KF/alumina catalyzed regioselective benzylation and benzoylation using solvent-free grind-stone chemistry†
Shreyans K.
Jain
a,
Samdarshi
Meena
a,
Baljinder
Singh
a,
Jaideep B.
Bharate
b,
Prashant
Joshi
b,
Varun P.
Singh
b,
Ram A.
Vishwakarma
*ab and
Sandip B.
Bharate
*b
aNatural Products Chemistry Division, Indian Institute of Integrative Medicine (CSIR), Canal Road, Jammu-180001, India
bMedicinal Chemistry Division, Indian Institute of Integrative Medicine (CSIR), Canal Road, Jammu-180001, India. E-mail: sbharate@iiim.ac.in; ram@iiim.ac.in; Fax: +91-191-2569333; Tel: +91-191-2569111
First published on 15th August 2012
Abstract
Potassium fluoride-impregnated on alumina catalyzes solvent-free regioselective O-benzylation, benzoylation and cinnamylation of phenols. Reaction proceeds simply by triturating together equivalent amounts of phenol and corresponding halide in the presence of 5 mol% of KF/alumina for 5–20 min with a mortar and pestle, without need for any additive such as phase-transfer catalyst or solvent. Key features of the protocol include its efficiency also for solid–solid precursors and regioselectivity for phenolic hydroxyls versus alcoholic hydroxyls. Utility of the protocol for N- and S-benzylation has also been explored. Products were obtained in excellent yields and the catalyst can be easily recycled several times without significant loss of activity.
Most of the existing processes in organic synthesis involve toxic and volatile organic solvents as reaction media which are environmentally unacceptable from a green chemistry viewpoint. Water or ionic liquids have been widely used as green solvents; however, “solvent-free” reaction conditions makes synthesis simpler, saves energy and prevents solvent waste, hazards, and toxicity. Together with “solvent-free” synthesis, heterogeneous catalysis1 have been applied as a powerful technique to make the process efficient and economical.2 The concept of “grind-stone chemistry”,3,4 which involves grinding solids together, has gained a tremendous amount of importance in organic chemistry5 as evidenced by its utility for a variety of organic transformations, for example, the Biginelli reaction,4 one-pot synthesis of spiro-indoline triones,6 Cannizzaro reaction,7 Aldol condensation,8 Claisen–Schmidt reaction9etc.
Potassium fluoride impregnated over aluminum oxide (KF/alumina) has been recognized as a remarkably useful heterogeneous surface to promote many base-catalyzed organic transformations such as O-alkylation of alcohols and phenols under solution phase,10 Michael addition,11 aldol condensation,12 Darzen's condensation13etc. Besides these classical organic reactions, KF/alumina has been successfully utilized as a heterogeneous basic surface in several metal-catalyzed coupling reactions such as Pd-catalyzed C–C bond-forming reactions – Heck, Stille, Suzuki, Trost–Tsuji reaction,14 C–N bond-forming Buchwald–Hartwig reactions and C–O bond-forming Baylis–Hillman type reactions.15
O-Alkylation/benzylation/benzoylation of phenols is one of the widest applied and routinely used organic reactions. Plenty of methods have been reported for this reaction involving heating reagents or submitting them to microwave irradiation,16 in the presence of a suitable base, in solvents such as DMF, acetone and dichloromethane.17 Many research groups have tried to improve the yield of this reaction by using various catalysts such as basic zeolites,18 oxides,19 DABCO,20etc. Jing and coworkers20 reported solvent-free O-alkylation of phenols by DABCO/K2CO3 using grinding, however the catalytic system was homogeneous. There also exist few reports21 on the use of KF/alumina for this reaction but these protocols involves use of a phase-transfer catalyst and many involve organic solvents. Ando and coworkers10 reported O-alkylation of phenols and alcohols using KF/alumina, however acetonitrile was used as a reaction medium and reaction times were up to 42 h. Further, selective O-benzylation/benzoylation of phenolic hydroxyls over aliphatic hydroxyls has never been reported under solvent-free conditions. Herein, we report for the first time, recyclable KF/alumina catalyzed “solvent-free” regioselective O-benzylation/benzoylation of phenols in shorter reaction times (5–20 min) without use of any additive such as a phase-transfer catalyst.
