Polymer-supported hexaethylene glycolic ionic liquid: efficient heterogeneous catalyst for nucleophilic substitutions including fluorinations†
Vinod H.
Jadhav
,
Hwan-Jeong
Jeong
,
Seok
Tae Lim
,
Myung-Hee
Sohn
and
Dong
Wook Kim
*
Department of Nuclear Medicine, Cyclotron Research Center, Research Institute of Clinical Medicine, Chonbuk National University Medical School, Jeonju, Jeonbuk 561-712, Korea. E-mail: kimdw@chonbuk.ac.kr; Fax: +82 63 255 1172; Tel: +82 63 250 2396
First published on 22nd June 2012
Abstract
Polymer-supported hexaethylene glycol substituted imidazolium salts (PS[hexaEGim][OMs] and PS[dihexaEGim][OMs]) were prepared, and were demonstrated to act as highly efficient heterogeneous catalysts in various nucleophilic substitution reactions using the corresponding alkali metal salts. Furthermore, it was observed that PS[hexaEGim][OMs] could indeed be reused repeatedly without the loss of its catalytic activity in the nucleophilic fluorination using KF, affording the corresponding product in a high yield (approximately 95%) in each cycle.
Introduction
Much attention has been paid recently to polymer-supported chemistry, mainly due to the inherent properties of the polymer backbone, which allow easy recovery by simple filtration, recycling, and reuse of the catalyst.1 Immobilization of the catalyst on a polymeric support such as silica gel, metal particle, and polystyrene resins have been widely-used for this purpose.2 Functionalized polystyrenes are especially favored due to their stability and chemical inertness.3 Moreover, the solvation property of polystyrenes is crucial for the reaction rate, and provides a microenvironment caused by the reactants within the polymeric support that is unique.3Nucleophilic substitution reactions employing alkali metal salts as nucleophile sources have occupied a privileged position in organic chemistry, although the low nucleophilicity and solubility of these metal salts in nonpolar organic solvents makes the process difficult.4 To overcome this problem, various phase transfer catalysts (PTCs) such as crown ether derivatives5 and quaternary ammonium salts6 systems have been generally used in polar aprotic solvents such as DMF and DMSO.7 In particular, polymer-supported PTCs have been developed for nucleophilic displacement for the easy isolation of products and reuse of these catalysts.8 However, these heterogeneous PTCs show low reactivity compared with their homogeneous form.8,9
Recent studies have revealed that protic solvent systems such as tert-alcohols10 and n-oligoethylene glycols11 are effective reaction media for nucleophilic substitutions, by virtue of their enhanced reactivity of alkali metal salts as a nucleophile source, in particular metal fluorides, by the formation of hydrogen (H)-bonds between nucleophiles and hydroxyl groups of these protic solvents.
Ionic liquids (ILs), especially imidazolium based ILs, play a crucial role in a variety of chemical reactions due to their unique chemical and physical properties.12 In particular, ILs and polymer-supported ILs enhance the reactivity of alkali metal salt nucleophiles in nucleophilic substitution reactions.13,14 More recently, we reported tailor-made oligoethylene glycolic ionic liquids (oligoEGILs) as organic catalysts designed for nucleophilic fluorination.15 For the recovery and reuse of these oligoEGILs as well as easy separation more in favor of “green chemistry”, we planned to immobilize oligoEGIL on polystyrene resin. Herein, we introduce polystyrene-supported oligoEGIL (PSoligoEGIL) as efficient heterogeneous catalysts for nucleophilic substitution reactions including fluorination with the corresponding alkali metal salts in a tert-alcohol medium.
Results and discussion
1. Synthesis of polystyrene-supported hexaethylene glycol substituted imidazolium salts – (a) PS[hexaEGim][OMs] and (b) PS[dihexaEGim][OMs]
