.8 and was fixed to that value for final refinement. In addition, there exist an intermolecular hydrogen bonding between the above discussed cationic cobalt complex with the respective anionic cobalt hydrazone complexes present on either side of the former species with bond lengths in the magnitude of [O4–H3W] 1.91 Å and [N1–H2W] 2.09 Å, as presented in Fig. 2. Similarly, independent hydrogen bonding was also observed between the free triphenylphosphonium oxide (O8) and water molecules (H1W) occupying the crystal lattice that were not shown in Fig. 2.
DNA binding studies
![Electronic absorption spectra of complexes 2 and 3 (25 μM) in the absence and presence of increasing amounts of CT DNA (2.5, 5.0, 7.5, 10.0, 12.5, 15.0, 17.5 and 20.0, 22.5 and 25 μM). Arrows show the changes in absorbance with respect to an increase in the DNA concentration. Inset: plot of [DNA] vs. [DNA]/(εa − εf).](/image/article/2012/RA/c2ra20597a/c2ra20597a-f3.gif) | ||
| Fig. 3 Electronic absorption spectra of complexes 2 and 3 (25 μM) in the absence and presence of increasing amounts of CT DNA (2.5, 5.0, 7.5, 10.0, 12.5, 15.0, 17.5 and 20.0, 22.5 and 25 μM). Arrows show the changes in absorbance with respect to an increase in the DNA concentration. Inset: plot of [DNA] vs. [DNA]/(εa − εf). | ||
Monitoring the changes in absorption spectra of the test compounds 1, 2 and 3 upon the incremental addition of DNA is one of the most widely used methods to determine overall binding constants. In general, a compound that binds to DNA through intercalation results in hypochromism and bathochromism involving strong stacking interaction between an aromatic chromophore and the base pairs of DNA. Upon the addition of calf-thymus DNA solution to the compounds 1, 2 and 3 a decrease in molar absorptivity (hypochromism, 13–61%) has been observed with respect to all of the above said absorption bands without any shift in wavelength. Among them, the hydrazone ligand (1) showed hypochromism of 13.36, 13.34 and 14.20% at 286, 295 and 322 nm, respectively, whereas the bands exhibited by complex 2 showed hypochromism of about 60.42 (304 nm) and 60.56% (374 nm) and 3 displayed about 18.57 and 19.08% at 327 and 384 nm, respectively, indicating the strong binding of them with DNA. In order to compare the DNA-binding affinity of these compounds quantitatively, their intrinsic binding constants were calculated by the changes monitored in absorption at higher energy band with increasing concentrations of DNA using eqn (1). The value of intrinsic binding constant (Kb) was calculated using eqn (1) and are found to be 1.4008 × 104 M−1, 1.3471 × 105 M−1 and 1.4812 × 104 M−1 corresponding to the hydrazone ligand 1 and complexes 2 and 3, respectively indicating their strong binding to DNA, that are comparable with other reported intercalating complexes.42–45 The magnitude of the binding constant value clearly showed that complex 2 bound more strongly with CT DNA than the free hydrazone ligand 1 and complex 3.
:[EB] = 2:1}, the DNA-induced emission intensity of EB was decreased. It is a well-established phenomenon that addition of a second DNA binding molecule would quench the EB emission by either replacing the EB molecules that were originally bound to DNA (if it binds to DNA more strongly than EB) or accepting an excited state electron from EB. Since complexes 2 and 3 possess Ni and Co ions coordinated with the ligand, they efficiently compete with EB for intercalative binding sites on DNA by replacing them, which was evidenced through quenching of the emission intensity of DNA-bound EB. The apparent binding constants, Kapp, have been calculated from the following equation,| KEB [EB] = Kapp[compound] |
The fluorescence spectra with Stern–Volmer plot (I0/I versus [Q]) of EB bound-DNA quenched by free ligand 1 and corresponding metal complexes 2 and 3 are shown in Fig. S2† and 4.
 | ||
| Fig. 4 Emission spectra of DNA–EB in the presence of 0, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140 and 150 μM of complexes 2 and 3. The arrow indicates the changes in the emission intensity as a function of complex concentration. Inset: the Stern–Volmer plot of the fluorescence titration data corresponding to complexes 2 and 3. | ||
The observed linearity in the plot supported the fact that the quenching of EB bound to DNA by the test compounds are in good agreement with the linear Stern–Volmer equation. The calculated apparent binding constant values, Kapp of compounds 1, 2 and 3 were 8.1188 × 104 M−1, 2.8117 × 105 M−1 and 1.9043 × 105 M−1, whereas the quenching constant Kq was determined to be 1.5965 × 103 M−1, 6.2052 × 103 M−1 and 3.4857 × 103 M−1. These data revealed the higher quenching efficiency of complex 2 than that of both free ligand 1 and complex 3, reflecting a strong binding between the nickel square planar complex 2 with DNA and supporting the outcome of the absorption spectral study discussed in the preceding section.