Rohitukine (1) is a naturally occurring anticancer and immunomodulatory chromone alkaloid isolated from Dysoxylum binectariferum and has also led to discovery of the clinically used anticancer drug flavopiridol.22,23 As a part of our natural product based drug discovery program,22,24 and development of synthetic methodologies for application in natural product chemistry,25 herein we have developed method for rapid and efficient regioselective O-benzylation/benzoylation of chromone alkaloid rohitukine (1) under solvent-free conditions (Scheme 1). Utility of a developed protocol for variety of aromatic and heteroaromatic substrates has also been investigated.
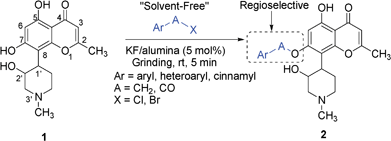 | ||
| Scheme 1 KF/Alumina catalyzed regioselective O-benzylation/benzoylation of chromone alkaloid under solvent-free conditions. | ||
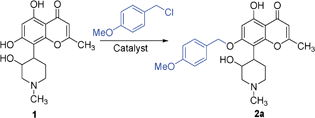
KF/alumina catalyst was prepared by impregnating potassium fluoride on basic alumina using a reported method.26 Reaction of rohitukine (1) with 4-methoxybenzyl bromide was chosen as a model reaction to regioselectively O-benzylate/benzoylate the phenolic hydroxyl. A set of reaction conditions varying in the nature of catalyst, amount of catalyst, reaction medium, temperature and reaction time were investigated as described in Table 1.
| Entry | Catalyst (mol%) | Solvent | Reaction conditions | Time (min) | Yieldb (%) |
|---|---|---|---|---|---|
| a Rohitukine (1, 1 mmol) and PMB–Cl (1 mmol) in presence/absence of catalyst/solvent was treated under mentioned reaction conditions/time; b isolated yield after column chromatography; c mixture of multiple products were obtained which were difficult to isolate; d TBAB (1 mol%) was used as phase transfer catalyst; e starting material was not completely consumed and mixture of multiple products were formed. | |||||
| 1. | None | acetone | reflux | 30 | 0 |
| 2. | None | DMF | reflux | 30 | 0 |
| 3. | K2CO3 (100) | acetone | rt | 60 | 0 |
| 4. | K2CO3 (100) | acetone | reflux | 60 | 0 |
| 5. | NaH (100) | DMF | reflux | 10 | —c |
| 6. | NaH (100) | DMF | reflux | 60 | —c |
| 7. | KF (50) | none | grinding | 10 | 0 |
| 8. | KF (50) | none | grinding | 10 | 0 |
| 9. | KF (50) | none | grinding | 10 | 0 |
| 10. | Alumina (50) | none | grinding | 10 | 0 |
| 11. | Alumina (50) | none | grinding | 20 | 0 |
| 12. | Alumina (50) | none | grinding | 30 | 0 |
| 13. | KF/Al2O3 (50) | none | grinding | 5 | 85 |
| 14. | KF/Al2O3 (10) | none | grinding | 5 | 70 |
| 15. | KF/Al2O3 (5) | none | grinding | 5 | 70 |
| 16. | KF/Al2O3 (5) + PTCd | none | grinding | 2 | 90 |
| 17. | KF/Al2O3 (5) | DMF | rt | 30 | 20e |
| 18. | KF/Al2O3 (5) | acetone | rt | 30 | 0 |
| 19. | KF/Al2O3 (5) | DMF | reflux | 30 | 30e |
| 20. | KF/Al2O3 (5) | acetone | reflux | 30 | 0 |
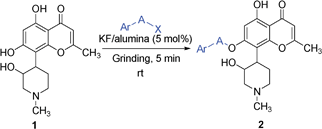
As expected, catalyst-free conditions were not successful (entries 1 and 2). Traditional alkylation conditions such as K2CO3/acetone, NaH/DMF were also found to be unsatisfactory (entries 3–6). In the case of K2CO3/acetone method, no conversion of starting material was observed even after reflux for several hours. The reason for this poor reactivity was thought to be due to poor solubility of 1 in acetone. In contrast, the NaH/DMF method produced multiple products, which were difficult to isolate (entries 5 and 6). Grinding together 1 mmol of 1 with 1 mmol of 4-methoxybenzyl chloride in the presence of 50 mol% of KF/alumina for 5 min resulted in formation of single mono-alkylated product 2a in 85% yield (entry 13). The chemical structure of 1 comprises three hydroxyls, two of which are aromatic (positions 5 and 7) and one is aliphatic (position 2′). Amongst the hydroxyl groups, 5-OH is H-bonded with the 4-carbonyl oxygen and therefore was not favored for benzylation or benzoylation. However, amongst the other two hydroxyls 7-OH (phenolic) and 2′-OH, 7-OH was benzylated selectively. Addition of 