The PSoligoEGILs PS[hexaEGim][OMs] (PS = polymer support; hexaEGim = 3-n-hexaethylene glycolic imidazolium cation; OMs = mesylate anion) and PS[dihexaEGim][OMs] (dihexaEGim = 1,3-n-dihexaethylene glycolic imidazolium cation) were prepared by the procedure shown in Scheme 1. The treatment of Merrifield resin161 (1% divinylbenzene, 3.9 mmol Cl/g) with imidazole in dried THF for 4 days afforded an imidazole substituted polystyrene resin 2. This resin 2 was then reacted with the mesylate 3 in CH3CN at 90 °C for 4 days to obtained PS[hexaEGim][OMs] (1.09 mmol of hexaEGim moiety per gram of polymer-supported product obtained). For preparation of PS[dihexaEGim][OMs], the treatment of the same Merrifield resin 1 with hexaethylene glycol (4) for 4 days yielded resin 5. The mesylation of the resin 5 with mesyl chloride provided a mesylated resin 6. Finally, the mesylated resin 6 was reacted with hexaethylene glycolic imidazole (7) for 4 days to obtain PS[dihexaEGim][OMs] (0.49 mmol of dihexaEGim/g). The PSoligoEGILs were characterized by 13C NMR (solid state) spectroscopy and by elemental analysis.![Preparation of polystyrene-supported hexaethylene glycol substituted imidazolium salts (a) PS[hexaEGim][OMs] and (b) PS[dihexaEGim][OMs].](/image/article/2012/RA/c2ra21142d/c2ra21142d-s1.gif) | ||
| Scheme 1 Preparation of polystyrene-supported hexaethylene glycol substituted imidazolium salts (a) PS[hexaEGim][OMs] and (b) PS[dihexaEGim][OMs]. | ||
2. Nucleophilic fluorination using alkali metal fluoride in the presence of PSoligoEGILs
Table 1 summarizes the data of the reactivity of PS[hexaEGim][OMs] and PS[dihexaEGim][OMs] for nucleophilic fluorination of 2-(3-methanesulfonyloxypropyl)naphthalene (8), as a model compound, with various alkali metal fluorides, together with those obtained in other polymer-supported systems reported previously by us.11b,15,17 Firstly, the nucleophilic fluorination reaction of 8 with KF in the presence of PS[hexaEGim][OMs] (0.5 equiv) in tert-amyl alcohol proceeded for 4 h at 100 °C, which produced the desired fluoro-product 9 in excellent yield (96%, entry 1), whereas the same reaction for 12 h in the absence of catalyst yielded only 10% of the fluorinated product 9 with 90% of unreacted starting material (entry 2). Encouragingly, the results of entries 1, 3 and 8 indicated that both PS[hexaEGim][OMs] and PS[dihexaEGim][OMs] as immobilized catalytic systems have a similar reactivity to a non-immobilized 3-n-hexaethylene glycolic methyl-imidazolium meylate [hexaEGmim][OMs] system. A comparison with entries 1 and 4 established that the hexaethylene glycol chain on PS[hexaEGim][OMs] allows its catalytic activity to increase compared with PS[hmim][BF4] (hmim = 1-n-hexyl-3-methylimidazolium). Furthermore, entry 5 shows that only 29% of the fluoro-product 9 was converted from the mesylate 8 even after a reaction time of 24 h by the same fluorination in the presence of polystyrene-supported pentaethylene glycol (PSpentaEG). These results indicates that the imidazolium salt core of PS[hexaEGim][OMs] plays a crucial role in its high catalytic reactivity in the reactions as shown in the comparison with entries 1 and 5. The other alkali metals, such as RbF and CsF, also can be efficiently activated by the PS[hexaEGim][OMs] heterogeneous catalyst in this substitution reaction (entries 6 and 7). PS[dihexaEGim][OMs], which have an additional hexaethylene glycol linker, displays better catalytic reactivity that PS[hexaEGim][OMs]; the same fluorination with KF or CsF in the presence of PS[dihexaEGim][OMs] proceeds rapidly and provides an excellent yield of the fluoroalkane 9 (97% in 3.5 h and 30 min for entries 8 and 9, respectively). To investigate how many times PS[hexaEGim][OMs] could be reused, the fluorination was performed repeatedly under the conditions given in entry 1 in Table 1. We observed that PS[hexaEGim][OMs] could indeed be reused repeatedly without the loss of its catalytic activity, and this reaction afforded product 9 in a high yield (approximately 95%) in each cycle.
| Entry | Catalyst or promoter (0.5 equivb) | MF | Time (h) | Yield (%)c |
|---|---|---|---|---|
| a All reactions were carried out on a 1.0 mmol reaction scale of mesylate 8 using 3 mmol of MF in 4.0 mL of solvent at 100 °C. b Equivalent amount of the ionic liquid portion, not PS[hexaEGim][OMs] or PS[dihexaEGim][OMs]. c Yields were determined by 1H NMR spectroscopy. d Isolated yields. e ref. 15. f ref. 17. g ref. 11b. | ||||
| 1 | PS[hexaEGim][OMs] | KF | 4 | 96 (93)d |
| 2 | — | KF | 12 | 10 |
| 3e | [hexaEGmim][OMs] | KF | 3 | 98 |
| 4f | PS[hmim][BF4] | KF | 7 | 91d |
| 5g | PSpentaEG (1 equiv) | KF | 24 | 29 |
| 6 | PS[hexaEGim][OMs] | RbF | 1.5 | 93d |
| 7 | PS[hexaEGim][OMs] | CsF | 45 min | 96 (94)d |
| 8 | PS[dihexaEGim][OMs] | KF | 3.5 | 97 (94)d |
| 9 | PS[dihexaEGim][OMs] | CsF | 30 min | 97 (93)d |
3. Various SN2 reactions in the presence of PS[hexaEGim][OMs]
