Facile synthesis of CuO nanorods with abundant adsorbed oxygen concomitant with high surface oxidation states for CO oxidation†
Kuan
Zhong
ab,
Jianjun
Xue
b,
Yanchao
Mao
a,
Chengsheng
Wang
a,
Teng
Zhai
a,
Peng
Liu
*a,
Xinde
Xia
b,
Haohua
Li
*c and
Yexiang
Tong
*a
aMOE of Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou 510275, P. R. China. Fax: +86 20 84112245; Tel: +86 20 84110071E-mail: chedhx@mail.sysu.edu.cn (Y.T.); ceslp@mail.sysu.edu.cn (P.L.)
bGuangzhou Great Power Energy & Technology Co., Ltd., Guangzhou 511483, P. R. China
cCenter for Photon Manufacturing Science and Technology, School of Materials Science and Engineering, Jiangsu University, Zhenjiang 212013, P. R. China. E-mail: lihaohua@ujs.edu.cn
First published on 1st October 2012
Abstract
Oxygen adsorption materials play an important role in catalysis. However, the conventional catalytic mechanism of CO oxidation over copper oxide-based catalysts is based on lattice-oxygen oxidation processes, which neglects the significance of the oxidizability of the copper component and the adsorbed oxygen. Herein, we propose that poorly-crystallized CuO nanorods are capable of adsorbing abundant oxygen along with increasing the Cu oxidation states to close to 3+, meaning that CO catalytic oxidation occurs directly on the adsorbed oxygen and that Cu oxidation states do not fall to 1+ during catalytic reactions. The rate-controlled step is the surface oxidizability of the CuO nanorods, which increases with increasing temperature and oxidizability of the environment involved. These catalytic processes are distinctly different from the conventional case. The unique oxygen adsorption and catalytic properties of the CuO nanorods originate from the increasing trend in Cu oxidation state in the p-type CuO, enhanced by the defect structures and coarse surfaces of the sample. Such structure and morphology characteristics are closely related to the liquid membrane growing environment, which induces poor crystallization of the nanorods. The characterization methods include scanning electron microscopy (SEM), transmission electron microscopy (TEM), high resolution transmission electron microscopy (HRTEM), X-ray photoelectron spectroscopy (XPS), Raman spectroscopy, and Fourier transformation infrared spectroscopy (FTIR).
1. Introduction
CuO is currently considered to be an excellent heterogeneous catalyst in CO oxidation, water–gas shift reaction, methanol steam reforming, and NO reduction,1–4 due to its intriguing catalytic properties in redox reactions, structural variety, stability, and environmental friendliness. Generally, oxygen is considered to be an important factor in the catalytic reactions occurring on CuO, and lattice oxygen, through which CuO nanostructures can adsorb abundant oxygen, is of particular importance.5 Oxygen adsorption on CuO nanostructures has been mentioned in other work, but without detailed studies.6,7 To the best of our knowledge, the catalytic activities of O2 adsorbed on metal oxide-based materials in the oxidation of CO and other reductive species have not been addressed.Low-temperature oxidation of CO is significant due to its promising application for the complete removal of residual CO during the production of H2.8–10 Simple oxidation of CO either with O2 or NO is also important for applications such as indoor air cleaning and automotive exhaust treatment, since CO is an atmospheric pollutant.11,12 However, it is reported that bulk CuO could not adsorb CO and thus did not present catalytic activity for CO oxidation; but that finely dispersed CuO species could adsorb CO, contributing to the catalytic activity of CuO/CeO2 catalysts.13 Nanostructured CuO has good activity in CO oxidation.14 This catalytic activity is generally attributed to reversible oxygen storage and release in CuO nanostructures via a chemical reaction of CuO ↔ Cu2O,15 and Cu oxidation states will drop to 1+ when this CO catalytic oxidation reaction occurs. In this case, a high O2 concentration is required to enable restoration of the surface oxygen, but this cannot exceed the amount at which O2 starts blocking the active surface Cu sites. The activation barrier of the CO oxidation reaction on transition metal surfaces is determined by the metal–oxygen bond breaking,16 where the reaction is an apparent structure-sensitive process and surface lattice oxygen is involved. However, in this paper, we will present that adsorbed oxygen, rather than lattice oxygen, is mainly involved during CO catalytic oxidation over CuO nanorods (NRs) and that the environmental O2 concentration should be as high as possible to enhance both oxygen adsorption and the oxidizability of the NRs. Moreover, the Cu oxidation states will not drop to 1+ during CO oxidation. Such peculiar properties are primarily attributed to the special properties of the prepared CuO NRs: abundant adsorbed oxygen, concomitant with high surface Cu oxidation states (close to 3+) promoted by the defect structures of the NRs and further promoted by an oxidizing environment, such as an O2-rich atmosphere, especially at high temperatures.
The properties of CuO nanostructures could be affected by their morphologies, sizes, surface states and structures, which are closely related to their preparation methods. Defect structures, small sizes and coarse surfaces are conducive to gas adsorption. Poorly-crystallized nanostructures present such structures and morphology features and so are promising potential gas adsorption materials. In general, a high temperature synthesis route (such as hydrothermal processes) leads to a well-crystallized sample, while poorly-crystallized nanostructures could be obtained via low temperature synthesis methods (wet chemical preparation pathway,17 for example). Doping can cause low crystallization of a sample.5 Additionally, constructing a metastable non-stoichiometric form of copper oxide via heating the catalyst in a redox environment greatly enhanced its activity in CO oxidation.18 A plasma treatment can also cause surface defect structures in CuO nanowires, resulting in enhanced catalytic activity in CO oxidation.19
In this study, we prepare poorly-crystallized CuO NRs grown directly from a sacrificial Cu foil via naturally-generated electrochemical corrosion, occurring in a liquid membrane self-constructing on the foil when wetted by a solution. The liquid membrane environment makes the CuO NRs grow at the gas–liquid interfaces and causes incomplete crystallization of the NRs, leading to large quantities of defect structures being generated in the resultant nanostructures.