1 mol% of tetrabutylammonium bromide (TBAB), a phase transfer catalyst, led to completion of the reaction in 2 min with 90% yield of the desired product (Table 1, entry 16). Reaction did not proceed at all in presence of either only KF or only alumina, even after triturating for longer duration (Table 1, entries 7–12). Reduction in catalyst loading to as low as 5 mol% also produced good yield (entry 15). Although addition of TBAB led to reduction of reaction time, in order to simplify the product isolation and recycle KF/alumina catalyst, use of only KF/alumina (5 mol%; entry 15 in Table 1) was chosen for further studies. Further, in order to compare the developed “solvent-free” protocol with the “reaction in presence of solvents”, a set of experiments in the presence of DMF or acetone at room temperature and at reflux conditions were performed. Reaction of 1 with PMB–Cl in the presence of 5 mol% of KF/alumina in acetone did not proceed at all (entries 18 and 20), whereas, in the presence of DMF, the desired product 2a was formed, but only in <30% yield. These results clearly indicate advantages of optimized “solvent-free” reaction condition (entry 15) over “reactions in presence of solvents” (entries 17–20).
Next, we investigated scope of this reaction for different benzyl/heteroaryl, cinnamyl halides under optimized reaction conditions. Results are summarized in Table 2.
| Entry | Ar–A–X | Product | Yieldb (%) |
|---|---|---|---|
| a Rohitukine (1, 1 mmol), aryl halide (1 mmol), KF/alumina (5 mol%) were triturated in mortar/pestle for 5 min; b isolated yield after chromatography. | |||
| 1. | Ph(4-OMe)–CH2–Cl | 2a | 70 |
| 2. | Ph–CH2–Br | 2b | 74 |
| 3. | Ph(4-Br)–CH2–Cl | 2c | 78 |
| 4. | Ph(4-NO2)–CH2Cl | 2d | 84 |
| 5. | Ph(2-Br)CH2Cl | 2e | 75 |
| 6. | Ph–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2Cl CH–CH2Cl |
2f | 70 |
| 7. | Benzimidazol-2-yl–CH2Cl | 2g | 72 |
| 8. | Fur-2-yl–CO–Cl | 2h | 88 |
| 9. | Ph(2-Me)–CO–Cl | 2i | 90 |
In general, benzyl bromides are preferred over benzyl chlorides. Benzyl halides substituted with electron donating groups (entries 3, 5 and 9) as well as electron withdrawing groups (entries 1 and 4) participated well in this reaction, producing excellent regioselective O-benzylation/benzoylation of phenolic hydroxyl. However, the method was not suitable for simple O-alkylation such as methylation and isopropylation (not shown in the table). Since the present reaction does not involve use of any solvent, we further investigated the efficiency of protocol if both starting materials are in solid form. To our surprise and further as a proof for green protocol, reaction of rohitukine (1) with 2-(chloromethyl)-1H-benzo[d]imidazole (solid powder) in presence of 5 mol% KF/alumina produced corresponding product 2g in 72% yield in 5 min under optimized reaction conditions (Table 2, entry 7).
The catalyst was reused repeatedly to prove its heterogeneous nature and its recyclability. Grinding together rohitukine (1), PMB-chloride and 5 mol% KF/alumina for 5 min produced 7-O-benzylated product 2a in 92%, 84% and 74% over three cycles, respectively. Thereafter, yields were significantly lowered such that for the fourth and fifth successive usages it gave only 50% and 36% yields, respectively. Probable reason for this may be twofold: (i) apparently, the amount of catalyst played an important role in the yield of products i.e. higher the amount of catalyst, the higher is the yield of products. During recovery, a loss of about 10–15% of catalyst was observed in every successive cycle and that would be one of the reasons for the lower in yield in every successive cycle; (ii) another plausible reason would be that the active sites on the surface of the catalyst might have slowly poisoned due to interaction with products. However, recyclability results clearly indicate heterogeneous nature of the catalyst with excellent recyclability up to three cycles.