The results of binding experiments clearly proved that among the free hydrazone ligand and its metal complexes, the latter showed stronger binding with DNA than the former that may be due to the chelation of the ligand with the metal ion that resulted in an alteration of electronic properties of the former. In general, the presence of an extended planarity in the chelated ligand would increase the strength of the interaction of the complexes with DNA. Changing the metal ions and the geometry of the complex (square planar, tetrahedral and octahedral) can also modify the binding properties and this may show variations upon interaction with nucleic acids. Between the complexes 2 and 3, complex 2 was identified to bind more strongly with DNA than the complex 3, which could be explained in terms of the planarity and geometry of the metal complexes. In this case, the higher binding affinity shown by complex 2 is due to the fact that it has square planar geometry, whereas complex 3 possesses octahedral geometry. The ability of the more planar structure of the former to penetrate deep into the DNA helix made complex 2 a good candidate for effective binding. However, the octahedral complex 3 with axial ligands finds it difficult to intercalate with DNA due to the steric inhibition. Moreover, in complex 3 a set of bulky triphenylphosponium oxide ligands also prevented the intercalation with DNA. The overall order of interaction of compounds 1, 2 and 3 with CT DNA, as well as BSA decreased in the order 2 > 3 > 1.
DNA cleavage
The interaction of pUC19 plasmid DNA with complexes 2 and 3 was evaluated in order to determine the efficiency with which they sensitize DNA cleavage by monitoring the transition from the naturally occurring, covalently closed circular form (Form I) to the open circular relaxed form (Form II). This transition normally occurs when one of the strands of the plasmid is nicked and can be determined by gel electrophoresis of the plasmid. The tests were performed under aerobic conditions with H2O2 as a co-oxidant using a complex concentration of 35 μM. Neither H2O2 nor the metal complexes 2 and 3 yielded significant strand scission when applied individually (figure not shown) demonstrating that one of the strands of the plasmidic DNA is required to cause the effective cleavage. Fig. 5 reveals that cleavage of pUC19 DNA induced by complexes 2 and 3 in the presence of H2O2 results in the conversion of Form I into Form II. The extent of Form I diminished gradually upon partial conversion to Form II with a simultaneous increase in the intensity of the latter form. However, the free hydrazone ligand completely failed to induce any cleavage (not shown in Fig. 5). | ||
| Fig. 5 Gel-electrophoresis pictures and bar diagram for the metal hydrazone complexes. A photograph showing the effects of transition metal hydrazones on DNA of pUC19: Lane 1: SC pUC19 DNA (0.5 μg) alone; Lane 2: SC pUC19 DNA (0.5 μg) + H2O2 (60 μM); Lane 3: SC pUC19 DNA (0.5 μg) + H2O2 (60 μM) + complex 2 (35 μM); Lane 4: SC pUC19 DNA (0.5 μg) + H2O2 (60 μM) + complex 3 (35 μM). | ||
Based upon the ability of complexes 2 and 3 to convert the supercoiled form (Form I SC) to the open circular form (Form II OC) given in Fig. 5 (bar diagram), it is obvious that the complex 2 with more planar square planar geometry has a strong capability to cleave the supercoiled plasmid DNA when compared to that of complex 3 that possess highly crowded octahedral geometry around the metal ion, which hinders the scissoring of DNA into the above said form.
Protein binding studies
 | ||
| Fig. 6 Emission spectra of BSA (1 × 10−6 M; λexi = 280 nm; λemi = 345 nm) as a function of concentration of the complexes 2 and 3 (0, 0.8, 1.6, 2.4, 3.2 and 4 × 10−6 M). The arrow indicates the effect of metal complexes 2 and 3 on the fluorescence emission of BSA. | ||
In addition, the UV-visible absorption spectra of BSA and free ligand 1 or metal hydrazone complexes 2 or 3 were measured to find the type of quenching exists. Addition of the above compounds to BSA obviously leads to an increase in BSA absorption intensity without affecting the position of absorption band indicating that the type of interaction between the test compounds and BSA was mainly a static quenching process.47 A representative absorption spectrum of pure BSA and BSA-complex 2 is shown in Fig. 7 (for compounds 1 and 2 see supplementary material Fig. S4†).
 | ||
| Fig. 7 The absorption spectra of BSA (1 × 10−5 M) and BSA–complex 2 (BSA= 1 × 10−5 M and complex 2 = 1 × 10−6 M). | ||
The fluorescence quenching is described by the Stern–Volmer relation,
| I0/I = 1 + KSV[Q] |
| log [(F0 − F)/F] = log K + nlog [Q] |
Thus, a plot of log[(F0 − F)/F] versus log [Q] (Fig. 8) can be used to determine the values of both K and n and such values calculated for compounds 1, 2 and 3 are listed in Table 3.
