Facile synthesis of microporous carbon spheres by selective pyrolysis†
Haemin
Yoo
and
Jun Hyuk
Moon
*
Department of Chemical and Biomolecular Engineering, Sogang University, 1 Sinsu-dong, Mapo-gu, Seoul, Republic of Korea. E-mail: junhyuk@sogang.ac.kr; Fax: +82 2 703 8971; Tel: +82 2 705 8921
First published on 10th August 2012
Abstract
Microporous carbon particles were obtained by pyrolyzing crosslinked poly(styrene-co-methyl methacrylate) (PS-PMMA) copolymer spheres. The micropores were randomly dispersed throughout the matrix. The surface area and pore volume increased as the fraction of MMA was increased to 20 wt%, reaching 1135 m2 g−1 and 0.40 cm3 g−1, respectively.
Microporous carbon particles or materials (pore width <2 nm, IUPAC) are attractive for their utility in certain applications, including biochemical sensors, electrochemical energy storage, CO2 or hydrogen adsorption, and catalysis.1–5 Micropores were generated by capturing templates, such as zeolites,2,6 gel particles,7 colloidal particles,8,9 clays,10 and metal–organic frameworks,11,12 in combination with liquid or gas-phase carbon precursors. Zeolites have been widely used as templates because they include micropore networks with a wall thickness of <1 nm and well-defined monodisperse pore diameters.13 However, templating processes require multiple tedious steps, including the injection of carbon precursors into a template, polymerization of the carbon precursor, and the removal of the template. Template removal often distorts or reduces the long-range order endowed by the template due to volume shrinkage of the carbon precursor during pyrolysis and template removal.13,14 Moreover, poor infiltration of the precursor into the micropores of zeolites can cause unfilled adjacent micropores to collapse, thereby forming unwanted mesopores upon template removal.14–17
In the present study, we demonstrated a novel approach to the synthesis of microporous carbon spheres. The approach is facile, low-cost and suitable for mass production. Briefly, microporous carbon particles were obtained by pyrolyzing and carbonizing crosslinked poly(styrene-co-methyl methacrylate) (PS-PMMA) copolymer spheres. Micropores were formed via the selective decomposition of methyl methacrylate (MMA) groups during pyrolysis. Here, the effect of MMA groups on the formation of micropores was investigated in detail.
The route used to prepare microporous carbon spheres was illustrated in Fig. 1: PS-PMMA spheres were synthesized by emulsifier-free emulsion polymerization, styrenes were selectively post-crosslinked via a Friedel–Crafts reaction,18 and MMA moieties in the crosslinked PS-PMMA spheres were selectively pyrolyzed while the post-crosslinked PS converted to carbon. Specifically, the randomly distributed MMA groups within the copolymer sphere selectively decomposed, leaving behind micropores. Here, PS-PMMA spheres containing 10 wt% and 20 wt% methyl methacrylate (MMA) were prepared along with PS spheres for comparison. The PS-PMMA particles containing >30 wt% MMA formed collapsed aggregates during the post-crosslinking reaction step (not shown). These morphologies may have resulted from a lower cross-linking density in the presence of high concentrations of MMA, which induced the melting of particles by the solvent (CHCl3).
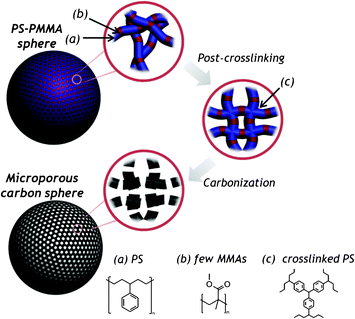 | ||
| Fig. 1 A schematic diagram showing the preparation of microporous carbon particles via selective pyrolysis of PS-PMMA spheres. | ||
The scanning electron microscopy (SEM) images of PS-PMMA particles (20 wt% MMA) and PS particles were shown in Fig. 2a and Fig. S1a (ESI†), respectively. Here, a Friedel–Crafts reaction was employed to achieve high crosslinking density of these particles, and thereby high conversion into carbon.19 The SEM images of PS-PMMA and PS particles after the post-crosslinking reaction and after pyrolysis at 700 °C are shown in Fig. 2b–c and Fig. S1b–c (ESI†), respectively. The results showed a volume shrinkage accompanying pyrolysis: the diameters of the PS-PMMA (20 wt% MMA) and PS particles were shrunk from 290 nm to 170 nm by 40% and from 310 nm to 200 nm by 36%, respectively.