Diverse nucleophilic transformations were also attempted with various alkali metal salts as nucleophile sources in the presence of 0.5 equiv. of PS[hexaEGim][OMs] in a tert-alcohol medium (Table 2). All transformation reactions – acetoxylation, thioacetoxylation, nitrilation, halogenations and azidation – of the mesylate 8 or bromo-substrate 10 with the corresponding alkali metal salts proceed nearly quantitatively (entries 1–7, 97–99%).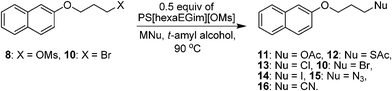
| Entry | X | MNu | Time (h) | Yield (%)b |
|---|---|---|---|---|
| a All reactions were carried out on a 1.0 mmol reaction scale of mesylate 8 or bromoalkane 10 using 3 mmol of MNu in the presence of 0.5 equiv of PS[hexaEGim][OMs] in 4.0 mL of solvent at 90 °C. b Isolated yields. | ||||
| 1 | OMs | KOAc | 1 | 99 |
| 2 | OMs | KSAc | 0.5 | 98 |
| 3 | OMs | KCl | 8 | 99 |
| 4 | OMs | KBr | 3 | 99 |
| 5 | OMs | KI | 1 | 98 |
| 6 | Br | NaN3 | 1.5 | 99 |
| 7 | Br | KCN | 2 | 97 |
4. Nucleophilic substitutions of various substrates in the presence of PS[hexaEGim][OMs]
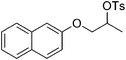
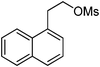
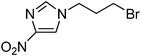
In order to demonstrate that this heterogeneous PS[hexaEGim][OMs] catalyst is generally applicable to the other transformation reactions of various substrates, the nucleophilic substitutions of various halide or sulfonate substrates in the presence of 0.5 equiv of PS[hexaEGim][OMs] were characterized (Table 3). Entries 1–5 show the scope of selectivity on nucleophilic substitutions. Initially, in entries 1–4, the fluorination of base sensitive substrates such as a sec-alkyl tosylate and 1-(2-mesylethyl)naphthalene with both KF and CsF in the presence of PS[hexaEGim][OMs] in tert-amyl alcohol proceeded in a highly chemoselective manner with the corresponding fluoro-products produced in excellent yields (90–92%). Moreover, the nucleophilic methoxylation of mesylate 8 even with strong basic KOCH3 nucleophile under this PS[hexaEGim][OMs]/tert-amyl alcohol condition provided the methoxylated product in a reasonable yield (entry 5, 63%). The fluorination of primary triflate of α-D-galactopyranose proceeded smoothly to afford the desired fluorosugar in 95% yield (entry 6). Entries 7–9 show that the transformation reactions such as fluorination, acetoxylation and azidation transformation reactions of nitroimidazolic mesylate or bromide substrate in the presence of PS[hexaEGim][OMs] were completed within 2 h and provided the corresponding nitroimidazolic derivatives in good yield (89–98%). 3-Chloro-picoline-N-oxide was converted to 3-fluoro-picoline-N-oxide with a yield of 69% (entry 10). In the final examples, biologically important drugs or drug intermediates such as 5,8-dimethoxy-4-fluoropropylquinoline18 and fluoropropyl ciprofloxacine19 were produced from their corresponding mesylate precursors in excellent yields (entries 11 and 12, 97 and 95%, respectively).| Entry | Substrate | MNu | Time (h) | T/°C | Yield (%)b | Comment |
|---|---|---|---|---|---|---|
| a All reactions were carried out on a 1.0 mmol scale of substrate with 3.0 equiv of MNu and 0.5 equiv of PS[hexaEGim][OMs] in 4.0 mL of t-amyl alcohol. b Yields were determined by 1H NMR spectroscopy. c 4.0 mL of CH3CN was used instead of 4.0 mL t-amyl alcohol. | ||||||
| 1 |
|
KF | 8 | 100 | 92 | 8% alkene |
| 2 | CsF | 3 | 100 | 91 | 9% alkene | |
| 3 |
|
KF | 5 | 100 | 90 | 10% alkene |
| 4 | CsF | 1 | 100 | 91 | 9% alkene | |
| 5 |

|
KOMe | 4 | 90 | 63 | 5% alkene 32% alcohol |
| 6 |

|
KF | 1 | 90 | 95 | — |
| 7 |

|
KF | 2 | 90 | 89 | 7% alkene |
| 8c |
|
KOAc | 0.7 | 90 | 98 | — |
| 9c | NaN3 | 1 | 90 | 97 | — | |
| 10 |
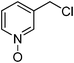
|
KF | 7 | 90 | 69 | 31% alcohol |
| 11 |
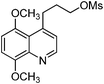
|
KF | 4 | 100 | 97 | — |
| 12 |
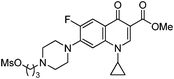
|
KF | 10 | 100 | 95 | — |
Conclusion
In summary, we have prepared new polystyrene-supported oligoethylene glycolic imidazolium salts (PSoligoEGIL) as highly efficient heterogeneous catalysts for nucleophilic fluorination and other substitutions using alkali metal nucleophile sources. These PSoligoEGILs are technically attractive in terms of simplicity of purification and the ready recovery and reuse of catalysts, and also significantly enhance the reactivity of alkali metal salts, in particular, a KF with tert-alcohol system. Further studies are currently in progress to develop more efficient heterogeneous ILs using various structural modifications for different organic transformations including 18F-labeling for PET applicationsExperimental