![Plot of log[(F0 − F)/F] versus log [Q].](/image/article/2012/RA/c2ra20597a/c2ra20597a-f8.gif) | ||
| Fig. 8 Plot of log[(F0 − F)/F] versus log [Q]. | ||
| Compound | K (M−1) | n |
|---|---|---|
| BSA + ligand 1 | 2.3085 × 103 | 0.6325 |
| BSA + complex 2 | 9.3856 × 104 | 0.8767 |
| BSA + complex 3 | 4.3829 × 103 | 0.6866 |
From the values of n, we inferred that there is only one independent class of binding site on BSA that was available for all of the compounds under investigation and a linear relation between the binding constant and number of binding sites was identified. These data showed the higher binding efficiency of complex 2 compared to that of the respective ligand 1 and complex 3 under investigation, reflecting a stronger binding of the former with albumin than the rest of the compounds, similar to the results observed in the DNA binding study.
It is well-known that the fluorescence of BSA may be due to the presence of tyrosine, tryptophan and phenylalanine residues and, hence, spectroscopic methods are usually applied to study the conformation of serum protein. According to Miller, in synchronous fluorescence spectroscopy, the difference between excitation and emission wavelength (Δλ = λemi − λexc) reflects the spectra of a different nature of chromophores, with large Δλ values, such as 60 nm, the synchronous fluorescence of BSA, is characteristic of tryptophan residues and a small Δλ value, such as 15 nm, is characteristic of tyrosine.51
 | ||
| Fig. 9 Synchronous spectra of BSA (1 × 10−6 M) as a function of concentration of complexes 2 and 3 (0, 0.8, 1.6, 2.4, 3.2 and 4 × 10−6 M) with a wavelength difference of Δλ = 15 nm (2a and 3a) and Δλ = 60 nm (2b and 3b) (a). The arrow indicates the change in emission intensity w.r.t various concentrations of complexes 2 and 3. | ||
To explore the structural changes of BSA due to the addition of compounds 1, 2 and 3, we measured synchronous fluorescence spectra of BSA with added test compounds at both Δλ = 15 nm and 60 nm. Upon increasing the concentration of compound 1, the intensity of emission corresponding to tyrosine was increased very slightly, whereas in the case of compounds 2 and 3 a slight decrease in the intensity without any change in the position of the wavelength was observed. However, the fluorescence emission of tryptophan showed a significant decrease in the intensity without any change in the position of the emission band by the addition of the ligand and its complexes. These experimental results indicate that the compounds affect the microenvironment of the tryptophan residues more effectively during the binding process. The spectra of compounds 1, 2 and 3 are given in Fig. S5† and 9.
Antioxidant activity
The antioxidant activity of free ligand 1 and metal complexes 2 and 3 were evaluated in a series of in vitro assays involving ABTS cationic radicals, superoxide and hydroxyl radicals in a dose-dependent manner. IC50 values of ligand 1 in relevance to ABTS cationic, O2− and OH assays were 38.87, 43.21 and >50 μM, respectively. The complexes 2 and 3 showed their IC50 values at 20.08, 24.42, 31.26, 25.11, 27.35 and 35.12 μM, respectively. The results of these experiments are shown in Fig. 10. | ||
| Fig. 10 Trends in the inhibition of ABTS (A), superoxide (B) and OH (C) radicals by ligand 1 and corresponding metal complexes 2 and 3 at variable concentrations. | ||
In general, the antioxidant activity of the ligand and its M(II) complexes (where M = Ni, and Co) against the free radicals i.e., ABTS cationic, O2− and OH was found to decrease in the order 2 > 3 > 1. The results indicated that the metal complexes exhibited greater antioxidant activity than the free ligand. Among the tested compounds, complex 2 displayed very high scavenging activity. Furthermore, the results obtained against the different radicals confirmed that all of the compounds are more effective to arrest the formation of the ABTS than the rest of the radicals. The significant aspect of the present investigation is that the newly synthesized complexes 2 and 3 displayed excellent antioxidant activities that are comparable to those of standard antioxidant butylated hydroxyl anisole (BHA).