2. Experimental
CuO NRs were grown in a liquid membrane supported on a sacrificial Cu foil, similar to the case of ZnO, reported elsewhere by our group.20 Concisely, a Cu foil wetted wholly by 1 M KOH solution was partially immersed in this solution and held there for 7 days under ambient conditions. NRs could then be obtained in the region above the water-line. CuO NRs were pretreated via a heat treatment at 200 °C for 5 h under ambient atmosphere unless specified otherwise.The morphology and structure of the sample were characterized by SEM (JSM-6330F, Shimadzu), TEM (JEM-2010HR, JEOL), and HRTEM. XPS (ESCALab 250), Raman spectroscopy (Renishaw inVia), and FTIR (EQUINOX 55) were used to study the surface states of the CuO NRs with different treatments. The XPS experiment was conducted with an Al Kα monochromator X-ray source operating at a power of 150 W. The XPS binding energies were internally referenced to the aliphatic C 1s peak at 284.8 eV. High-resolution spectra were resolved by fitting each peak with a combination Gaussian-Lorentzian function, after subtracting the background, conducted in XPS Peak 41 software. The Lorentzian-Gaussian ratio was set to be 20%, and the FWHM (full width at half maximum) were all less than 2.0 eV in peak fitting. The XPS experiments for the CuO NRs were carried out shortly after annealing and catalytic reactions (within 5 h) to avoid or decrease air contamination. FTIR spectra were recorded with diffuse reflectance mode at room temperature. The sizes of the CuO films were larger than those of the IR incident light (10 mm in diameter), so the band areas reflected the relative quantities of the species adsorbed on the samples. CuO samples were characterized directly without removing the substrate, except in the TEM experiment (the samples were treated by dispersing the NRs into ethanol via ultrasonic followed by collecting the dispersed NRs with carbon copper grids) and the CO conversion experiment (the samples were removed from the Cu substrate by carefully shaving with a blade).
The O2 or CO environment was obtained via aerifying pure O2 or CO into a small container. Octadecane could be adsorbed on CuO NRs by bubbling pure Ar into the melted octadecane (50 °C) for 2 min (CuO NRs were placed above the melted octadecane). After CO exposure or octadecane adsorption for a certain period, the surface states of the NRs were studied via FTIR and XPS.
The catalytic activity of the CuO NRs was further tested using a fixed bed flow microreactor under atmospheric pressure. The amount of catalyst powder was 80 mg. The catalyst was pretreated by heating at 200 °C for 2 h in an O2 atmosphere prior to catalytic measurement. In the CO conversion experiment, the gases used were 15 vol% CO + 30 vol% O2 balanced with He, and the total gas flow rate was 20 mL min−1. The reaction time was 60 min. Determination of the reactant and product compositions was carried out with a GC-508A gas chromatograph equipped with a thermal conductivity detector.
3. Results and discussion
3.1. Preparation of CuO NRs
A liquid membrane will be naturally formed when a metal foil is wetted by a solution. A Cu foil wetted by 1 M KOH solution was partially immersed in this solution under ambient conditions, then electrochemical corrosion occurred on the Cu foil (see Electronic Supplementary Information, ESI† for detail). Black species were obtained in the region above the water-line after one week. The morphologies of the black species were observed via SEM, and the result shows that sharp-topped NRs of ca. 70 nm in diameter and ca. 500 nm in length are obtained (Fig. 1a). Detailed observation of the surface microstructures of the NRs via TEM reveals that the surfaces are very coarse and there are many small bright areas (Fig. 1b), revealing the formation of many micropores in this sample, which indicates its poor crystallization. The needle-like shapes of the NRs also imply difficulty in the crystal growth of the NRs in the liquid membrane. The diffraction spots of the selected area electron diffraction (SAED) pattern (Fig. 1b inset) are ordered, but most have tail-like shapes. Additionally, the lattice fringes shown in the HRTEM image (Fig. 1c) are continuous as a whole but with interruption in some regions; furthermore, the HRTEM image also demonstrates the coarse surfaces of the NRs (Fig. 1c). The above three points together suggest the formation of single crystalline NRs, but with defect structures (especially in the surfaces) and low crystallization (though with heat treatment). Such surface and structure characteristics are greatly beneficial for gas adsorption. In addition, indexing the SAED pattern (Fig. 1b inset) shows that the NRs are constructed as CuO monoclinic structures and elongated along the (020) planes. The two planes with the same interplanar distance of 0.28 nm shown in Fig. 1c are attributed to the (110) and (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10) planes of the monoclinic CuO crystal from top to bottom, respectively, and the (020) planes showing the growth direction for the NRs can also be seen.
10) planes of the monoclinic CuO crystal from top to bottom, respectively, and the (020) planes showing the growth direction for the NRs can also be seen.
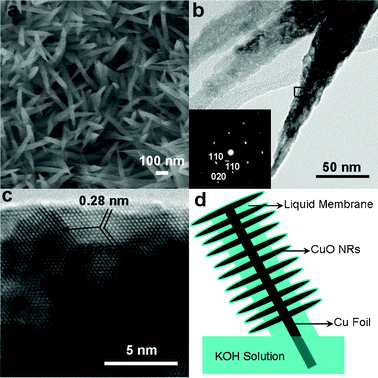 | ||
| Fig. 1 Morphology and structure characterizations of the obtained sample. (a) SEM image. (b) TEM image and the SAED pattern recorded at the rightmost NR. (c) HRTEM image recorded from the box in (b). The arrow indicates the growth direction of the NRs. (d) Schematic of CuO NRs grown in a liquid membrane. | ||
The growth mechanism of the NRs can be described as primary CuO colloids preferentially growing along the (020) planes.21 The occurrence of many grain boundaries in the NRs before annealing (Fig. S1 in ESI†) and the sharp tops of the NRs indicate that the primary CuO colloids diffused along the NRs and integrated with the NRs at their tops. Such growth processes can be assigned to the well-known oriented-attachment. The intrinsic potential difference between the outer part (above the water-line) and the immersed part (below the water-line) of the Cu foil facilitated the preferential growth, as it was found that the whole Cu foil wetted only by the liquid membrane did not produce NRs (Fig. S2 in ESI†). The CuO colloids were generated by self-degradation of Cu(OH)2 nanowires formed via alkaline corrosion of Cu foil (Fig. S3 in ESI†). This structure transformation is similar to a case reported elsewhere.22 However, for our case, the structure reconstruction proceeded in a liquid membrane at room temperature, and thus the NRs were grown at the gas–liquid interfaces, which caused difficult crystallization of the CuO NRs, as can be seen from the formation of the coarse surfaces and large quantities of defects shown in Fig. 1 and Fig. S1.† The NRs grown in the liquid membrane are schematically illustrated in Fig. 1d.