General
Unless otherwise noted all reagents and solvents were commercially available. Reaction progress was followed by TLC on 0.25 mm silica gel glass plates containing F-254 indicator. Visualization on TLC was monitored by UV light. Flash chromatography was performed with 230–400 mesh silica gel. 1H and 13C NMR spectra were recorded on a 400 or 600 MHz spectrometer, and chemical shifts were reported in δ units (ppm) relative to tetramethylsilane. Solid-state 13C NMR spectra were recorded on a 600 MHz spectrometer at RT. Low- and high-resolution electron impact (EI, 70 eV) spectra were obtained. Elemental analyses were performed by the Microanalytical Service Laboratory of the University of Illinois.Resin 2
Imidazole (2.7 g, 39.8 mmol, 10 equiv) was added to a suspension of NaH (60% in mineral oil, 0.955 mg, 39.8 mmol, 10 equiv) in dried THF at 0 °C, and the mixture was stirred for 30 min at 0 °C. Merrifield peptide resin (1.0 g, 3.9 mmol, 1.0 equiv, 1% DVB, 3.9 mmol Cl/g) was then added to the reaction mixture followed by tetra-n-butylammonium iodide (1.5 g, 4.0 mmol, 1.0 equiv), and the reaction mixture was stirred over 4 days at 25 °C. After filtration, the resin was washed repeatedly with THF, water, acetone, water, methanol, and finally with dichloromethane. After drying under high vacuum, 1.2 g of resin 2 was obtained and identified by solid state NMR and elemental analysis: 13C NMR (solid state) δ 41 (aliphatic polystyrene skeleton), 120–146 (aromatic polystyrene skeleton and 2 carbon of imidazole), 181 (carbon from imidazole). Anal. N, 9.36 (3.2 mmol imidazole portion/g); Cl, 0.86.PS[hexaEGim][OMs]
17-Hydroxy-3,6,9,12,15-pentaoxaheptadecyl methanesulfonate (3), (7.49 g, 22.54 mmol) was added dropwise to the mixture of resin 2 (1 g, 3.22 mmol) in CH3CN (25 mL). The reaction mixture was stirred at 90 °C for 4 days. After filtration, the resin was washed repeatedly with THF, water, acetone, water, methanol, and finally with dichloromethane. After drying under high vacuum, 1.3 g of PS[hexaEGim][OMs] was obtained and identified by solid state NMR and elemental analysis: 13C NMR (solid state) δ 50 (-CH3 from mesylate), 71 (ether carbons), 123–137 (aromatic polystyrene skeleton). Anal. N, 3.92. (1.09 mmol ionic liquid portion/g).Resin 5
The preparation followed the procedure of resin 2 except using hexaethylene glycol (11.2 g, 39.8 mmol, 10 equiv.) instead of using imidazole and 1.5 g of resin 5 was obtained and identified by solid state NMR and elemental analysis: 13C NMR (solid state) δ 41 (aliphatic polystyrene skeleton), 62 (terminal hydroxy methylene carbons), 71–74 (ether carbons) 128–154 (aromatic polystyrene skeleton). Anal. Cl, 0.77.Resin 6
Methanesulfonyl chloride (1.4 mL, 17.0 mmol) was added to a mixture of the resin 5 (1 g 1.7 mmol hexaEG portion) and triethylamine (2.4 mL, 17.0 mmol) in methylene chloride (50 mL) at 0 °C and allowed to come to room temperature for stirring overnight. After filtration, the resin was washed repeatedly with THF, water, acetone, water, methanol, and finally with dichloromethane. After drying under high vacuum, 1.1 g of resin 6 was obtained and identified by solid state NMR and elemental analysis: 13C NMR (solid state) δ 38 (–CH3 from mesylate), 41 (aliphatic polystyrene skeleton), 71 (ether carbons) 128–146 (aromatic polystyrene skeleton). Anal. Cl, 0.17.PS[dihexaEGim][OMs]
17-(1H-Imidazol-1-yl)-3,6,9,12,15-pentaoxaheptadecan-1-ol (7) (3.47 g, 10.43 mmol) was added dropwise to a mixture of resin 6 (1 g, 1.49 mmol) in CH3CN (25 mL). The reaction mixture was stirred at 90 °C for 4 days. After filtration, the resin was washed repeatedly with THF, water, acetone, water, methanol, and finally with dichloromethane. After drying under high vacuum, 1.1 g of PS[dihexaEGim][OMs] was obtained and identified by solid state NMR and elemental analysis: 13C NMR (solid state) δ 41 (aliphatic polystyrene skeleton), 50 (–CH3 from mesylate), 71 (ether carbons) 128–146 (aromatic polystyrene skeleton). Anal. N, 1.76 (0.49 mmol ionic liquid portion/g).Typical procedure of the fluorination in Table 1