In vitro cytotoxicity
The hydrazone ligand, starting precursor complex and its metal hydrazone complexes were screened for in vitro cytotoxicity towards NIH 3T3 normal cell lines and HeLa, HepG-2 and A431 tumour cell lines as a preliminary screening technique using a MTT assay (cell viability test) for their potential cytotoxic activity, in which mitochondrial dehydrogenase activity is measured as an indication of cell viability.52 The test compounds were dissolved in DMSO and blank samples containing the same amount of DMSO are taken as controls. The effect of the ligand and its complexes on the viability of these cells was evaluated after an exposure period of 48 h. Results of the in vitro cytotoxicity of the compound with their corresponding IC50 value, related to inhibition of tumour cell growth at the 50% level, are presented in Fig. 11. | ||
| Fig. 11 Cell growth inhibition % of NIH 3T3, HepG-2 and A431 cell lines as a function of concentration of complex 2. | ||
From the cytotoxicity results, it was found that the tested samples 1 and 3 and starting precursor complex were completely inactive against HeLa, HepG-2 and A431 cell lines under the investigated concentration range (15–500 μM) and can therefore be considered as non-cytotoxic. However, sample 2 has displayed moderate cytotoxic potentials against HepG-2 and A431 cell lines with IC50 values of 168 and 84 μM, respectively (Fig. 11). From the IC50 values, it can be said that complex 2 showed more cytotoxic potential towards A431 cells when compared to that of HepG-2 cells. In parallel, the influence of the widely used anticancer drug cisplatin was also assayed as standard (IC50 value of 18 μM), which is not shown in Fig. 11. A simple structure–activity relationship suggests that among the investigated samples, complex 2 that contains a square geometry is more responsible for the potential inhibition against the tumour cell, whereas an octahedrally coordinated complex was inactive in this respect. In addition, it was proved that complex 2 did not cause any damage to the healthy cells (NIH 3T3) (IC50 = 467 μM) (Fig. 11), indicating that they are non-toxic to normal cells as expected for a better drug.
Conclusion
A set of new nickel(II) and cobalt(II)/(III) hydrazone complexes 2 and 3 supported by a tridentate NNO donor hydrazone 1 derived from benzhydrazide and salicylaldehyde were synthesized and characterized using FT-IR, UV-visible and 1H NMR spectroscopic techniques. Among them, there exists a 1:1 metal-to-ligand stoichiometric ratio in the case of complex 2, whereas a 1:2 ratio was found in the case of complex 3. Single crystal XRD results of complex 2 revealed a distorted square planar geometry around the nickel ion, but the structure of complex 3 was quite different, in which the cobalt ion was found to possess mixed oxidation states of +2 and +3 for cationic and anionic complexes, respectively, with distorted octahedral geometry. However, in both of the complexes, 2 and 3, the hydrazone ligand coordinated with metal ion in a dibasic tridentate fashion comprising azomethine nitrogen, an enolic oxygen and a phenolic oxygen atom. Studies on the binding of the titled complexes with both DNA and BSA reflected that they behaved as intercalators towards the former and selectively interacted with micro environmental residues of the latter. The results of a cleavage experiment showed that complex 2 significantly converts the supercoiled pUC19 DNA into its open circular form. The ligand 1 and both nickel or cobalt complexes 2 and 3 displayed significant scavenging activity towards the ABTS cationic, O2− and OH radicals. In vitro cytotoxicity results showed that compounds 1 and 3 were inactive against the selected cell lines, whereas complex 2 displayed moderate activity against A431 and lesser activity against HepG-2 cells. From the biological activity experiments, we concluded that complex 2 containing a nickel metal ion with more planar square planar structure exhibited higher potential than the free ligand and complex 3 containing a cobalt ion with a more steric octahedral environment.Experimental section
Materials and reagents
Reagent grade chemicals NiCl2·6H2O, CoCl2·6H2O, triphenylphosphine, salicylaldehyde and benzhydrazide were purchased from Sigma-Aldrich company and used as received. Calf thymus DNA (CT DNA) and bovine serum albumin (BSA) were purchased from Himedia Company, India. The plasmid supercoiled (SC) pUC19 DNA was purchased from Bangalore GeNei, Bangalore, India. The human cervical cancer cell line (HeLa), human liver hepatocellular carcinoma cells (HepG-2), human skin cancer cell line (A431) and immortalized NIH 3T3 cells were obtained from National Centre for Cell Science (NCCS), Pune, India. All other chemicals and reagents used for DNA binding/cleavage, protein binding, antioxidant and cytotoxicity assays were of high quality.Physical measurements and instrumentation
Elemental analyses (% C, H & N) were performed on a Vario EL III CHNS analyzer and IR spectra of the ligand and its complexes were recorded as KBr pellets on a Nicolet Avatar instrument in the frequency range 400–4000 cm−1. The electronic absorption and emission spectra of the ligand and its complexes were recorded in DMSO:buffer solution using a Jasco V-630 spectrophotometer and Jasco FP 6600 spectrofluorometer, respectively. The 1H NMR spectrum of the ligand in CDCl3 was recorded on a Bruker AMX 500 MHz spectrometer using tetramethylsilane as an internal standard. The X-ray diffraction data of the complexes 2 and 3 were collected at 293 K with Mo-Kα radiation (λ = 0.71073 Å) using a Bruker Smart APEX II CCD diffractometer equipped with a graphite monochromator. The structure was solved by direct methods and refined using full-matrix least squares (on F2) SHELXS97 and SHELXL9753 and the graphics were produced using PLATON97.54 All of the non-hydrogen atoms were refined anisotropically and the hydrogen atoms were positioned geometrically and refined as a riding model.Synthesis of ligand and metal complexes
 | ||
| Scheme 1 | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1571(CN), 1074 (NN), 1203 (phenolic, C–OH). 1H NMR δH (500 MHz; CDCl3): 12.08 (1H, s, OH), 11.27 (1H, s, NH), 8.63 (1H, s, CHN), 6.90–7.94 (9H, m, Ar–H).