3.2. Oxygen adsorption on CuO NRs concomitant with high surface oxidation states characterized by XPS
The surface states of the CuO NRs were analyzed via XPS and the results are shown in Fig. 2. The Cu 2p3 XPS shows a main peak at 934.7 eV (Fig. 2a), shifted up relative to that of bulk CuO (933.8 eV).23 Additionally, the Cu LMM Auger peak is centered at 917.6 eV (Fig. S4 in ESI†), so the Auger parameter is 1852.3 eV, larger than that of commercial CuO.24 Thus, the surfaces of the CuO NRs exhibit relatively high oxidation states. However, these comparisons are not very exact, as bulk CuO and the NRs are greatly different in size, and generally binding energy decreases in the nanostructure (especially with defect structures) compared to the bulk counterpart (further discussed below).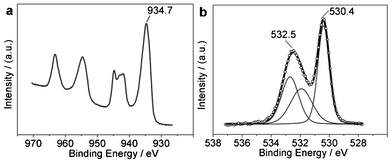 | ||
| Fig. 2 XPS of CuO NRs (with heat treatment). (a) Cu 2p photoelectron line. (b) O 1s photoelectron line. | ||
The O 1s XPS of the NRs contains two peaks, a main peak at 530.4 eV and a broad peak at 532.5 eV (Fig. 2b). The main peak is undoubtedly derived from the lattice oxygen O2−. The intensity ratio of this peak to Cu 2p3 is calculated to be 1.06 (close to 1), consistent with the stoichiometric composition of CuO. For the broad peak at 532.5 eV, possible assignments include hydroxyl group,25 oxygen defect sites (oxygen ions with unusual coordination in oxygen defect sites, which can be caused by the defect structures and/or by the physicochemical properties of a sample, such as CeOx (where x < 2)),26,27 carbonate species,28 and adsorbed oxygen (from the environment).6 The analyzed sample was pretreated by heat treatment, thus the broad band at 532.5 eV could not arise from hydroxyl (demonstrated further later). In addition, the carbon contaminants adsorbed on the NRs are very few (only 5.9 at% obtained by XPS, but the excessive oxygen takes up 34.7 at%, excluding the stoichiometric O in CuO) with hydrocarbon as the main form (Fig. S4 in ESI†). Hence, the assignment of carbonates to the broad O 1s band at 532.5 eV can be ruled out. On the other hand, the broad O 1s band disappeared when the sample was treated by ion sputtering prior to XPS determination (Fig. S5 in ESI†), indicating that the broad O 1s band in Fig. 2b is derived from surface adsorbed species. Therefore, the broad O 1s band arises from adsorbed oxygen. CuO tends to adsorb oxygen to increase its oxidation states due to its p-type oxide semiconducting property. The trend of increasing valency will cause oxygen vacancies in CuO, and such vacancies induce oxygen adsorption from the environment.26 Substantial adsorbed oxygen simultaneously enhances the valence increase in the copper component. Nanoscale sizes, coarse surfaces, and defect structures promote this cycle. Therefore, the formation of the broad O 1s band has resulted from the co-action of oxygen vacancies (associated with Cu valence increase) and oxygen adsorption.13,27 These exist symbiotically on the surfaces of the CuO.
The adsorption of oxygen generates an oxygen adsorption bond where oxygen vacancies are located. It is known that O2 adsorbs on an absorbent with either a side-on or end-on model. The chemical environments of the two O atoms in the adsorbed O2 are the same for the side-on type adsorption, resulting in an O 1s XPS peak composed of only one contribution, but are different for the end-on type adsorption. Such a broad O 1s XPS band as is seen at 532.5 eV is unlikely to be derived from only one contribution. Thus, O2 appear to be adsorbed on the NRs with end-on type (further explained later). Consequently, the fitting results are obtained as shown in Fig. 2b.
The types of the adsorbed oxygen on metal oxide are rather complex, such as O2−, O22−, and O−.29 However, generally, only O2− will be stable at room temperature and other oxygen species will be formed at elevated temperatures. Therefore, the oxygen adsorbed on the NRs can be described as Cu–[O(1)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(2)]ads (where “[O(1)O(2)]ads” is the adsorbed oxygen O2−, and the numbers denote the different chemical environments of the O atoms). Thus, for the broad O 1s band, the peak at ca. 533 eV is derived from O(2), and the other peak at ca. 532 eV is attributed to the O(1) associated with oxygen vacancies. On the basis of the fitting results, the O(2) 1s/Cu 2p3 intensity ratio is calculated to be as large as 0.76, which means that large quantities of oxygen are adsorbed on the CuO NRs, demonstrating high oxygen adsorption capacity (OAC). Here the OAC is recorded as 0.76 (referenced to the oxygen storage capacity for oxygen storage materials). Similarly, the O(1) 1s/Cu 2p3 intensity ratio is measured to be 0.72 (which reflects the quantities of the effective generated oxygen vacancies), approximately equal to the OAC. This relationship mainly results from the O2 linearly (i.e. end-on) adsorbing on the surface Cu sites. This also indicates that the peak simulation is reasonable. In this case, a feedback bond between Cu 3d orbital and O(1) 2p orbital can be formed, which stabilizes the Cu–Oads bond, while simultaneously weakening the adsorbed OO bond, thus enhancing the activity of the adsorbed oxygen. In addition, on the basis of the above calculations for the fitting peaks, we can calculate that over 70 at% of the detected Cu atoms are adsorbed by oxygen (deduced from the value of OAC, 0.76). However, it is notable that the adsorption of oxygen only occurs on the outermost surfaces of the NRs, but the Cu atoms detected by XPS can be several nanometers thick. Thus, the outermost layer of Cu atoms is all adsorbed by oxygen. This is reasonable because CuO exhibits the trend of increasing valency in the Cu component, which induces oxygen adsorption, and this is enhanced by the coarse and porous surfaces, nanometer sizes, and defect structures of the NRs. Furthermore, the mutual promotion of the valence-increase trend and oxygen adsorption enhances the OAC. Additionally, the obtained NRs predominantly display (01)-exposed surfaces (Fig. 1c), which adsorb O2 more easily than other exposed planes such as (001) or (111).30 The adsorbed oxygen on the CuO NRs is rather stable, as it can be observed that a strong broad O 1s XPS band in Fig. 2b is obtained under an UHV (ultra high vacuum) condition. The total O/Cu ratio is calculated to be 1.06 + 0.76 + 0.72 = 2.54, larger than the latest value (1.72) reported for CuO nanoparticles,5 implying the existence of excess oxygen adsorbed on the surfaces of the sample. The abundant adsorbed oxygen is a great advantage for the oxidation of reductive carbon species such as CO.