 | ||
| Fig. 2 SEM images of (a) PS-PMMA, (b) post-crosslinked PS-PMMA, (c) carbonized PS-PMMA and (inset) TEM image of carbonized particles. | ||
FT-IR spectra were used to analyze the post-crosslinking reaction of styrenes and pyrolysis. In the Friedel–Crafts alkylation, nucleophilic aromatic substitution by the alkyl halides activated areas in the PS, and subsequent polyalkylation induced crosslinking between neighboring PS chains.19Fig. 3a shows that a strong peak at 700 cm−1 (out-of-plane C–H bending in the benzene ring) and a weak peak at 3025 cm−1 (C–H stretching in the benzene ring) largely decreased after the reaction, indicating the loss of hydrogen atoms due to the formation of a phenylmethane linkage between the benzene rings (i.e., crosslinking between the benzene rings). An intense peak at 1731 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) and a broad peak over the range 1260–1000 cm−1 (C–O stretching), attributed to the presence of MMA, remained unchanged during crosslinking. This result confirmed that the PS was selectively crosslinked whereas the MMA was not affected. The PS-PMMA pyrolyzed at 700 °C did not display such peaks, suggesting the conversion into pure carbon. Moreover, Raman spectrum was measured to characterize the microstructure of the carbon particles (see Fig. S2, ESI†). The intensity ratios of D/G were 0.76 for 700 °C and 0.94 for 900 °C. This result indicated the formation of amorphous carbon and an increase in the number of smaller graphite domains with increasing pyrolysis temperature.
O stretching) and a broad peak over the range 1260–1000 cm−1 (C–O stretching), attributed to the presence of MMA, remained unchanged during crosslinking. This result confirmed that the PS was selectively crosslinked whereas the MMA was not affected. The PS-PMMA pyrolyzed at 700 °C did not display such peaks, suggesting the conversion into pure carbon. Moreover, Raman spectrum was measured to characterize the microstructure of the carbon particles (see Fig. S2, ESI†). The intensity ratios of D/G were 0.76 for 700 °C and 0.94 for 900 °C. This result indicated the formation of amorphous carbon and an increase in the number of smaller graphite domains with increasing pyrolysis temperature.
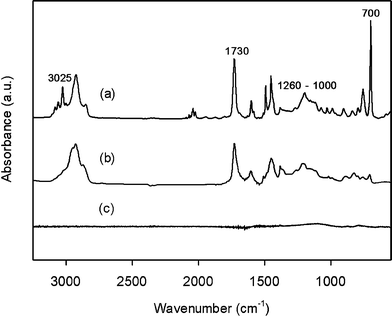 | ||
| Fig. 3 FT-IR spectra of (a) PS-PMMA particles, (b) post-crosslinked PS-PMMA particles, and (c) PS-PMMA after heat treatment at 700 °C. | ||
The compositional change of PS-PMMA spheres during pyrolysis was further investigated by comparing the weight loss of PS-PMMA to the one of PS particles. The thermogravimetric (TGA) analysis showed that the initial weight of the PS-PMMA decreased by 25 wt% at 700 °C, whereas the weight of the PS spheres decreased only by 5 wt% (Fig. S3, ESI†). This result confirmed that the MMA moieties selectively decomposed during the pyrolysis while the crosslinked PS was converted into carbon.
Meanwhile, it should be noted that, as seen in SEM analysis, PS-PMMA particles showed larger weight loss than PS particles, whereas all particles yielded comparable volume shrinkages. We therefore expected that air cavities were incorporated into the crosslinked PS matrix. Brunauer–Emmett–Teller (BET) nitrogen adsorption isotherms were measured to characterize the porous structure in the carbonized PS-PMMA particles. Specifically, we analyzed the isotherms of PS-PMMA particles with 10 wt% and 20 wt% MMA fraction, and also compared them to the isotherm of PS particles (Fig. 4). The isotherms displayed a large degree of nitrogen adsorption below P/P0 = 0.1, which was attributed to capillary filling of N2 into the uniform micropores. Moreover, as the MMA fraction increased, the adsorption volume (or micropore volume) increased significantly.20 The isotherm was used to calculate the corresponding surface areas, micropore volumes, and average pore diameters for the PS and PS-PMMA samples, as shown in Table 1. The presence of MMA largely enhanced the creation of micropores of around 1.6 nm. The BET surface area was increased up to 1135 m2 g−1 for 20 wt% MMA fraction and the micropore volume was 0.4 cm3 g−1. The surface area of the PS-PMMA particles was 5 times greater than that of the PS particles. Fig. 4b clearly shows a narrow size distribution of micropores. Moreover, it should be noted that the micropore diameter remained nearly unchanged as the MMA content in the PS-PMMA spheres increased. This suggested that the micropores were created uniformly throughout the carbonized PS-PMMA spheres, and furthermore the MMA was distributed randomly as a result of emulsion copolymerization of styrene and MMA. This could be explained by comparing the reactivity ratios of styrene and MMA during copolymerization. The reactivity ratio of the styrene or MMA during copolymerization was defined by the ratio of the reaction rate constants for the styrene–styrene or MMA–MMA polymerizations and the styrene–MMA polymerization.20 The reactivity ratios for the styrene or MMA during emulsion copolymerization were reported to be 0.44 and 0.46, respectively.21 Because the reactivity ratios were below 1 and were comparable, styrene and MMA were expected to form alternating copolymers rather than homopolymers.