O), 1571(CN), 1074 (NN), 1203 (phenolic, C–OH). 1H NMR δH (500 MHz; CDCl3): 12.08 (1H, s, OH), 11.27 (1H, s, NH), 8.63 (1H, s, CHN), 6.90–7.94 (9H, m, Ar–H).Synthesis of metal precursor complexes
The precursor metal complexes [NiCl2(PPh3)2] and [CoCl2(PPh3)2] were prepared according to the literature method.55,56 | ||
| Scheme 2 | ||
Color: red. Yield: Complex 2 (65%, 0.363 g), mp 184 °C. Found C, 68.02; H, 4.12; N, 4.52% Calc. for NiC32H25N2O2P: C, 68.73; H, 4.51; N, 5.01%. Selected IR bands (νmax/cm−1): 1598 and 1520 (CN–NC), 1340 (phenolic C–O), 1298 (enolic C–O), 1096 (N–N). 1H NMR δH (500 MHz; CDCl3): 8.36 (1H, s, HCN), 7.86–6.53 (24H, m, Ar–H).
Color: reddish brown. Yield: Complex 3 (29%, 0.440 g), mp 198 °C. Found C, 64.93; H, 4.68; N, 3.52% Calc. for Co2C82H75N4 O12P3: C, 64.83; H, 4.98; N, 3.69%. Selected IR bands (νmax/cm−1): 1600 and 1519 (CN–NC), 1339 (phenolic C–O), 1267 (enolic C–O), 1094 (NN).
DNA binding studies
| [DNA]/[εa − εf] = [DNA]/[εb − εf] + 1/Kb [εb − εf] | (1) |
| I0/I = Kq [Q] + 1 |
| KEB [EB] = Kapp [complex]. |
The complex concentration was obtained from the value at a 50% reduction of the fluorescence intensity of EB and KEB = 1.0 × 107 M−1, [EB] = 5 μM.
DNA cleavage experiment
The extent of DNA cleavage induced by the test compounds was monitored by agarose gel electrophoresis. A solution containing 25 μL of pUC19 DNA (1 μg), HCl (50 mM, pH 7.5), NaCl (50 mM), the metal complex (35 μM) and H2O2 (60 μM) was incubated at 37 °C for 1h. Subsequently, 4 μL of 6× DNA loading buffer containing 0.25% bromophenol blue, 0.25% xylene cyanol and 60% glycerol was added to the test solution and then mixed with 1% agarose gel containing 1.0 μg mL−1 of ethidium bromide. Electrophoresis was performed at 5 V cm−1 for 2 h in a TBE buffer and the bands were visualized under UV light and photographed. The cleavage efficiencies were measured by determination of the ability of each complex to convert the super coiled DNA (SC) to the open circular form (OC). After electrophoresis, the proportion of both the cleaved and uncleaved DNA in each fraction was quantitatively estimated on the basis of the band intensities using the BIORAD Gel Documentation System. The intensity of each band relative to that of the plasmid supercoiled form was multiplied by 1.43 to take account of the reduced affinity for ethidium bromide.59Protein binding studies
Binding of the free hydrazone ligand 1 and metal complexes 2 and 3 with bovine serum albumin (BSA) was studied using fluorescence spectra recorded by excitation at 280 nm and corresponding emission at 345 nm assignable to that of free bovine serum albumin (BSA). The excitation and emission slit widths and scan rates were constantly maintained for all of the experiments. Samples were carefully degassed using pure nitrogen gas for 15 min using quartz cells (4 × 1 × 1 cm) with high vacuum Teflon stopcocks. A stock solution of BSA was prepared in 50 mM phosphate buffer (pH = 7.2) and stored in the dark at 4 °C for further use. Concentrated stock solutions of the test compounds were prepared by dissolving them in DMSO:phosphate buffer (1:99) and diluted suitably with phosphate buffer to get the required concentrations. 2.5 ml of BSA solution (1 μM) was titrated by successive additions of a 2 μL stock solution of compounds 1, 2 and 3 (10−3 M) using a micropipette. Synchronous fluorescence spectra was also recorded using the same concentration of BSA and the synthesised compounds as mentioned above with two different Δλ (difference between the excitation and emission wavelengths of BSA) values, such as 15 and 60 nm.Antioxidant assays
The total antioxidant activity assay using ABTS cationic radical was studied according to the following procedure.60 ABTS (2,2′-azino-3-ethylbenzthiazoline-6-sulfonic acid diammonium salt) was dissolved in water to a 5 mM concentration and its cationic radical was produced by reacting with 5 mM potassium persulfate. The resulting mixture was kept in the dark at room temperature for 12–16 h before use. Prior to assay, the solution was diluted in ethanol (about 1:79 v/v) and equilibrated to 30 °C to give an absorbance of 0.70 ± 0.02 at 734 nm. After the addition of 2 mL of diluted ABTS cationic radical solution to different concentration (10–50 μM) of the compounds, the absorbance was taken at 30 °C exactly 30 min after the initial mixing and the reaction mixture without test sample was used as the control.The superoxide (O2−) radical scavenging assay was done based on the capacity of the compounds to inhibit formazan formation by scavenging the superoxide radicals generated in the riboflavin-light-NBT system.61 Each 3 mL reaction mixture contained 50 mM sodium phosphate buffer (pH 7.6), 20 μg riboflavin, 12 mM EDTA and 0.1 mg NBT. The reaction was started by illuminating the reaction mixture with different concentrations of the test compounds (10–50 μM) for 90 s. Immediately after illumination, the absorbance was measured at 590 nm. The entire reaction assembly was enclosed in a box lined with aluminum foil. The above reaction mixture without test sample was used as the control.