O(2)]ads (where “[O(1)O(2)]ads” is the adsorbed oxygen O2−, and the numbers denote the different chemical environments of the O atoms). Thus, for the broad O 1s band, the peak at ca. 533 eV is derived from O(2), and the other peak at ca. 532 eV is attributed to the O(1) associated with oxygen vacancies. On the basis of the fitting results, the O(2) 1s/Cu 2p3 intensity ratio is calculated to be as large as 0.76, which means that large quantities of oxygen are adsorbed on the CuO NRs, demonstrating high oxygen adsorption capacity (OAC). Here the OAC is recorded as 0.76 (referenced to the oxygen storage capacity for oxygen storage materials). Similarly, the O(1) 1s/Cu 2p3 intensity ratio is measured to be 0.72 (which reflects the quantities of the effective generated oxygen vacancies), approximately equal to the OAC. This relationship mainly results from the O2 linearly (i.e. end-on) adsorbing on the surface Cu sites. This also indicates that the peak simulation is reasonable. In this case, a feedback bond between Cu 3d orbital and O(1) 2p orbital can be formed, which stabilizes the Cu–Oads bond, while simultaneously weakening the adsorbed OO bond, thus enhancing the activity of the adsorbed oxygen. In addition, on the basis of the above calculations for the fitting peaks, we can calculate that over 70 at% of the detected Cu atoms are adsorbed by oxygen (deduced from the value of OAC, 0.76). However, it is notable that the adsorption of oxygen only occurs on the outermost surfaces of the NRs, but the Cu atoms detected by XPS can be several nanometers thick. Thus, the outermost layer of Cu atoms is all adsorbed by oxygen. This is reasonable because CuO exhibits the trend of increasing valency in the Cu component, which induces oxygen adsorption, and this is enhanced by the coarse and porous surfaces, nanometer sizes, and defect structures of the NRs. Furthermore, the mutual promotion of the valence-increase trend and oxygen adsorption enhances the OAC. Additionally, the obtained NRs predominantly display (01)-exposed surfaces (Fig. 1c), which adsorb O2 more easily than other exposed planes such as (001) or (111).30 The adsorbed oxygen on the CuO NRs is rather stable, as it can be observed that a strong broad O 1s XPS band in Fig. 2b is obtained under an UHV (ultra high vacuum) condition. The total O/Cu ratio is calculated to be 1.06 + 0.76 + 0.72 = 2.54, larger than the latest value (1.72) reported for CuO nanoparticles,5 implying the existence of excess oxygen adsorbed on the surfaces of the sample. The abundant adsorbed oxygen is a great advantage for the oxidation of reductive carbon species such as CO.
CuO NRs exhibit relatively high oxidation states, accompanied by abundant adsorbed oxygen, as mentioned above. The newly-prepared CuO NRs without heat treatment still present OAC of 0.30 (Fig. S6 in ESI†). The Cu 2p3 binding energy and Cu Auger parameter of this sample were determined to be 933.1 and 1850.3 eV, respectively (Fig. S6 in ESI†), smaller by 1.6 and 2.0 eV, respectively, than those of the NRs with heat treatment. The Cu 2p3 binding energy is expected to be further decreased for the sample with a lower OAC. The CuO sample exhibiting an OAC of near zero is unavailable due to the intrinsic properties of the CuO, that is, valence-increase trend leading to oxygen adsorption. However, near zero OAC can be obtained for the sample with ion sputtering treatment in XPS experiment, but leading to the reduction of the Cu(II) to Cu(I) (Fig. S5 in ESI†). It has been reported that Cu(III) compounds show a 2 eV shift to higher energy relative to Cu(II) species.31 Thus, the surfaces of the CuO NRs exhibit high, close to 3+ Cu oxidation states. The high Cu oxidation states in the NRs are mainly attributed to the abundant adsorbed oxygen, which can stabilize copper species with high oxidation states. From the above comparisons, we also realize that heat treatment enhances oxygen adsorption, which is one of the properties of the p-type oxide semiconductor (i.e. inclining to adsorb oxygen to increase its oxidation states). Additionally, the O 1s broad band for the annealed NRs is at 532.5 eV (Fig. 2b), shifted up from that of the newly-prepared NRs (531.4 eV) (Fig. S6 in ESI†), also manifesting the enhancement of oxygen adsorption after heat treatment.
3.3. Oxygen adsorption on CuO NRs characterized by Raman spectrum
To further confirm the occurrence of oxygen adsorption on CuO NRs, we carried out a Raman experiment and the result is shown in Fig. 3. The three Raman peaks at 300, 349, and 630 cm−1 are derived from the Ag, B1g, and B2g modes of the monoclinic CuO, respectively.32 The most prominent feature of this Raman spectrum is that the B2g peak (at 630 cm−1) has a shoulder peak at 592 cm−1 (not reported by other work to the best of our knowledge). We found that the sample with less adsorbed oxygen (the newly-prepared NRs) did not obviously demonstrate this shoulder peak (Fig. S7 in ESI†), while the CuO NRs after 5 days exposure to O2 at room temperature showed this shoulder peak more distinctly than the one in Fig. 3 (Fig. S7 in ESI†). Thus, the shoulder peak in Fig. 3 is associated with adsorbed oxygen. CeO2 also displays this peak, which is assigned to oxygen vacancies.33 Hence, the shoulder peak in Fig. 3 is assignable to oxygen vacancies. Therefore, the Raman experiment also reveals the oxygen adsorption occurring on CuO NRs.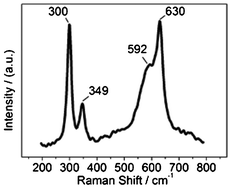 | ||
| Fig. 3 Raman spectrum of CuO NRs. | ||
In addition, we find that there is no obvious peak of OH groups above 3600 cm−1 for the Raman spectrum of the annealed NRs (Fig. S8 in ESI†), indicating that the broad O 1s XPS band in Fig. 2b does not arise from OH groups.
3.4. Catalytic oxidation reactions occurring on adsorbed oxygen
When the newly-prepared NRs were stored under dry air conditions at room temperature for at least three months (we denote this class of sample as the aged CuO NRs), the Cu 2p3 binding energy increased to 933.5 eV, and a broad O 1s XPS band is also obtained (Fig. 4a). Meanwhile, this sample has a large amount of surface adsorbed carbon species (61.2 at% determined via XPS), mainly composed of hydrocarbon species (corresponding to the C 1s XPS peak at 284.8 eV) and CO-like species (C 1s XPS peak at 288.2 eV), such as carbonate or carboxylate (Fig. 4b). However, the newly-prepared NRs show a C 1s XPS peak at 286.1 eV, which can be assigned to C–O-like species (such as alkoxide), excepting hydrocarbon and CO-like species (Fig. S6 in ESI†). Thus, the adsorbed C–O-like species on the newly-prepared NRs can be oxidized to CO-like species during ageing, indicating the catalytic oxidation performance of the adsorbed oxygen on CuO NRs. For further investigation of the catalytic activity of the adsorbed oxygen, we stored the aged NRs in an O2 atmosphere at room temperature for 5 days. The XPS results show that the Cu 2p3 binding energy is further increased to 933.8 eV, and an obvious shoulder peak above 533.7 eV occurs in the resulting O 1s photoelectron line (Fig. 4c). Additionally, upon careful observation we found that the profile of the main C 1s XPS peak (at 284.8 eV) is asymmetric and that there is a peak at 285.6 eV, which can be obtained by peak simulation (Fig. 4d). It is not difficult to understand that the adsorption states and intermolecular interaction can affect the binding energies of the constituted elements. Thus, the carbonyl ligand-containing species,34,35 such as those similar to Ni(CO)4, may be formed on the surfaces of the CuO NRs, which also suggests that catalytic oxidation of the surface adsorbed carbon species on the adsorbed oxygen occurs. In this case, catalytic oxidation means the further oxidation of the –CO-like species to the (–CO–)n-like carbonyl ligand-containing species (where n = 2, 3, 4…), which is reasonable due to the high O2-concentration environment to which the NRs are exposed. For the detailed molecular structures of the formed carbonyl ligand-containing species, further study is required. The above comparisons also indicate that the catalytic activity of the CuO NRs is enhanced with enhancing the oxidizability of the environment involved, which leads to the enhancement of the oxidizability of the NRs.