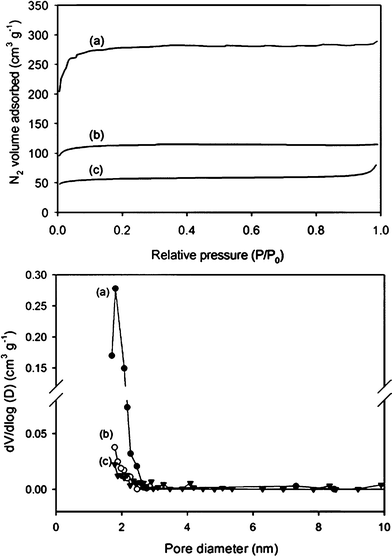 | ||
| Fig. 4 Nitrogen adsorption isotherms and pore size distribution (BJH method) curves for PS-PMMA particles containing (a) 20 wt% , (b) 10 wt% MMA and (c) the carbonized PS. | ||
| Sample | BET surface area (m2 g−1) | Micropore volume (cm3 g−1) | Avg. pore diameter (nm) |
|---|---|---|---|
| PS | 212 | 0.08 | 2.3 |
| PS-PMMA (MMA 10 wt%) | 450 | 0.16 | 1.6 |
| PS-PMMA (MMA 20 wt%) | 1135 | 0.40 | 1.6 |
The microporous carbon spheres are expected to be promising electrode materials for supercapacitors. In Fig. S4 (ESI†), the capacitive behaviours of carbonized PS and carbonized PS-PMMA spheres were compared by measuring the galvanostatic charge-discharge at a current density of 0.5 A g−1. The specific capacitances of PS-PMMA (20 wt% MMA) carbonized particles were estimated to be 50.5 F g−1, and it was about 10 times higher than the value of PS carbonized spheres (5.7 F g−1). The enhancement of specific capacitance was attributed to the large micropore volume and surface area of PS-PMMA carbonized spheres.
Conclusion
In conclusion, we employed PS-PMMA copolymer particles synthesized by emulsion copolymerization for one-step synthesis of microporous particles. Our novel approach to the generation of micropores involved the selective removal of MMA during pyrolysis of the crosslinked PS matrix. The selective decomposition of MMA and the subsequent pore generation was confirmed by the high weight loss observed for higher fractions of MMA, whereas the diameters of the spheres with higher fractions of MMA remained unchanged upon pyrolysis. BET isotherms showed a pore size of 1.6 nm with a narrow size distribution. The uniform distribution of micropores could be explained by random copolymerization of styrene and MMA. The surface area increased dramatically as the fraction of MMA increased, reaching 1135 m2 g−1. The specific area in this result is similar or even higher value to activated carbon materials. We are currently investigating the control of pore size by using other PS copolymer particles. We believe that our result will lead to more efficient production of microporous carbon particles and practical applications in electrochemical electrode, adsorption and catalysis.Experimental
Monodisperse PS-PMMA spheres were synthesized by emulsifier-free emulsion polymerization of styrene (99.9%, Aldrich) and MMA (98.5%, Aldrich). Briefly, deionized water was used as a reaction medium, and potassium persulfate was applied to initiate polymerization. Divinyl benzene (Aldrich) was added to crosslink the styrene groups. The reaction was carried out at 70 °C for 24 h. The post-crosslinking of PS was required to enhance the conversion of PS components into carbon. PS-PMMA was separated from the aqueous solution by centrifugation and was added to a flask containing CHCl3 (99.8%, Aldrich) for the post-cross-linking reaction. A 0.3 g sample of PS-PMMA particles was placed in the flask, and CHCl3 and 0.1 M AlCl3 were added with vigorous stirring (300 rpm). The reaction was heated to 50 °C for 18 h. Subsequently, the PS-PMMA particles were purified by centrifugation, washing with 30 mL ethanol three times, and dried in a convection oven (60 °C) overnight. The pyrolysis of the post-crosslinked PS-PMMA spheres was achieved in a tube furnace. The temperature was steadily increased to 700 °C over 2 h and was maintained at 700 °C for another 2 h under nitrogen flow.The surface morphologies were measured by SEM (Hitachi, Carl Zeiss) and TEM (Carl Zeiss). FT-IR and Raman spectra was taken using, respectively, a Nicolet FT-IR spectrometer and a Horiba Jobin Yvon LabRAM HR equipped with an air-cooled Ar-ion laser working at 541 nm. Nitrogen adsorption isotherms were recorded on a Micromeritics ASAP 2405N. The specific surface area was calculated using the BET method applied to the nitrogen adsorption data.