The hydroxyl (OH) radical scavenging activity of compounds 1–3 were investigated using the Nash method.62In vitro hydroxyl radicals were generated by a Fe3+/ascorbic acid system. The detection of hydroxyl radicals was carried out by measuring the amount of formaldehyde formed from the oxidation reaction with DMSO. The formaldehyde produced was detected spectrophotometrically at 412 nm. A mixture of 1.0 mL of iron–EDTA solution (ferrous ammonium sulphate (0.331 mM) and EDTA (0.698 mM), 0.5 mL of EDTA solution (0.048 mM) and 1.0 mL of DMSO (10.83 mM) DMSO (v/v) in 0.1 M phosphate buffer, pH 7.4) were sequentially added to the test tubes containing the test compounds with different concentrations in the range of 10–50 μM. The reaction was initiated by adding 0.5 mL of ascorbic acid (1.25 mM) and incubated at 80–90 °C for 15 min in a water bath. After incubation, the reaction was terminated by the addition of 1.0 mL of ice-cold TCA (107 mM). Subsequently, 3.0 mL of Nash reagent was added to each tube and left at room temperature for 15 min. The reaction mixture without sample was used as the control. The intensity of the colour formed was measured spectrophotometrically at 412 nm against reagent blank.
In the case of the above three assays, all of the tests were run in triplicate. The percentage of activity was calculated using the formula, % of activity = [(A0 − AC)/A0] × 100. A0 and AC are the absorbance in the absence and presence of the tested compounds, respectively. The IC50 can be calculated using the percentage of activity results.
Cytotoxicity studies
The in vitro cytotoxicity assay (IC50) was performed on the human cervical cancer cell line (HeLa), human liver hepatocellular carcinoma cells (HepG-2), human skin cancer cell line (A431) and immortalized NIH 3T3 cells. The tumor cell lines used in this work were grown in Eagles Minimum Essential Medium containing 10% fetal bovine serum (FBS) and the NIH 3T3 fibroblasts were grown in Dulbeccos Modified Eagles Medium (DMEM) containing 10% FBS. For the screening experiments, the cells were seeded into 96-well plates in 100 mL of the respective medium containing 10% FBS, at a plating density of 10000 cells/well. The cells were incubated at 37 °C in 5% CO2 and 95% air at a relative humidity of 100% for 24 h prior to the addition of the complexes. The complexes were solubilized in dimethylsulfoxide and diluted in the respective serum free medium. After 24 h, 100 mL of the medium containing the test compounds with various concentrations (e.g. 15, 30, 60, 120, 250 and 500 μM) was added and incubated at 37 °C in an atmosphere of 5% CO2 and 95% air with 100% relative humidity for 48 h. All measurements were made in triplicate and the medium containing no test complexes served as the control. After 48 h, 15 mL of MTT (5 mg mL−1) in phosphate buffered saline (PBS) was added to each well and incubated at 37 °C for 4 h. The medium with MTT was then flicked off and the formazan crystals that had formed were solubilized in 100 mL of DMSO and the absorbance at 570 nm was measured using a micro plate reader. The % cell inhibition was determined using the following formula, | % Cell inhibition = 100 − Abs(sample)/Abs(control) × 100 |
The IC50 values were calculated from the graph plotted between % cell inhibition and concentration of the test compounds.