Encouraging results of regioselective solvent-free O-benzylation/benzoylation of phenolic chromone alkaloid, prompted us to further investigate utility of this protocol for O/N/S-benzylation/benzoylation of simple substituted phenols or heterocycles. As shown in Table 3, a variety of phenolic substrates substituted with electron withdrawing groups (e.g. OMe, CHO; entry 6) as well as electron donating groups (e.g. CH3 and other alkyl chains, halogens; entries 2–5) participated well in this reaction and produced excellent yields of corresponding O-substituted products.

| Entry | R | R′ | 4 | Time (min) | Yieldb (%) | Ref. |
|---|---|---|---|---|---|---|
| a Phenol (3, 1 mmol), aryl halide (1 mmol), KF/alumina (5 mol%) were triturated for 5–15 min; b the isolated yield after chromatography; c new compounds. | ||||||
| 1. | H | Ph | 4a | 15 | 92 | 27 |
| 2. | 2-Cl | Ph | 4b | 15 | 72 | 28 |
| 3. | 4-Me | Ph | 4c | 15 | 50 | 29 |
| 4. | 4-CH(CH3)Et | Ph | 4d | 15 | 92 | Nc |
| 5. | 4-CH(CH3)2, 3-Me | Ph | 4e | 15 | 80 | Nc |
| 6. | 2-OMe,4-CHO | Ph | 4f | 15 | 80 | 30 |
| 7. | H | Ph(4-OMe) | 4g | 10 | 85 | 31 |
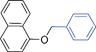
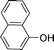
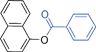
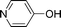
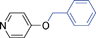
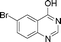
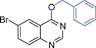
Bicyclics such as 1-naphthol produced corresponding O-benzyl and O-benzoyl substituted products 6a and 6b in good yields (Table 4, entries 1 and 2). Heteroaryl hydroxyls, for example, pyridine-4-ol (entry 3) and 6-bromoquinazolin-4-ol (entry 4) also underwent O-benzylation using optimized reaction conditions producing corresponding products 6c and 6d in 92 and 78% yield, respectively. Next we explored N- and S-benzylation of aromatics and heteroaromatics (Table 4).
| Entry | 5 | Product | 6 | Time (min) | Yieldb (%) | Ref. |
|---|---|---|---|---|---|---|
| a Hydroxy aromatics and heteroaromatics/aniline/NH-heterocycle/thiophenol (1 mmol), benzyl or benzoyl halide (1 mmol), KF/alumina (5 mol%) were triturated for 10–15 min; b the isolated yield after chromatography; c new compounds. | ||||||
| 1. |
|
|
6a | 15 | 92 | 32 |
| 2. |
|
|
6b | 15 | 80 | 33 |
| 3. |
|
|
6c | 15 | 92 | 34 |
| 4. |
|
|
6d | 10 | 78 | Nc |
| 5. |
|
|
6e | 15 | 45 | 35 |
| 6. |
|
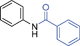
|
6f | 15 | 92 | 36 |
| 7. |
|
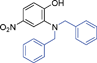
|
6g | 15 | 40 | Nc |
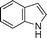
| 8. |
|
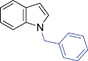
|
6h | 10 | 90 | 37 |
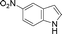
| 9. |
|
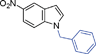
|
6i | 10 | 80 | 38 |
| 10. |
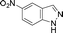
|
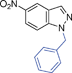
|
6ja | 10 | 45 | 39 |
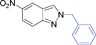
|
6jb | 30 | Nc | |||
| 11. |
|
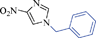
|
6k | 15 | 70 | 40 |
| 12. |
|
|
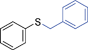
6l | 10 | 85 | 41 |
| 13. |
|
|
6m | 15 | 80 | 42 |
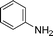
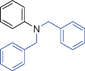
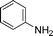
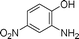
Grinding together aniline with benzyl bromide/benzoyl chloride in the presence of 5 mol% of KF/alumina under optimized reaction conditions produced the N,N-dibenzylated product 6e35 and N-benzoyl product 6f36 in 45% and 92% yield, respectively (entry 5 and 6). Interestingly, when the substrate 4-nitro-2-aminophenol (which contains OH as well as NH2 group) was reacted with benzyl bromide under the optimized reaction conditions, N-alkylation was favored, yielding the N,N-dibenzylated product 6g in 40% yield (entry 7). We could get up to 75% yield of 6g when 2.5 equiv of benzyl bromide was used. Next we investigated the scope of N-benzylation