Acknowledgements
This work was supported by grants from the National Research Foundation of Korea (2010-0028961). The Korea Basic Science Institute is also acknowledged for the SEM measurements.References
- M. Inagaki, H. Orikasa and T. Morishita, RSC Adv., 2011, 1, 1620 RSC.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073 CrossRef CAS.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169 CrossRef CAS.
- P. X. Hou, H. Orikasa, T. Yamazaki, K. Matsuoka, A. Tomita, N. Setoyama, Y. Fukushima and T. Kyotani, Chem. Mater., 2005, 17, 5187 CrossRef CAS.
- A. Almasoudi and R. Mokaya, J. Mater. Chem., 2012, 22, 146–152 RSC.
- J. Zhou, W. Li, Z. S. Zhang, W. Xing and S. P. Zhuo, RSC Adv., 2012, 2, 161 RSC.
- H. M. Cheng, D. W. Wang, F. Li, M. Liu and G. Q. Lu, Angew. Chem., Int. Ed., 2008, 47, 373 CrossRef.
- W. Fan, M. A. Snyder, S. Kumar, P.-S. Lee, W. C. Yoo, A. V. McCormick, R. Lee Penn, A. Stein and M. Tsapatsis, Nat. Mater., 2008, 7, 984 CrossRef CAS.
- Z. B. Lei, Y. G. Zhang, H. Wang, Y. X. Ke, J. M. Li, F. Q. Li and J. Y. Xing, J. Mater. Chem., 2001, 11, 1975 RSC.
- J. Pires, A. C. Araujo, A. P. Carvalho, M. L. Pinto, J. M. Gonzalez-Calbet and J. Ramirez-Castellanos, Microporous Mesoporous Mater., 2004, 73, 175 CrossRef CAS.
- H. L. Jiang, B. Liu, Y. Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Q. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854 CrossRef CAS.
- D. S. Zhang, Z. Chang, Y. B. Lv, T. L. Hu and X. H. Bu, RSC Adv., 2012, 2, 408 RSC.
- S. A. Johnson, E. S. Brigham, P. J. Ollivier and T. E. Mallouk, Chem. Mater., 1997, 9, 2448 CrossRef CAS.
- T. Kyotani, T. Nagai, S. Inoue and A. Tomita, Chem. Mater., 1997, 9, 609 CrossRef CAS.
- J. S. Yu, S. B. Yoon, Y. J. Lee and K. B. Yoon, J. Phys. Chem. B, 2005, 109, 7040 CrossRef CAS.
- A. Garsuch, O. Klepel, R. R. Sattler, C. Berger, R. Glaser and J. Weitkamp, Carbon, 2006, 44, 593 CrossRef CAS.
- C. Ducrot-Boisgontier, J. Parmentier and J. Patarin, Microporous Mesoporous Mater., 2009, 126, 101 CrossRef CAS.
- D. O. Jordan and A. R. Mathieson, J. Chem. Soc., 1952, 611–620 RSC.
- W. R. Bussing and N. A. Peppas, Polymer, 1983, 24, 209 CrossRef CAS.
- S. J. Gregg and K. S. W. Sing, Adsorption, surface area, and porosity, Academic Press, London ; New York, 1982 Search PubMed.
- J. M. Goldwasser and A. Rudin, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 1993 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Additional experimental results. See DOI: 10.1039/c2ra21079g |
| This journal is © The Royal Society of Chemistry 2012 |