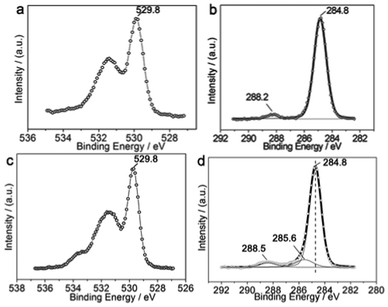 | ||
| Fig. 4 XPS of the aged CuO NRs without heat treatment. (a) O 1s and (b) C 1s XPS of the aged CuO NRs, respectively. (c) O 1s and (d) C 1s XPS of the aged CuO NRs with 5 days O2 exposure, respectively. | ||
3.5. Oxygen adsorption and CO catalytic oxidation on CuO NRs characterized via FTIR
We used FTIR to further study the oxygen adsorption on CuO NRs and investigate their catalytic activity in CO oxidation. FTIR spectra of the CuO NRs with various surface states are shown in Fig. 5. The spectra can be basically divided into three zones, 400–650 cm−1, 650–1500 cm−1, and 1500–3000 cm−1, respectively. In the following, we will first discuss the FTIR spectra of the annealed CuO NRs with and without CO exposure, and the FTIR spectrum of the octadecane-adsorbed sample will be discussed independently.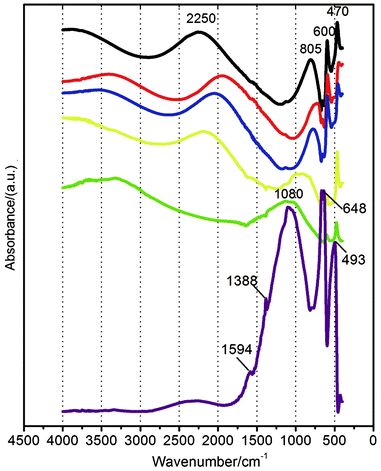 | ||
| Fig. 5 FTIR spectra of the CuO NRs with heat treatment before (black line) and after 1 h (red line), 5 h (blue line), 1 day (yellow line), and 5 days (green line) exposure to CO at 60 °C, respectively. Purple line: FTIR spectrum of the octadecane-adsorbed NRs after 5 days exposure to O2 at room temperature. | ||
The two sharp IR peaks at 470 and 600 cm−1 are derived from CuO NRs, reflecting the surface fine structures of the NRs since the bulk CuO only displays a broad band above 580 cm−1.
The broad FTIR bands in the zones of 650–1500 cm−1 and 1500–3000 cm−1 reflect the existence of surface adsorption states on the CuO NRs. It is known that the surface adsorbed reductive carbon species can be oxidized by/desorbed from CuO at elevated temperatures (such as in heat treatment). In addition, the adsorption state of O2 (with end-on type adsorption) makes the OO stretching vibration infrared active. In the course of O2 adsorption, an oxygen adsorption bond is generated which is also infrared active. Thus, for the NRs with heat treatment, the two broad bands at 2250 and 805 cm−1 (Fig. 5, black line) are both associated with adsorbed oxygen. The large band area means a large quantity of oxygen adsorbed on the sample, which is consistent with the results of XPS.
The covalent bond of O2 is stronger than the oxygen adsorption bond, and the oxygen adsorption bond (Cu–Oads) is slightly similar to (though the bond strength is weaker than that of) a C–O single bond which induces a ca. 1000 cm−1-centered IR band. So the broad band at 805 cm−1 is ascribed to Cu–Oads oxygen adsorption bond stretching vibration, and the other broad band (above 2250 cm−1) is derived from OO stretching vibration. The OO IR band occurring at 2250 cm−1 is possibly derived from the adsorption state of O2 which is similar to that of CO (which shows the IR bands above ca. 2100 cm−1),36 as they both have transition states between triple bond and double bond (the detailed mechanism requires further study).
In addition, there is no obvious peak of OH groups above 3600 cm−1 for the FTIR spectrum of the annealed NRs (black line in Fig. 5), which means that the broad O 1s XPS band in Fig. 2b is indeed derived from oxygen adsorption rather than OH groups.
When the annealed NRs were exposed to CO at 60 °C, the two broad IR bands were initially red shifted from 2250 to 1940 cm−1 and from 805 to 730 cm−1, respectively (from black line to red line in Fig. 5). These two broad bands then blue shift with increasing interaction time with CO and the adsorbed O2 as a whole (from red line to green line in Fig. 5). In addition, the intensity of the broad band at a relatively high wavenumber position decreases on prolonging the duration of the NRs in the CO environment, and this band even disappears (green line in Fig. 5). On the other hand, the intensity of the other broad band located at a relatively low wavenumber position first decreases and then increases during storage of the NRs in the CO environment (Fig. 5). These phenomena indicate the evolution of the surface states of the CuO NRs, that is, the evolution of the interaction between adsorbed oxygen and adsorbed CO. It is known that the IR peaks derived from adsorbed CO are at ca. 2050 and ca. 2110 cm−1,36 and CO2 IR peaks are at ca. 2360 and ca. 2340 cm−1.37 Furthermore, the intensities and widths of these two kinds of peaks are much weaker and narrower than those of our case. Thus, the broad band at the high wavenumber position occurring on the NRs after CO exposure can be assigned to the transition between CO and CO2, that is, CO adsorption on the adsorbed O2. The surfaces of CuO NRs are all adsorbed by O2 due to the strong OAC of the sample as shown by XPS above. In this case, CO must be adsorbed on the adsorbed O2 prior to catalytic reaction, similarly to a case reported elsewhere.38 Additionally, both CO and CO2 are linear molecules. Thus, CO will adsorb on the adsorbed O2 at the C end when the NRs are exposed to CO, likely forming Cu–(OO)ads–(CO)ads. It is expected that the distribution of the strengths of the transition bonds and the electron delocalization state in the transition molecule are facilely generated, which gives birth to broad IR bands. The gradual decrease in the intensity of the broad band in the high wavenumber zone accompanied by the gradual increase in the intensity of the broad band in the low wavenumber zone implies the transition molecule transforms, at least partially, to C–O-like species. There is no direct evidence proving the generation of CO2 at this stage. However, it is reported that the formed CO2 is facilely desorbed from copper oxide,39 and the presence of C–O-like adsorbed species and the remaining adsorbed O2 can hinder CO2 adsorption, causing the unclear occurrence of the CO2 IR band. However, we will prove the generation of CO2 by other experiments later.