for heterocycles. N-Benzylation of indole (entry 8) and substituted indole (entry 9) worked well producing ≥80% yield of products 6h and 6i. Reaction of another heterocycle 5-nitro-1H-indazole with benzyl bromide produced two products 6ja and 6jb (a pair of regioisomers) in 45 and 30% yields, respectively (entry 10).43 Since indazole exists in the form of pair of resonance structures 5ja and 5jb, we observed formation of two benzylated products, as depicted in Fig. 1. The pair of regioisomers were distinguished from each other with the help of HMBC correlations and further by comparison with literature 1H NMR values of one of the isomers, 6ja.39
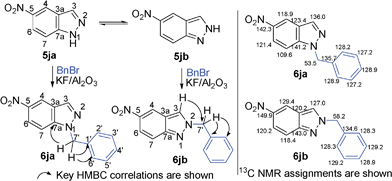 | ||
| Fig. 1 Formation of a pair of regioisomers 6ja/6jb during benzylation of 5-nitro-indazole (5j). Key HMBC correlations and 13C NMR assignments for structures 6ja/6jb are also shown. | ||
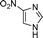
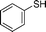
Another NH-heterocycle viz. 4-nitro-1H-imidazole was reacted with benzyl bromide under optimized reaction conditions; however, unlike indazole, only one product 6k was formed (entry 11). Similarly thiophenols also participated well in this reaction producing corresponding thio-ethers 6l and 6m in ≥80% yield (entries 12 and 13). All products were characterized by melting point, MS and 1H NMR data and their physical data were similar to those reported in the literature27–32,34,35,44 (spectral data is provided in the ESI†).
In conclusion, we have reported a faster and simpler method for selective O-benzylation/O-benzoylation of phenols, other aromatics and heteroaromatics in comparison with previous methods. The method is selective for phenols versus alcohols. Most importantly, the developed protocol is consistent with a green chemistry approach since no solvent is needed and it involves use of a recyclable heterogeneous catalyst. The catalyst used is easy to prepare from inexpensive chemicals that are commonly found in organic chemistry laboratories, thus this procedure can be easily adopted in process chemistry research. Apart from its green chemistry aspects, this method is convenient and time-saving in comparison to routine reflux reactions. Routine methods of O-alkylation and acylation such as NaH/DMF, K2CO3/acetone were found to be inefficient for O-benzylation/benzoylation of rohitukine (1). However, KF-alumina catalyzed grind-stone chemistry led to completion of the reaction within 5 min producing solely mono-benzylated/mono-benzoylated products in excellent yields. Thus, results obtained clearly indicate KF/alumina catalyzed solvent-free regioselective O-benzylation and O-benzoylation of rohitukine (1). Results obtained herein further guarantees utility of this protocol for benzylation of structurally diverse phenols over alcohols. Furthermore, the developed protocol also showed its utility for N- and S-benzylation.
Acknowledgements
SKJ and PJ are thankful to CSIR and BS is thankful to ICMR for award of Senior Research Fellowship. SM is thankful to UGC for Junior Research Fellowship. Authors thank analytical department, IIIM for analytical support. IIIM publication number IIIM/1479/2012.References
- (a) M. A. Chari, G. Karthikeyan, A. Pandurangan, T. S. Naidu, B. Sathyaseelan, S. M. J. Zaidi and A. Vinu, Tetrahedron Lett., 2010, 51, 2629–2632 Search PubMed; (b) R. R. Yadav, N. Battini, R. Mudududdla, J. B. Bharate, N. Muparappu, S. B. Bharate and R. A. Vishwakarma, Tetrahedron Lett., 2012, 53, 2222–2225 Search PubMed.
- K. Tanaka, Solvent-Free Organic Synthesis, Wiley-VCH, GmbH and KgaA, Weinheim, Germany, 2004 Search PubMed.
- (a) F. Toda and K. Okuda, J. Chem. Soc., Chem. Commun., 1991, 1212–1214 RSC; (b) F. Toda, H. Takumi and M. Akehi, J. Chem. Soc., Chem. Commun., 1990, 1270–1271 RSC; (c) F. Toda, K. Tanaka and A. Sekikawa, J. Chem. Soc., Chem. Commun., 1987, 279–280 RSC.