Acknowledgements
We thank the University Grants Commission (UGC), New Delhi, India, for the award of Fellowship in Science for Meritorious Students (RFSMS) to one of the authors (P. Krishnamoorthy) under the UGC-SAP-DRS programme. PTM is thankful to DST-India (FIST Programme at School of Chemistry, Bharathidasan University, Tiruchirappalli, India) for the use of the Bruker Smart APEX II diffractometer.References
- L. J. K. Boerner and J. M. Zaleski, Curr. Opin. Chem. Biol., 2005, 9, 135 CrossRef CAS.
- Q. Jiang, N. Xiao, P. F. Shi, Y. G. Zhu and Z. J. Guo, Coord. Chem. Rev., 2007, 251, 1951 CrossRef CAS.
- T. Ito, S. Thyagarajan, K. D. Karlin and S. E. Rokita, Chem. Commun., 2005, 4812 RSC.
- T. Storr and K. H. Thompson, Chem. Soc. Rev., 2006, 35, 534 RSC.
- J. Reedijk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(7), 3611 CrossRef CAS.
- T. W. Hambley, Dalton Trans., 2007, 4929 RSC.
- H. J. Yu, S. M. Huang, L. Y. Li, H. N. Jia, H. Chao, Z. W. Mao, J. Z. Liu and N. Ji, J. Inorg. Biochem., 2009, 103, 881 CrossRef CAS.
- V. G. Vaidyanathan and B. U. Nair, J. Inorg. Biochem., 2003, 95, 334 CrossRef CAS.
- Y. Sun, S. N. Collins and L. E. Joyce, Inorg. Chem., 2010, 49, 4257 CrossRef CAS.
- V. G. Vaidyanathan and B. U. Nair, Dalton Trans., 2005, 2842 RSC.
- V. Uma, A. Castineiras and B. U. Nair, Polyhedron, 2007, 26, 3008 CrossRef CAS.
- V. G. Vaidyanathan and B. U. Nair, Eur. J. Inorg. Chem., 2003,(19), 3633 CrossRef CAS.
- R. Indumathy, T. Weyhermuller and B. U. Nair, Dalton Trans., 2010, 39, 2087 RSC.
- J. K. Barton, A. Danishefsky and J. Goldberg, J. Am. Chem. Soc., 1984, 106, 2172 CrossRef CAS.
- J. M. Kelly, M. J. Murphy, D. J. Mc Conell and C. Oh Uigin, Nucleic Acids Res., 1985, 13, 167 CrossRef CAS.
- R. Hartmann, A. Robert, V. Duarte, B. K. Keppler and B. Meunier, JBIC, J. Biol. Inorg. Chem., 1997, 2, 427 CrossRef.
- S. R. Rajski and R. M. Williams, Chem. Rev., 1998, 98, 2723 CrossRef CAS.
- M. V. Keek and S. J. Lippard, J. Am. Chem. Soc., 1992, 114, 3386 CrossRef.
- J. R. J. Sorenson, in Handbook on Metal–Ligand Interactions in Biological Fluids, Bioinorganic Medicine, ed G. Berthon, New York, 1995, pp. 1128–1139 Search PubMed.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051 CrossRef CAS.
- A. M. Badawi, M. A. S. Mohamed, M. Z. Mohamed and M. M. Khowdairy, J. Cancer Res., 2007, 3, 198 CAS.
- S. Bhattacharya and S. S. Mandal, Biochim. Biophys. Acta, Biomembr., 1997, 1323, 29 CrossRef CAS.
- Y. Ni, D. Li and S. Kokot, Anal. Biochem., 2006, 352, 231 CrossRef CAS.
- D. C. Carter, B. Chang, J. X. Ho, K. Keeling and Z. Krishnasami, Eur. J. Biochem., 1994, 226, 1049 CrossRef CAS.
- Y. J. Hu, Y. Liu, W. Jiang, R. M. Zhao and S. S. Qu, J. Photochem. Photobiol., B, 2005, 80, 235 CrossRef CAS.
- J.N. Tian, J.Q. Liu, X. Tian, Z.D. Hu and X.G. Chen, J. Mol. Struct., 2004, 691, 197 CrossRef CAS.
- J. Seetharamappa and B. P. Kamat, Chem. Pharm. Bull., 2004, 52, 1053 CrossRef CAS.
- H. X. Zhang, X. Huang, P. Mei and S. Gao, J. Solution Chem., 2008, 37, 631 CrossRef CAS.
- J. Yang, Z. H. Jing, J. J. Jie and P. Guo, J. Mol. Struct., 2009, 920, 227 CrossRef CAS.
- L. F. Wang, Y. Zhu, Z. Y. Yang, J. G. Wu and Q. Wang, Polyhedron, 1991, 10, 2477 CrossRef CAS.
- S. Y. Yu, S. X. Wang, Q. H. Luo, L. F. Wang and R. P. Zhou, Polyhedron, 1993, 12, 1093 CrossRef CAS.