Furthermore, we found that the Cu 2p3 binding energy of the sample after 5 days storage in the CO environment has decreased to 933.3 eV and the Cu Auger parameter has also declined to 1850.5 eV (Fig. S9 in ESI†), suggesting that the surface Cu atoms have a reduction trend as compared to those of the annealed NRs. Such a reduction trend indicates the occurrence of the interaction between adsorbed O2 and CO. However, the surface Cu still primarily exhibits a 2+ oxidation state. CO is thus not adsorbed on the Cu site, because CO will transfer an electron to Cu2+ to form Cu+ if it adsorbs on the Cu site. Therefore, CO is still adsorbed on the adsorbed oxygen after 5 days storage of the sample in the CO environment. The type of oxygen adsorbed by CO is mainly the single O(1) atom (the other O(2) atom in the previous adsorbed O2 is likely reduced by CO to CO2 and then removed), as can be seen from the FTIR of this sample presenting an unclear IR band for adsorbed O2 (in the range of 1500–3000 cm−1, green line in Fig. 5), resulting in the formation of Cu–Oads–(CO)ads which contributes to the occurrence of a strong broad IR band above 1080 cm−1 (green line in Fig. 5). The non-occurrence of further reaction in Cu–Oads–(CO)ads to generate CO2 is mainly due to the insufficiently strong oxidation capacity of the Cu atom and the Oads still exhibiting oxidizability. It is imagined that CO2 will be generated in Cu–Oads–(CO)ads at elevated temperatures due to the increase in the oxidation capacity of the Cu atom with increasing temperatures (as will be demonstrated by the CO conversion experiment discussed below). In addition, this experiment (i.e., storage of CuO NRs in CO environment) also indicates that CuO could not transform to Cu2O, even under the CO reducing environment, as long as the CuO had previously adsorbed oxygen.
Since relatively higher temperatures (such as 60 °C) enhance oxygen adsorption on the p-type CuO and the poorly-crystallized NRs with coarse surfaces also favor oxygen adsorption, the adsorbed O2 on CuO NRs could not suffer from desorption during storage at 60 °C (it is also reported that the adsorbed O2 on CuO nanostructures can be stable at 60 °C5) unless the adsorbed O2 has reacted with CO to produce CO2. We can also understand this point from the assumption that CO will adsorb on the Cu sites, causing the reduction of the CuO to Cu2O, if the desorption of the adsorbed O2 takes place at 60 °C. However, the real case is that the NRs still exhibited the CuO composition after 5 days CO exposure, as indicated previously. Thus, the decrease in the relative intensity of the O 1s XPS peaks can throw light on the generation of CO2via the catalytic oxidation of CO by adsorbed O2. On the basis of this consideration, the O 1s/Cu 2p intensity ratio for the CuO NRs after 5 days CO exposure is calculated to be 2.15, smaller by 15.4% than that of the annealed NRs (2.54), indicating CO2 was generated. In addition, the O/Cu atomic ratio will not change if all of the Cu–Oads are adsorbed by CO. Thus, the Cu–Oads species is also formed which also contributes to the IR band above 1080 cm−1. Meanwhile, the generation of the Cu–Oads species means that CO is relatively difficult to adsorb on the Oads in that species, possibly because the oxidizability of its Cu component is not strong enough, as indicated by the relatively low Cu 2p3 binding energy (933.3 eV, Fig. S9 in ESI†).
Therefore, we can conclude from the FTIR results assisted by XPS data that the CuO NRs show catalytic activity in the oxidation of CO, mainly arising from the strong OAC and high Cu oxidation states of the sample.
To further confirm the catalytic activity of the CuO NRs with abundant adsorbed oxygen and high surface oxidation states, we studied the case of octadecane. The octadecane was adsorbed on the NRs in the solid state. After 5 days storage of the octadecane-adsorbed CuO NRs in an O2 atmosphere at room temperature, the resulting FTIR spectrum (purple line in Fig. 5) shows, similarly to the case of CO, a strong broad band at 1086 cm−1, mainly derived from the C–O-like species. Besides, no obvious alkane IR peaks appear above 2900 cm−1. These two points imply that the adsorbed octadecane has been oxidized. This broad band is much stronger than that arising from CO adsorption (green line in Fig. 5), which is mainly due to the reaction products of the oxidation of octadecane formed as a solid state. The two weak peaks at 1594 and 1388 cm−1 can be ascribed to carbonate or carboxylates associated with the support,40,41 which likely result from the oxidation of octadecane, since the annealed NRs did not obviously show such peaks.
The two sharp peaks at 648 and 493 cm−1 (purple line in Fig. 5) are derived from CuO NRs and are blue shifted compared with the CuO IR peaks recorded on other samples, which is mainly due to the relatively higher reduction degree of the CuO induced by octadecane oxidation, as shown by the decrease in the Cu 2p3 binding energy (933.0 eV) and Cu Auger parameter (1850.4 eV) (Fig. S10 in ESI†), leading to the change (increase) in the strength of the Cu–Olattice bond.
Though the CuO NRs exhibit a reduction trend after oxidation of the adsorbed octadecane, the NRs still have the CuO characteristics, indicating the catalytic activity is mainly attributed to the adsorbed oxygen and the resulting electronic structure changes in the NRs. However, the reduction degree of the CuO adsorbed by octadecane is larger than that of adsorbed by CO as can be observed from the Cu 2p3 binding energy for the former (933.0 eV) being lower compared to the latter (933.3 eV), which is mainly due to the relatively stronger reducibility of octadecane compared with that of CO and the presence of the adsorbed octadecane as a solid state rather than a gaseous phase. This indicates that the reduction degree of CuO NRs increases with increasing the reducibility of the environment involved. In contrast, enhancing the oxidizability of the environment can strengthen the oxidizability of the CuO NRs, where the adsorbed oxygen rather than the lattice oxygen plays an important role in the processes of catalytic oxidation reaction.
From the above, we know that the CuO NRs with abundant adsorbed O2 and high surface oxidation states can catalyze the CO oxidation at low temperatures via the reaction between adsorbed oxygen (rather than lattice oxygen) and CO, and the high oxidation states of the Cu component do not fall to monovalence after catalytic reactions.