- A. K. Bose, S. Pednekar, S. N. Ganguly, G. Chakraborty and M. S. Manhas, Tetrahedron Lett., 2004, 45, 8351–8353 CrossRef CAS.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS.
- R. Ghahremanzadeh, S. Ahadi, G. I. Shakibaei and A. Bazgir, Tetrahedron Lett., 2010, 51, 499–502 CrossRef CAS.
- A. F. M. M. Rahman and A. A. Kadi, Arabian J. Chem., 2012 DOI:10.1016/j.arabjc.2012.1002.1010.
- C. L. Raston and J. L. Scott, Green Chem., 2000, 2, 49–52 RSC.
- A. F. Rahman, R. Ali, Y. Jahng and A. A. Kadi, Molecules, 2012, 17, 571–583 Search PubMed.
- T. Ando, J. Yamawaki, T. Kawate, S. Sumi and T. Hanafusa, Bull. Chem. Soc. Jpn., 1982, 55, 2504–2507 CAS.
- T. Ando, S. J. Brown, J. H. Clark, D. G. Cork, T. Hanafusa, J. Ichihara, J. M. Miller and M. S. Robertson, J. Chem. Soc., Perkin Trans. 2, 1986, 1133–1139 RSC.
- W.-x. Lu, C.-g. Yan and R. Yao, Synth. Commun., 1996, 26, 3719–3723 Search PubMed.
- J. Yamawaki, T. Kawate, T. Ando and T. Hanafusa, Bull. Chem. Soc. Jpn., 1983, 56, 1885–1887 CAS.
- F. Bernhardt, R. Trotzki, T. Szuppa and A. S. B. Ondruschka, Beilstein J. Org. Chem., 2010, 6, 1–9 Search PubMed.
- B. Basu, P. Das and S. Das, Curr. Org. Chem., 2008, 12, 141–158 CrossRef CAS.
- G. Keglevich, E. Bálint, É. Karsai, A. Grün, M. Bálint and I. Greiner, Tetrahedron Lett., 2008, 49, 5039–5042 CrossRef CAS.
- (a) P. G. M. Wuts and T. W. Greene, Greene's protective groups in organic synthesis: Protection for phenol and catechols, John Wiley and Sons, 2007 Search PubMed; (b) S. B. Bharate, S. I. Khan, N. A. M. Yunus, S. K. Chauthe, M. R. Jacob, B. L. Tekwani, I. A. Khan and I. P. Singh, Bioorg. Med. Chem., 2007, 15, 87–96 CrossRef CAS.
- S. C. Lee, S. W. Lee, K. S. Kim, T. J. Lee, D. H. Kim and J. C. Kim, Catal. Today, 1998, 44, 253–258 CrossRef CAS.
- V. V. Rao, K. V. R. Chary, V. Durgakumari and S. Narayanan, Appl. Catal., 1990, 61, 89–97 Search PubMed.
- X. Bu, H. Jing, L. Wang, T. Chang, L. Jin and Y. Liang, J. Mol. Catal. A: Chem., 2006, 259, 121–124 CrossRef CAS.
- (a) B. E. Benjamin, Tetrahedron, 2002, 58, 9301–9320 CrossRef; (b) B. Basu and B. Mandal, Curr. Org. Chem., 2011, 15, 3870–3893 Search PubMed.
- S. K. Jain, S. B. Bharate and R. A. Vishwakarma, Mini-Rev. Med. Chem., 2012, 12, 632–649 Search PubMed.
- (a) A. D. Harmon, U. Weiss and J. V. Silverton, Tetrahedron Lett., 1979, 8, 721–724 Search PubMed; (b) R. G. Naik, S. L. Kattige, S. V. Bhat, B. Alreja, N. J. de Souza and R. H. Rupp, Tetrahedron, 1988, 44, 2081–2086 CrossRef CAS.
- (a) S. B. Bharate, S. Manda, P. Joshi, B. Singh and R. A. Vishwakarma, Med. Chem. Commun., 2012, 3, 1098–1103 RSC; (b) S. B. Bharate, S. Manda, N. Mupparapu, N. Battini and R. A. Vishwakarma, Mini Rev. Med. Chem., 2012, 12, 650–664 CAS; (c) S. B. Bharate, R. R. Yadav, S. Battula and R. A. Vishwakarma, Mini Rev. Med. Chem., 2012, 12, 618–631 Search PubMed.