- S. Padhy and G. B. Kauffman, Coord. Chem. Rev., 1985, 63, 127 CrossRef.
- J. G. Wu, R. W Deng and Z. N. Chen, Transition Met. Chem., 1993, 18, 23 CrossRef CAS.
- S. Chandra and K. K. Sharma, Transition Met. Chem., 1984, 9, 1 CrossRef CAS.
- Z. Y. Yang, R. D. Yang, Q. Li and F. S. Li, Synth. React. Inorg. Met.-Org. Chem., 1999, 29, 205 CrossRef CAS.
- Z. Y. Yang, Synth. React. Inorg. Met.-Org. Chem., 2000, 30, 1265 CrossRef CAS.
- P. Sathyadevi, P. Krishnamoorthy, R. R. Butorac, A. H. Cowley, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2011, 40, 9690 RSC.
- S. Banerjee, A. Ray, S. Sen, S. Mitra, D. L. Hughes, R. J. Butcher, S. R. Batten and D. R. Turner, Inorg. Chim. Acta, 2008, 361, 2692 CrossRef CAS.
- K. Chichak, U. Jacquenard and N. R. Branda, Eur. J. Inorg. Chem., 2002, 357 CrossRef CAS.
- T. Ghosh, B. Mondal, T. Ghosh, M. Sutradhar, G. Mukherjee and M. G. B. Drew, Inorg. Chim. Acta, 2007, 360, 1753 CrossRef CAS.
- M. Kandaz, I. Yilmaz, S. Keskin and A. Koca, Polyhedron, 2002, 21, 825 CrossRef CAS.
- P. Krishnamoorthy, P. Sathyadevi, A. H. Cowley, R. R. Butorac and N. Dharmaraj, Eur. J. Med. Chem., 2011, 46, 3376 CrossRef CAS.
- T. R. Li, Z. Y. Yang, B. D. Wang and D. D. Qin, Eur. J. Med. Chem., 2008, 43, 1688 CrossRef CAS.
- Y. Xiong, X. F. He, X. H. Zou, J. Z. Wu, X. M. Chen, L. N. Ji, R. H. Li, J. Y. Zhou and K. B. Yu, Dalton Trans., 2003, 114 Search PubMed.
- B. Y. Wu, L. H. Gao, Z. M. Duan and K. Z. Wang, J. Inorg. Biochem., 2005, 99, 1685 CrossRef CAS.
- M. Bhattacharyya, U. Chaudhuri and R. K. Poddar, Biochem. Biophys. Res. Commun., 1990, 167, 1146 CrossRef CAS.
- D. Senthil Raja, G. Paramaguru, N. S. P. Bhuvanesh, J. H. Reibenspies, R. Renganathan and K. Natarajan, Dalton Trans., 2011, 40, 4548 RSC.
- J. R. Lakowicz, Fluorescence Quenching: Theory and Applications. Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 1999, 53–127 Search PubMed.
- X. Z. Feng, Z. Yang, L. J. Wang and C. Bai, Talanta, 1998, 47, 1223 CrossRef CAS.
- Y. Z. Hu, Y. Liu, Z. B. Pi and S. S. Qu, Bioorg. Med. Chem., 2005, 13, 6609 CrossRef CAS.
- J. N. Miller, Proc. Anal. Div. Chem. Soc., 1979, 16, 203 CAS.
- M. Blagosklonny and S. EI-Diery, Int. J. Cancer, 1996, 67, 386 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, D65, 148 CrossRef CAS.
- L. M. Venanzi, J. Chem. Soc., 1958, 719 RSC.
- G. N. Rao, C. Janardhana, K. Pasupathy and P. Mahesh Kumar, Ind. J. Chem., 2000, 39B, 151 CAS.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392 CrossRef CAS.
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forrow and J. A. Hartley, Biochemistry, 1993, 32, 4237 CrossRef CAS.
- J. Bernadou, G. Pratviel, F. Bennis, M. Girardet and B. Meunier, Biochemistry, 1989, 28, 7268 CrossRef CAS.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231 CrossRef CAS.
- C. Beauchamp and I. Fridovich, Anal. Biochem., 1971, 44, 276 CrossRef CAS.
- T. Nash, Biochem. J., 1953, 55, 416 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electronic absorption spectra of binding of ligand 1 with DNA (Fig. S1†), Emission spectra of DNA- EB in the presence of ligand 1 (Fig. S2†), Emission spectra of binding of ligand 1 with BSA (Fig. S3†), Absorption spectra of binding of ligand 1 and complex 3 with BSA (Fig. S4†), Synchronous spectra (Δλ = 15 and 60 nm) of binding of ligand 1 with BSA (Fig. S5†). CCDC reference numbers 751230 and 896151 for complexes 2 and 3. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20597a |
| This journal is © The Royal Society of Chemistry 2012 |