3.6. Catalytic study of CO conversion over CuO NRs.
The catalytic activity of the CuO NRs was further investigated by performing a CO conversion experiment, and the result is shown in Fig. 6. It can be clearly observed that the CO conversion rate increases with increasing reaction temperature. It is discernible that the catalytic oxidation of CO occurs at 60 °C (inset in Fig. 6), and a 100% CO conversion is achieved at 160 °C. It is known that bulk CuO does not show activity in CO oxidation below 200 °C. Hitherto, the involved CO catalytic oxidation on CuO-based materials is mainly based on the lattice-oxygen oxidation processes, that is, the well-known nucleophilic interfacial Mars–van Krevelen oxidation mechanism.42 In such catalytic processes, CO is adsorbed on a Cu site, and the catalytic activity depends on the Cu–Olattice bond breaking.16 Too much oxygen is detrimental to the catalytic processes, since it blocks the active surface Cu sites.15 However, it also shows that, compared to CO, O2 can preferentially adsorb on the Cu sites at a high O2 concentration, which is exactly the case in our paper. The high O2 concentration enhances the oxidizability of the CuO NRs, leading to improved catalytic activities in the oxidation of the surface adsorbed carbon species, as indicated in Fig. 4. Heat treatment also leads to the enhancement of oxygen adsorption as mentioned above, resulting in high, close to 3+ surface oxidation states for the CuO NRs. CO catalytic oxidation reactions could then occur on the NRs and the high surface oxidation states did not fall to 1+ during the catalytic reactions, as mentioned in Fig. 5. Additionally, increasing temperature increases the catalytic activity for CO oxidation, as shown in Fig. 6. It is known that increasing temperature increases the trend in oxygen adsorption, which increases the oxidation states of a p-type oxide semiconductor such as CuO, thus enhancing the catalytic activity toward CO oxidation on the CuO NRs. Therefore, the rate-determining step can be assigned to the oxidizability of the Cu in CuO NRs in our case. The higher the oxidizability of the sample, the stronger the catalytic activity for CO oxidation. Abundant adsorbed O2 is one of the routes to reaching higher oxidizability of CuO NRs. The poorly-crystallized structures, small sizes, and coarse surfaces enhance the adsorption of O2 at elevated temperatures and so release of the adsorbed O2 does not occur (another paper also reported that adsorbed oxygen on CuO nanostructures could be stable at 160 °C5), which simultaneously enhances the oxidizability of the CuO NRs. From this point of view, the oxidizability is associated with the OAC of the CuO NRs. Increasing the oxidizability of the NRs enhances the dissociation of the adsorbed O2, as well as that of the Cu–Oads bond. (The breaking of the Cu–Oads bond proceeds with the prerequisite of the generation of CO2, that is, the Cu–Oads–(CO)ads transforms to Cu–Ovacancy and CO2.) Thus, the abundant adsorbed oxygen and the high Cu oxidation states are mainly responsible for the observed catalytic activity, which is distinctly different from the conventional case where the lattice oxygen plays a critical role in CO oxidation.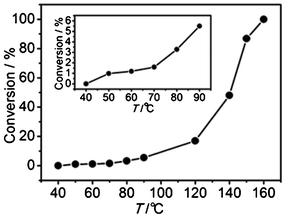 | ||
| Fig. 6 Conversion of CO over CuO NRs as a function of reaction temperature. Inset is a detailed curve of the relatively lower temperature range. | ||
4. Conclusions
In summary, poorly-crystallized CuO NRs were prepared via electrochemical corrosion and oriented-attachment occurring in a KOH liquid membrane supported on a sacrificial Cu foil. The CuO NRs grown via such a pathway were characterized by substantial defect structures and coarse surfaces. Such structure and morphology features, together with the trend for increased valency of Cu in CuO, led to the NRs exhibiting strong OAC and high oxidation states (close to 3+), especially at high temperatures and in a high O2 concentration environment. The NRs could catalyze CO oxidation, and the Cu oxidation states did not drop to 1+ during catalytic reactions. The adsorbed oxygen rather than the lattice oxygen was mainly involved in the catalytic processes, and the rate-controlled step was proposed to be the oxidizability of the CuO NRs, enhanced by high temperatures and an oxidizing environment, such as an O2-rich atmosphere, due to a higher OAC available. The special properties of the poorly-crystallized CuO NRs are greatly desirable for the complete oxidation of various reductive species. Futher enhancing the oxidizability of the NRs via modifying the structure of the sample (such as by doping) is of great significance. This related study is under way.Acknowledgements
This work was supported by the National Nature Science Foundations of China (Grant 20973205 and 90923008), the Natural Science Foundations of Guangdong Province (Grant 8151027501000095, 2008B010600040, and 9251027501000002), Senior Intellectuals Fund of Jiangsu University (Grant No.11JDG100), and the Program of Cooperation of Industry, Education and Academy of Guangdong Province (2011B090400618). In addition, the authors acknowledged Dr Danyan Feng and Dr Zebao Rui for the help in CO conversion experiment, Dr Fangyan Xie in analyses of XPS data, and Shing Chi Ian Wang (at Shenzhen Middle School in Shenzhen) in sample preparation.References
- X. Wang, J. A. Rodriguez, C. Hanson, D. Gamarra, A. Martinez-Arias and M. Fernandez-Garcia, J. Phys. Chem. B, 2006, 110, 428–434 CrossRef CAS.
- R. Kydd, W. Y. Teoh, K. Wong, Y. Wang, J. Scott, Q.-H. Zeng, A.-B. Yu, J. Zou and R. Amal, Adv. Funct. Mater., 2009, 19, 369–377 CrossRef CAS.
- S. Schuyten, P. Dinka, A. S. Mukasyan and E. Wolf, Catal. Lett., 2008, 121, 189–198 CrossRef CAS.
- P. Bera, K. R. Priolkar, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro and N. P. Lalla, Chem. Mater., 2002, 14, 3591–3601 CrossRef CAS.
- H. Zhang, J.-L. Cao, G.-S. Shao and Z.-Y. Yuan, J. Mater. Chem., 2009, 19, 6097–6099 RSC.
- J. F. Xu, W. Ji, Z. X. Shen, S. H. Tang, X. R. Ye, D. Z. Jia and X. Q. Xin, J. Solid State Chem., 1999, 147, 516–519 CrossRef CAS.
- W. Wang, Z. Liu, Y. Liu, C. Xu, C. Zheng and G. Wang, Appl. Phys. A: Mater. Sci. Process., 2003, 76, 417–420 CrossRef CAS.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS.