KF (174 mg, 3.0 mmol) was added to the mixture solution of mesylate 8 (281 mg, 1.0 mmol) and PS[hexaEGim][OMs] (500 mg, 0.5 mmol) in tert-amyl alcohol (4 mL). The reaction mixture was stirred for 4 h at 100 °C. We determined the reaction time by checking TLC. The reaction mixture was filtered and washed with diethyl ether. The filtrate was evaporated under reduced pressure. Flash column chromatography (10% EtOAc/hexanes) of the filtrate afforded 191 mg (0.93 mmol, 93%) of 2-(3-fluoropropoxy)naphthalene (9) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 2.14–2.39 (m, 2H), 4.24 (t, J = 6.2 Hz, 2H), 4.72 (dt, J = 46.8, 5.8 Hz, 2H), 7.16–7.22 (m, 2H), 7.34–7.53 (m, 2H), 7.76–7.83 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 30.4 (d, J = 20.1 Hz), 63.6 (d, J = 25.3 Hz), 80.8 (d, J = 163.9 Hz), 106.8, 118.8, 123.6, 126.4, 126.7, 127.6, 129.1, 129.4, 134.6, 156.7; 19F NMR (400 MHz, CDCl3) δ −24.80; MS (EI) m/z 204 (M+); HRMS (EI) m/z calcd for C13H13FO(M+) 204.0950, Found 204.0932.Typical procedure for acetoxylation
KOAc (295 mg, 3 mmol) was added to a mixture solution of mesylate 8 (281 mg, 1.0 mmol) and PS[hexaEGim][OMs] (500 mg, 0.5 mmol) in CH3CN (4 mL) in a reaction vial. The reaction mixture was stirred for 1 h at 90 °C. The reaction mixture was filtered and washed with diethyl ether. The filtrate was evaporated under reduced pressure. Flash column chromatography (10% EtOAc/hexanes) of the filtrate afforded 241 mg (0.99 mmol, 99%) of 2-(3-acetoxypropoxy)naphthalene (11); 1H NMR (600 MHz, CDCl3) δ 2.08 (s, 3H), 2.18–2.22 (m, 2H), 4.17 (t, J = 6.18 Hz, 2H), 4.31 (t, J = 6.18 Hz, 2H), 7.10–7.15 (m, 2H), 7.33 (t, J = 7.56 Hz, 1H), 7.43 (t, J = 6.90 Hz, 1H), 7.71–7.77 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 21.1, 28.7, 61.5, 64.4, 106.6, 118.9, 123.7, 126.5, 126.8, 127.7, 129.0, 129.5, 134.6, 156.8, 171.2; MS (EI) m/z 244 (M+); HRMS (EI) m/z calcd for C15H16O3 (M+) 244.1099. Found 244.1096.2-(3-Thioacetoxypropoxy)naphthalene (12)
The preparation followed the typical procedure of the acetoxylation except that KSAc (424 mg, 3.0 mmol) was used and 255 mg (0.98 mmol, 98%) of 2-(3-thioacetoxypropoxy)naphthalene (12) was obtained; 1H NMR (600 MHz, CDCl3) δ 2.11–2.16 (m, 2H), 2.34 (s, 3H), 3.10 (t, J = 6.84 Hz, 2H), 4.13 (t, J = 6.18 Hz, 2H), 7.11–7.15 (m, 2H), 7.33 (t, J = 6.84 Hz, 1H), 7.44 (t, J = 6.90 Hz, 1H), 7.71–7.76 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 26.1, 29.3, 30.8, 66.2, 106.7, 118.9, 123.7, 126.5, 126.8, 127.7, 129.0, 129.5, 134.6, 156.8, 195.9; MS (EI) m/z 260 (M+); HRMS (EI) m/z calcd for C15H16O2S (M+) 260.0871. Found 260.0874.Procedure of the halogenation (entries 3–4 in Table 2)
The preparation followed the typical procedure of acetoxylation except that KX (X = Cl, Br, or I) was used and 217 mg (0.99 mmol, 99%) of 2-(3-chloropropoxy)naphthalene (13), 262 mg (0.99 mmol, 99%) of 2-(3-bromopropoxy)naphthalene (10), or 305 mg (0.98 mmol, 98%) of 2-(3-iodopropoxy)naphthalene (14) was obtained, respectively.2-(3-Chloropropoxy)naphthalene (13)
1H NMR (600 MHz, CDCl3) δ 2.28–2.32 (m, 2H), 3.79 (t, J = 6.2 Hz, 2H), 4.23 (t, J = 6.2 Hz, 2H), 7.12–7.15 (m, 2H), 7.34 (t, J = 6.8 Hz, 1H), 7.44 (t, J = 6.8 Hz, 1H), 7.72–7.77(m, 3H); 13C NMR (100 MHz, CDCl3) δ 32.17, 41.60, 64.17, 106.56, 118.76, 123.65, 126.38, 126.71, 127.61, 128.94, 129.41, 134.44, 156.57; MS (EI) m/z 220 (M+), 144, 115; HRMS (EI) m/z calcd for C13H13OCl (M+) 220.0655. Found 220.0667.2-(3-Bromopropoxy)naphthalene (10)
1H NMR (400 MHz, CDCl3) δ 2.36–2.43 (m, 2H), 3.67 (t, J = 6.6 Hz, 2H), 4.23 (t, J = 5.6 Hz, 2H), 7.14–7.17 (m, 2H), 7.34–7.49 (m, 2H), 7.74–7.80 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 30.1, 32.2, 65.2, 106.6, 118.8, 123.7, 126.4, 126.7, 127.6, 128.9, 129.4, 134.4, 156.5; MS (EI) m/z 264 (M+), 266 (M+), 144 (100), 115; HRMS (EI) m/z calcd for C13H13O79Br (M+) 264.0150. Found 264.0151.2-(3-Iodopropoxy)naphthalene (14)
1H NMR (400 MHz, CDCl3) 2.32–2.38 (m, 2H), 3.43 (t, J = 6.6 Hz, 2H), 4.16 (t, J = 5.8 Hz, 2H), 7.15–7.17 (m, 2H), 7.35–7.49 (m, 2H), 7.49–7.80 (m, 3H); 13C NMR (100 MHz, CDCl3) 2.7, 32.8, 67.1, 106.6, 118.8, 123.6, 126.4, 126.7, 127.6, 128.1, 129.4, 134.4, 156.5; MS (EI) m/z 312 (M+), 185, 144, 115 (100); HRMS (EI) m/z Calcd for C13H13OI (M+) 312.0011. Found 312.0006.2-(3-Azidopropoxy)naphthalene (15)