- S. B. Bharate, R. Mudududdla, J. B. Bharate, N. Battini, S. Battula, R. R. Yadav, B. Singh and R. A. Vishwakarma, Org. Biomol. Chem., 2012, 10, 5143–5150 RSC.
- I. P. Singh, S. K. Jain, A. Kaur, S. Singh, R. Kumar, P. Garg, S. S. Sharma and S. K. Arora, Eur. J. Med. Chem., 2010, 45, 3439–3445 Search PubMed.
- S. G. Powell and R. Adams, J. Am. Chem. Soc., 1920, 42, 646–658 Search PubMed.
- J. Forrester, R. V. H. Jones, L. Newton and P. N. Preston, Tetrahedron, 2001, 57, 2871–2884 Search PubMed.
- K. Okuma, O. Sakai and K. Shioji, Bull. Chem. Soc. Jpn., 2003, 76, 1675–1676 CrossRef CAS.
- D. A. Learmonth, M. A. Vieira-Coelho, J. Benes, P. C. Alves, N. Borges, A. P. Freitas and P. Soares-da-Silva, J. Med. Chem., 2002, 45, 685–695 Search PubMed.
- M. Arisawa, Y. Igarashi, H. Kobayashi, T. Yamada, K. Bando, T. Ichikawa and M. Yamaguchi, Tetrahedron, 2011, 67, 7846–7859 Search PubMed.
- M. I. F. Mohamed and S. Arunadevi, J. Chem. Pharm. Res., 2010, 2, 296–300 Search PubMed.
- A. K. Chakraborti and S. V. Chankeshwara, J. Org. Chem., 2009, 74, 1367–1370 CrossRef CAS.
- E. Shaw, J. Am. Chem. Soc., 1949, 71, 67–70 CrossRef CAS.
- F. M. Moghaddam, S. M. DokhtTaimoory, H. Ismaili and G. R. Bardajee, Synth. Commun., 2006, 36, 3599–3607 CrossRef CAS.
- H. Charville, D. A. Jackson, G. Hodges, A. Whiting and M. R. Wilson, Eur. J. Org. Chem., 2011, 5981–5990 CrossRef CAS.
- T. Kiguchi, N. Kuninobu, Y. Takahashi, Y. Yoshida, T. Naito and I. Ninomiya, Synthesis, 1989, 10, 778–781 Search PubMed.
- V. Gasparotto, I. Castagliuolo and M. G. Ferlin, J. Med. Chem., 2007, 50, 5509–5513 Search PubMed.
- R. C. Wheeler, E. Baxter, I. B. Campbell and S. J. F. Macdonald, Org. Process Res. Dev., 2011, 15, 565–569 Search PubMed.
- M. Mgkosza and Z. Owczarczyk, J. Org. Chem., 1989, 54, 5094–5100 CrossRef CAS.
- L. D. S. Yadav and R. Kapoor, Synth. Commun., 2011, 41, 100–112 Search PubMed.
- Q. Ding, B. Cao, J. Yuan, X. Liu and Y. Peng, Org. Biomol. Chem., 2011, 9, 748–751 RSC.
- Reaction between 5-nitro-1H-indazole with benzyl bromide produced two products 6ja and 6jb in 45 and 30% yields, respectively. These two products have close Rf values viz. 0.60 and 0.50 (EtOAc
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane – 9:1, triple run) and were separated by silica gel (#230–400) column chromatography using EtOAc:hexane as mobile phase. Isolated isomers were characterized by 1H, 13C NMR, DEPT-135 and 2D-NMR (HMBC, HSQC) data.
:hexane – 9:1, triple run) and were separated by silica gel (#230–400) column chromatography using EtOAc:hexane as mobile phase. Isolated isomers were characterized by 1H, 13C NMR, DEPT-135 and 2D-NMR (HMBC, HSQC) data. - H.-L. Dai, W.-Q. Liu, H. Xu, L.-M. Yang, M. Lv and Y.-T. Zheng, Chem. Pharm. Bull., 2009, 57, 84–86 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data scans. See DOI: 10.1039/c2ra21154h |
| This journal is © The Royal Society of Chemistry 2012 |