- C.-J. Jia, Y. Liu, H. Bongard and F. Schüth, J. Am. Chem. Soc., 2010, 132, 1520–1522 CrossRef CAS.
- P. Sangeetha, B. Zhao and Y.-W. Chen, Ind. Eng. Chem. Res., 2010, 49, 2096–2102 CrossRef CAS.
- G. Qiu, S. Dharmarathna, Y. Zhang, N. Opembe, H. Huang and S. L. Suib, J. Phys. Chem. C, 2012, 116, 468–477 CAS.
- S. H. Taylor, G. J. Hutchings and A. A. Mirzaei, Chem. Commun., 1999, 1373–1374 Search PubMed.
- M.-F. Luo, Y.-J. Zhong, X.-X. Yuan and X.-M. Zheng, Appl. Catal., A, 1997, 162, 121–131 CrossRef CAS.
- U. R. Pillai and S. Deevi, Appl. Catal., B, 2006, 64, 146–151 CrossRef CAS.
- Z. Zhong, V. Ng, J. Luo, S.-P. Teh, J. Teo and A. Gedanken, Langmuir, 2007, 23, 5971–5977 CrossRef CAS.
- S. Wendt, M. Knapp and H. Over, J. Am. Chem. Soc., 2004, 126, 1537–1541 CrossRef CAS.
- X. Song, S. Sun, W. Zhang, H. Yu and W. Fan, J. Phys. Chem. B, 2004, 108, 5200–5205 CrossRef CAS.
- U. R. Pillai and S. Deevi, Appl. Catal., B, 2006, 64, 146–151 CrossRef CAS.
- Y. Feng and X. Zheng, Nano Lett., 2010, 10, 4762–4766 CrossRef CAS.
- K. Zhong, J. Xia, H. H. Li, C. L. Liang, P. Liu and Y. X. Tong, J. Phys. Chem. C, 2009, 113, 15514–15523 CAS.
- Y. Chang and H. C. Zeng, Cryst. Growth Des., 2004, 4, 397–402 CAS.
- H. Xu, W. Wang, W. Zhu, L. Zhou and M. Ruan, Cryst. Growth Des., 2007, 7, 2720–2724 CAS.
- C. C. Chusuei, M. A. Brookshier and D. W. Goodman, Langmuir, 1999, 15, 2806–2808 CrossRef CAS.
- A. Hornés, P. Bera, A. L. Cámara, D. Gamarra, G. Munuera and A. Martínez-Arias, J. Catal., 2009, 268, 367–375 CrossRef.
- M. S. P. Francisco, V. R. Mastelaro, P. A. P. Nascente and A. O. Florentino, J. Phys. Chem. B, 2001, 105, 10515–10522 CrossRef CAS.
- M. Iwamoto, Y. Yoda, N. Yamazoe and T. Seiyama, J. Phys. Chem., 1978, 82, 2564–2570 CrossRef CAS.
- J. P. Holgado, G. Munuera, J. P. Espinos and A. R. Gonzalez-Elipe, Appl. Surf. Sci., 2000, 158, 164–171 CrossRef CAS.
- A. R. Gonzalez-Elipe, J. P. Espinos, A. Fernandez and G. Munuera, Appl. Surf. Sci., 1990, 45, 103–108 CrossRef CAS.
- Y. Chi and S. S. C. Chuang, J. Phys. Chem. B, 2000, 104, 4673–4683 CrossRef CAS.
- K. Zhou, R. Wang, B. Xu and Y. Li, Nanotechnology, 2006, 17, 3939–3943 CrossRef CAS.
- J. L. DuBois, P. Mukherjee, T. D. P. Stack, B. Hedman, E. I. Solomon and K. O. Hodgson, J. Am. Chem. Soc., 2000, 122, 5775–5787 CrossRef CAS.
- S. Guha, D. Peebles and T. J. Wieting, Phys. Rev. B: Condens. Matter, 1991, 43, 13092–13101 CrossRef CAS.
- M.-F. Luo, Z.-L. Yan, L.-Y. Jin and M. He, J. Phys. Chem. B, 2006, 110, 13068–13071 CrossRef CAS.
- D. F. Van De Vondel, L. F. Wuyts, G. P. Van Der Kelen and L. Bevernage, J. Electron Spectrosc. Relat. Phenom., 1977, 10, 389–392 CrossRef CAS.
- H. Willemen, L. F. Wuyts, D. F. Van De Vondel and G. P. Van Der Kelen, J. Electron Spectrosc. Relat. Phenom., 1977, 11, 245–250 CrossRef CAS.
- G. Samjeské, K. Komatsu and M. Osawa, J. Phys. Chem. C, 2009, 113, 10222–10228 Search PubMed.
- J. Zhuang, C. N. Rusu and J. J. T. Yates, J. Phys. Chem. B, 1999, 103, 6957–6967 CrossRef CAS.
- Y.-N. Sun, L. Giordano, J. Goniakowski, M. Lewandowski, Z.-H. Qin, C. Noguera, S. Shaikhutdinov, G. Pacchioni and H.-J. Freund, Angew. Chem., Int. Ed., 2010, 49, 4418–4421 CrossRef CAS.
- B. White, M. Yin, A. Hall, D. Le, S. Stolbov, T. Rahman, N. Turro and S. O'Brien, Nano Lett., 2006, 6, 2095–2098 CrossRef CAS.
- T. Shishido, T. Miyatake, K. Teramura, Y. Hitomi, H. Yamashita and T. Tanaka, J. Phys. Chem. C, 2009, 113, 18713–18718 CAS.
- A. Yee, S. J. Morrison and H. Idriss, J. Catal., 2000, 191, 30–45 CrossRef CAS.
- G. I. Golodets, Heterogeneous Catalytic Reactions Involving Molecular Oxygen (Elsevier, Amsterdam, 1983, p. 280) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Proof of the occurrence of electrochemical corrosion. HRTEM image of the CuO NRs without annealing. SEM image showing a floc-like structure. Morphology and structure transformation from Cu(OH)2 nanowires to CuO NRs. Cu LMM Auger line and C 1s XPS peak of annealed NRs. XPS of the CuO NRs with ion sputtering surface treatment. XPS of the newly-prepared NRs without annealing. Raman spectra of the newly-prepared CuO NRs and the 5-day O2 exposed CuO NRs. Full scan Raman spectrum of the annealed CuO NRs. Cu 2p and O 1 s photoelectron lines and Cu LMM Auger line of the NRs after exposure to CO at 60 °C for 5 days. Cu 2p photoelectron line and Cu LMM Auger line of the octadecane-adsorbed CuO NRs after 5 days exposure to O2 at room temperature. See DOI: 10.1039/c2ra21149a |
| This journal is © The Royal Society of Chemistry 2012 |