NaN3 (195 mg, 3.0 mmol) was added to a mixture solution of 2-(3-bromopropoxy)naphthalene (10) (264 mg, 1.0 mmol) and PS[hexaEGim][OMs] (500 mg, 0.5 mmol) in CH3CN (4 mL). The reaction mixture was stirred for 1.5 h at 90 °C. The reaction mixture was filtered and washed with diethyl ether. The filtrate was evaporated under reduced pressure. Flash column chromatography (10% EtOAc/hexanes) of the filtrate afforded 224 mg (0.99 mmol, 99%) of 2-(3-azidopropoxy)naphthalene (15); 1H NMR (600 MHz, CDCl3) δ 2.10–2.14 (m, 2H), 3.57 (t, J = 6.9 Hz, 2H), 4.17 (t, J = 6.2 Hz, 2H), 7.13–7.16 (m, 2H), 7.32–7.35 (m, 1H), 7.42–7.45 (m, 1H), 7.71–7.77 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 28.9, 48.4, 64.6, 106.7, 118.8, 123.8, 126.5, 126.8, 127.7, 129.1, 129.5, 134.6, 156.7; MS (EI) m/z 227 (M+), 169, 143(100), 115; HRMS (EI) m/z calcd for C13H13N3O (M+) 227.1059. Found 227.1060.2-(3-Cyanopropoxy)naphthalene (16)
The preparation followed the procedure for the preparation of 2-(3-azidopropoxy)naphthalene (15) except using KCN (195 mg, 3.0 mmol) instead of using NaN3 and 204 mg (0.97 mmol, 97%) of 2-(3-cyanopropoxy)naphthalene (16) was obtained; 1H NMR (600 MHz, CDCl3) δ 2.18–2.23 (m, 2H), 2.63 (t, J = 6.90 Hz, 2H), 4.19 (t, J = 6.18 Hz, 2H), 7.12–7.14 (m, 2H), 7.35 (t, J = 6.90 Hz, 1H), 7.45 (t, J = 8.22 Hz, 1H), 7.72–7.78 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 14.4, 25.6, 65.4, 106.9, 118.8, 119.3, 124.0, 126.7, 126.9, 127.7, 129.2, 129.6, 134.5, 156.4; MS (EI) m/z 211 (M+); HRMS (EI) m/z calcd for C14H13NO (M+) 211.0997. Found 211.0998.2-(2-Fluoropropoxy)naphthalene
The preparation followed the typical procedure of fluorination and 187 mg (0.92 mmol, 92%) of 2-(2-fluoropropoxy)naphthalene was obtained; 1H NMR (600 MHz, CDCl3) δ 1.43 (dd, J = 30.6, 7.2 Hz, 3H), 4.03–4.15 (m, 2H), 4.93–4.99 (m, 0.5H), 5.03–5.07 (m, 0.5H), 7.05 (s, 1H), 7.12 (dd, J = 12, 3 Hz, 1H), 7.27 (t, J = 15 Hz, 1H), 7.37 (t, J = 15 Hz, 1H), 7.64–7.70 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 17.6 (d, J = 86.2 Hz), 70.8 (d, J = 97.62), 88.5 (d, J = 672.1), 106.8, 118.9, 123.9, 126.6, 126.9, 127.8, 129.3, 129.7, 134.5, 156.5; MS (EI) m/z 204 (M+); HRMS (EI) m/z calcd for C13H13FO(M+) 204.0950, found 204.0952.1-(2-Fluoroethyl)naphthalene
The preparation followed the typical procedure of fluorination and 156 mg (0.90 mmol, 90%) of 1-(2-fluoroethyl)naphthalene was obtained; 1H NMR (600 MHz, CDCl3) δ 3.44 (dt, J = 13.7, 6.9 Hz, 2H), 4.75 (dt, J = 47.5, 6.9 Hz, 2H), 7.30–7.37 (m, 2H), 7.41–7.49 (m, 2H), 7.70 (d, J = 8.2 Hz, 1H), 7.80 (d, J = 8.2 Hz, 1H), 7.95 (d, J = 8.2 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 33.9 (d, J = 20.1 Hz), 83.6 (d, J = 168.0 Hz), 123.4, 125.6, 125.7, 126.3, 127.3, 127.7, 128.9, 132.0, 132.8 (d, J = 8.5 Hz), 133.9; MS (EI) m/z 174 (M+), 141 (100); Anal. calcd: C, 82.73; H, 6.36, found: C, 82.63; H, 6.34.2-(3-Methoxypropoxy)naphthalene
KOMe (211 mg, 3 mmol) was added to the mixture solution of mesylate 8 (281 mg, 1.0 mmol) and PS[hexaEGim][OMs] (500 mg, 0.5 mmol) in tert-amyl alcohol (4 mL). The reaction mixture was stirred for 4 h at 90 °C. We determined the reaction time by checking TLC. The reaction mixture was filtered and washed with diethyl ether (15 mL). The filtrate was evaporated under reduced pressure. Flash column chromatography (10% EtOAc/hexanes) of the filtrate afforded 136 mg (0.63 mmol, 63%) of 2-(3-methoxypropoxy)naphthalene; 1H NMR (600 MHz, CDCl3) δ 2.09–2.16 (s, 2H), 3.37 (S, 3H), 3.60 (t, J = 6.18 Hz, 2H), 4.18 (t, J = 6.18 Hz, 2H), 7.13–7.15 (m, 2H), 7.31 (t, J = 6.84 Hz, 1H), 7.42 (t, J = 6.84 Hz, 1H), 7.71–7.77 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 29.7, 58.9, 64.9, 69.4, 106.7, 119.0, 123.6, 126.4, 126.8, 127.7, 128.9, 129.4, 134.7, 157.0; MS (EI) m/z 216 (M+); HRMS (EI) m/z calcd for C14H16O2 (M+) 216.1150, found 216.1149.1,2:3,4-Di-O-isopropylidene-6-fluoro-6-deoxy-α-D-galactopyranose
The preparation followed the typical procedure of fluorination and 237 mg (0.95 mmol, 95%) of 1,2:3,4-di-O-isopropylidene-6-fluoro-6-deoxy-α-D-galactopyranose was obtained; 1H NMR (600 MHz, CDCl3) δ 1.34 (s, 6H), 1.45 (m, 3H), 1.55 (m, 3H), 4.06–4.10 (m, 1H), 4.26–4.28 (m, 1H), 4.34–4.37 (m, 1H), 4.48–4.65 (m, 3H), 5.56 (d, J = 5.5 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 24.48, 24.98, 25.98, 26.10, 66.69 (d, J = 21.5 Hz), 70.47, 70.59 (d, J = 5.8 Hz), 70.64, 82.15 (d, J = 168.00 Hz), 96.25, 108.88, 109.73; HRMS (ESI+) m/z calcd for C12H20FO5 + H (M+ + H) 263.1295, found 263.1298.1-(3-Fluoropropyl)-4-nitroimidazole
The preparation followed the typical procedure of fluorination and 153 mg (0.89 mmol, 89%) of 1-(3-fluoropropyl)-4-nitroimidazole was obtained; 1H NMR (600 MHz, CDCl3) δ 2.20–2.28 (m, 2H), 4.24 (t, J = 6.8 Hz, 2H), 4.50 (dt, J = 46.68 and 5.52 Hz, 2H), 7.49 (s, 1H), 7.81 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 31.46 (d, J = 20.1 Hz), 44.42, (d, J = 4.3 Hz), 79.64 (d, J = 166.6 Hz), 119.20, 136.23, 148.44; MS (EI) m/z 173 (M+, 100), 127, 61; HRMS (EI) calcd for C6H8FN3O2: (M+) 173.0601, found 173.0633.1-(3-Acetoxypropyl)-4-nitroimidazole
The preparation followed the typical procedure of acetoxylation and 208 mg (0.98 mmol, 98%) of 1-(3-acetoxypropyl)-4-nitroimidazole was obtained; 1H NMR (600 MHz, CDCl3) δ 2.08 (s, 3H), 2.19–2.23 (m, 2H), 4.13–4.18 (m, 4H), 7.49 (s, 1H), 7.84 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 20.86, 29.99, 45.43, 60.53, 119.25, 136.15, 148.34, 170.80; MS (EI) m/z 213 (M+), 153, 140 (100), 68; HRMS (EI) calcd for C8H11N3O4: (M+) 213.0750, found 213.0721.1-(3-Azidopropyl)-4-nitroimidazole
The preparation followed the procedure for the preparation of 2-(3-azidopropoxy)naphthalene (15) and 191 mg (0.97 mmol, 97%) of 1-(3-azidopropyl)-4-nitroimidazole was obtained; 1H NMR (600 MHz, CDCl3) δ 2.08–2.13 (m, 2H), 3.41 (t, J = 6.2 Hz, 2H), 4.17 (t, J = 6.8 Hz, 2H), 7.48 (s, 1H), 7.81 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 29.95, 45.24, 47.60, 119.23, 136.22, 148.40; MS (FAB) m/z 197 (M+); HRMS (FAB) calcd for C6H8N6O2: (M+) 197.0787, found 197.0785.3-Fluoropicoline N-oxide
The preparation followed the typical procedure of fluorination and 87 mg (0.69 mmol, 69%) of 3-fluoropicoline N-oxide was obtained; 1H NMR (600 MHz, CDCl3) δ 5.38 (d, J = 46.7 Hz, 2H), 7.25–7.28 (m, 1H), 7.32 (t, J = 6.2 Hz, 1H), 8.20 (d, J = 6.2 Hz, 1H), 8.26 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 80.62 (d J = 170.9 Hz), 123.95 (d, J = 5.8 Hz), 126.10, 135.93 (d, J = 18.7 Hz), 137.66 (d, J = 7.2 Hz), 139.16; MS (EI) m/z 127 HRMS (EI) calcd for C6H6FNO: (M+) 127.0433, found 127.0432.4-(3-Fluoropropyl)-5,8-dimethoxyquinoline
The preparation followed the typical procedure of fluorination and 241 mg (0.97 mmol, 97%) of 4-(3-fluoropropyl)-5,8-dimethoxyquinoline was obtained; 1H NMR (600 MHz, CDCl3) δ 1.95–2.03 (m, 2H), 3.30 (t, J = 7.5 Hz, 2H), 3.84 (s, 3H), 3.96 (s, 3H), 4.43 (dt, J = 47.4, 5.8 Hz, 2H), 6.72 (d, J = 8.2 Hz, 1H), 6.87 (d, J = 8.2 Hz, 1H), 7.13 (d, J = 4.1 Hz, 1H), 8.70 (d, J = 9.6 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 32.5 (d, J = 4.3 Hz), 33.0 (d, J = 18.7 Hz), 55.6, 56.2, 83.6 (d, J = 165.2 Hz), 104.9, 106.7, 121.0, 123.8, 141.8, 148.5, 150.0, 150.4; MS (EI) m/z 249 (M+), 234 (100); HRMS (EI) m/z calcd for C14H16NO2F (M+) 249.1165, found 249.1162.N 4'-3-Fluoropropylciprofloxacin methyl ester
The preparation followed the typical procedure of fluorination and 385 mg (0.95 mmol, 95%) of N4'-3-fluoropropylciprofloxacin methyl ester was obtained; 1H NMR (600 MHz, CDCl3) δ 1.12–1.15 (m, 2H), 1.30–1.33 (m, 2H), 1.89–1.98 (m, 2H), 2.57 (t, J = 14.4 Hz, 2H), 2.67–2.69 (m, 4H), 3.28–3.29 (m, 4H), 3.40–3.44 (m, 1H), 3.91 (s, 3H), 4.55 (dt, J = 46.7, 5.8 Hz, 2H), 7.27 (d, J = 7.5 Hz, 1H), 8.05 (d, J = 13.1 Hz, 1H), 8.55 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 8.20, 27.96 (d, J = 20.1 Hz), 34.57, 50.05 (d, J = 4.3 Hz), 52.12, 53.04, 54.25 (d, J = 5.7 Hz), 84.41 (d, J = 163.7 Hz), 104.80 (d, J = 3.8 Hz), 110.17, 113.37 (d, J = 23.0 Hz), 123.08 (d, J = 7.2 Hz), 138.10, 144.65 (d, J = 11.5 Hz), 148.41, 153.51 (d, J = 248.0 Hz), 166.57, 173.15; MS (EI) m/z 405 (M+), 358 (100); HRMS (EI) m/z calcd for C21H25N3O3F2 (M+) 405.1864, found 405.1868.Acknowledgements
This work was supported by the Nuclear Research & Development Program of the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (grant code: 2011-0006322 and 2011-0030952), the Conversing Research Center Program through the MEST (grant code: 2011K000705).References
- See: (a) P. Seneci ,Solid-Phase Synthesis and Combinatorial Technologies, Wiley InterScience, New York, 2000, pp. 1-164 CrossRef; (b) A. Kirsching, in Immobilized Catalysts, Topics in Current Chemistry, Springer, Berlin, 2004 Search PubMed.
- (a) C. A. McNamara, M. J. Dixon and M. Bradley, Chem. Rev., 2002, 102, 3275 CrossRef CAS; (b) N. E. Leadbeater and M. Macro, Chem. Rev., 2002, 102, 3217 CrossRef CAS; (c) R. Colombo, Tetrahedron Lett., 1981, 22, 4129 CrossRef CAS.
- (a) M. Benaglia, A. Puglisi and S. Cozzi, Chem. Rev., 2003, 103, 3401 CrossRef CAS; (b) B. M. L. Dioos, I. F. J. Venkelecom and P. A. Jacobs, Adv. Synth. Catal., 2006, 348, 1413 CrossRef CAS.
- (a) M. D. Smith, J. March in Advanced Organic Chemistry, Wiley-Interscience, New York, 5th edn, 2001, 389 Search PubMed; (b) M. Makosza and K. Wojciechowski, Chem. Rev., 2004, 104, 2631 CrossRef CAS; (c) O. A. Mascaretti, Aldrichimica Acta, 1993, 26, 47 CAS.
- (a) CRC Handbook of Chemistry and Physics, CRC Press, Boston, 71st edn, 1990-1991 Search PubMed; (b) G. W Gokel, in Crown Ethers and Cryptands, Royal Society of Chemistry, Cambridge, 1991 Search PubMed.
- (a) E. V. Dehmlow and S. S. Dehmlow, Phase Transfer Catalysis, VCH Ltd, New York, 3rd edn, 1993 Search PubMed; (b) A. S. Pilcher, H. L. Ammon and P. Deshong, J. Am. Chem. Soc., 1995, 117, 5166 CrossRef CAS; (c) H. Sun and S. G. DiMango, J. Am. Chem. Soc., 2005, 127, 2050 CrossRef CAS.
- (a) C. Reichardt, Solvents and Solvents Effect in Organic Chemistry, VCH (UK) Ltd, Cambridge, U. K, 2nd edn, 1998 Search PubMed; (b) D. Martin, A. Weise and H. Niclas, Angew. Chem., Int. Ed. Engl., 1967, 6, 318 CrossRef CAS.
- (a) M. Cinquini, S. Colonna, H. Molinari, F. Montanari and P. Tundo, J. Chem. Soc., Chem. Commun., 1976, 394 RSC; (b) S. L. Regen, J. Am. Chem. Soc., 1975, 97, 5956 CrossRef CAS; (c) S. L. Regen and D. P. Lee, J. Am. Chem. Soc., 1974, 96, 294 CrossRef CAS.
- H. Molinari, F. Montanari and P. Tundo, J. Chem. Soc., Chem. Commun., 1977, 639 RSC.
- (a) D. W. Kim, D. -S. Ahn, Y. -H. Oh, S. Lee, S. J. Oh, S. J. Lee, J. S. Kim, J. -S. Ryu, D. H. Moon and D. Y. Chi, J. Am. Chem. Soc., 2006, 128, 16394 CrossRef CAS; (b) D. W. Kim, H.-J. Jeong, S. T. Lim, M.-H. Sohn, J. A. Katzenellenbogen and D. Y. Chi, J. Org. Chem., 2008, 73, 957 CrossRef CAS; (c) D. W. Kim ,H.-J Jeong, S. T. Lim and M.-H Sohn, Angew. Chem. Int. Ed., 2008, 47, 8404 Search PubMed.
- (a) J. W. Lee, H. Yan, H. B. Jang, H. K. Kim, S.-W. Park, S. Lee, D. Y. Chi and C. E. Song, Angew. Chem., Int. Ed., 2009, 48, 7683 CrossRef CAS; (b) V. H. Jadhav, S. H. Jang, H.-J. Jeong, S. T. Lim, M.-H. Sohn, D. Y. Chi and D. W. Kim, Org. Lett., 2010, 12, 3740 CrossRef CAS.
- (a) H. Zhao and S. V. Malhotra, Anal. Chim. Acta, 2002, 35, 75 CrossRef CAS; (b) P. Wasserscheid and W. Kein, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef CAS; (c) T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS; (d) C. C. Tzschucke, C. Markert, W. Bannwarth, S. Roller, A. Hebel and R. Haag, Angew. Chem., Int. Ed., 2002, 41, 3964 CrossRef CAS; (e) A. Tesfai, B. El-Zahab, D. K. Bwambok, G. A. Baker, S. O. Fakayode, M. Lowry and I. M. Warner, Nano Lett., 2008, 8, 897 CrossRef CAS.
- (a) D. W. Kim, C. E. Song and D. Y. Chi, J. Am. Chem. Soc., 2002, 124, 10278 CrossRef CAS; (b) D. W. Kim, C. E. Song and D. Y. Chi, J. Org. Chem., 2003, 68, 4281 CrossRef CAS.
- (a) D. W. Kim, D. J. Hong, K. S Jang and D. Y. Chi, Adv. Synth. Catal., 2006, 348, 1719 CrossRef CAS; (b) D. W. Kim and D. Y. Chi, Angew. Chem., Int. Ed., 2004, 43, 483 CrossRef CAS.
- V. H. Jadhav, H.-J. Jeong, S. T. Lim, M.-H. Sohn and D. W. Kim, Org. Lett., 2011, 13, 2502 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS.
- D. W. Kim, H.-J. Jeong, S. T. Lim, M.-H. Sohn and D. Y. Chi, Tetrahedron, 2008, 64, 4209 CrossRef CAS.
- K. Sachin, H.-J. Jeong, S. T. Lim, M.-H. Sohn, D. Y. Chi and D. W. Kim, Tetrahedron, 2011, 67, 1763 CrossRef CAS.
- K. Sachin, E.-M. Kim, S.-J. Cheong, H.-J. Jeong, S. T. Lim, M.-H. Sohn and D. W. Kim, Bioconjugate Chem., 2010, 21, 2282 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all compounds and 13C solid state NMR spectra of all solid-phase compounds. See DOI: 10.1039/c2ra21142d |
| This journal is © The Royal Society of Chemistry 2012 |