An ELF analysis of the C–C bond formation step in the N-heterocyclic carbene-catalyzed hydroacylation of unactivated C–C double bonds
Luis R.
Domingo
*,
Jose A.
Saéz
and
Manuel
Arnó
Departamento de Química Orgánica, Universidad de Valencia, Dr Moliner 50, E-46100 Burjassot, Valencia, Spain. E-mail: domingo@utopia.uv.es
First published on 9th July 2012
Abstract
The changes in electron-density along the C–C bond-formation step in the N-heterocyclic carbene-catalyzed hydroacylation of unactivated double bonds has been studied by an electron localization function (ELF) analysis at the B3LYP/6-31G** level in order to characterize the reaction mechanism. Analysis of DFT reactivity indices and the natural bond orbital and ELF analysis at the most relevant points of the intrinsic reaction coordinate indicate that the reaction path takes place through a two-stage one-step mechanism with non-polar character. In the first stage a hydrogen atom is transferred from the hydroxyl group of Breslow intermediate 12 to the terminal olefinic carbon atom, to yield a pseudodiradical species. The barrierless C–C bond formation in this species constitutes the second stage of the reaction allowing the formation of the corresponding alcohoxy intermediate 13. The present theoretical study establishes that this mechanism is completely different to that involved in the intramolecular Stetter reaction, which is initialized by a polar Michael-type addition.
Introduction
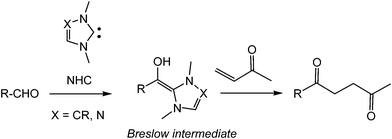
The umpolung reactivity of aldehydes promoted by N-heterocyclic carbenes (NHCs) constitutes an important class of organocatalysis and has found a broad range of applications in synthetic organic chemistry.1 The corresponding acyl anions or homoenolates equivalent intermediates nucleophilically attack various electrophiles, such as aldehydes,2 ketones,3 imines,4 and even activated polarized C–C double bonds.5The latter class of reactions is especially interesting, since olefins are ubiquitous in organic chemistry. In the Stetter reaction, a Breslow intermediate, which inverts the normal reactivity mode of an aldehyde, provokes a Michael-type addition to an electrophilically activated C–C double bond (see Scheme 1).6
 | ||
| Scheme 1 | ||
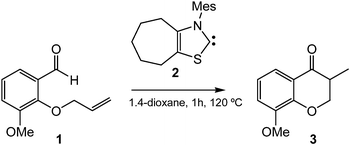
Although umpolung reactivity over activated olefins is quite common, the use of unactivated olefins in organocatalyzed reactions is very rare due to the nucleophilic character of both the homoenolate and the olefin (see later). In spite of this behavior, Glorius et al. recently reported the hydroacylation of unactivated olefins (see Scheme 2).7 In this work, the treatment of O-allylated vanillin 1 with a 20 mol% of NHC 2 provided the desired chroman-4-one 3 in 69% yield. This method was applied to a wide range of substituted aromatic compounds.
 | ||
| Scheme 2 | ||
These NHC-catalyzed hydroacylations of unactivated olefins are initialized by the formation of the corresponding Breslow intermediate8 between the formyl framework and the NHC. Glorius et al.7 proposed that, as in a concerted Conia-ene-type transition state structure (TS),9 the homoenolate adds to the olefin of the unactivated allyl moiety. They suggested that this is possible because as a negative charge develops at the end of the olefin, it is stabilized by the acidic alcohol hydrogen atom of the Breslow intermediate.7
Therefore, unactivated olefins such as 1 show a different behavior with regard to those involved in the Stetter reaction, which have an electrophilic character. While in the Stetter reaction, the hydrogen-bond (HB) formation is able to activate electrophilically the C–C double bond, in the case of unactivated olefins it is not.
Very recently, Glorius et al. reported an experimental study on the highly asymmetric NHC-catalyzed hydroacylation of unsaturated alkenes such as 4 (see Scheme 3).10 These reactions allow an enantioselective synthesis (99% ee) of product 5. Density functional theory (DFT) calculations were performed to disclose whether the proton migration or the C–C bond formation takes place first. Analysis of the bond lengths and the corresponding bond orders of the O–H breaking and H–C forming-bond at the unique TS associated with the one-step mechanism showed that O–H is half broken and the new H–C bond is half formed.10
 | ||
| Scheme 3 | ||
Our interest in organocatalysis, more specifically in the participation of NHCs as catalysts in the umpolung reactivity of aldehydes, recently prompted us to perform some theoretical studies about the molecular mechanisms of these interesting reactions.11 Thus, very recently, we studied the mechanism of the NHC-catalyzed intramolecular Stetter reaction of salicylaldehyde 6 to yield chromanone 11 (see Scheme 4).12 This NHC-catalyzed reaction takes place through five elementary steps, which involve: i) formation of the Breslow intermediate 8; ii) an intramolecular Michael-type addition in 8 to form the new C–C σ bond; and iii) extrusion of the NHC catalyst from the Michael adduct 10 to yield chromanone 11. Analysis of the relative free energies in toluene indicated that while the formation of Breslow intermediate 8 involves the rate-determining step of the catalytic process, the intramolecular Michael-type addition is the stereoselectivity-determining step responsible for the configuration of the stereogenic carbon α to the carbonyl carbon of chromanone 11. An electron localization function (ELF) bonding analysis at TSs and intermediates involved in this intramolecular Michael-type addition allowed for the characterization of the bond formation. While at TS1 the new C3–C4 σ bond is already formed, at TS2 the hydroxyl H1 hydrogen is transferred to the olefinic C5 carbon. Finally, analysis of the reactivity indices defined within the conceptual DFT verified the high reactivity of Breslow intermediate 8.
 | ||
| Scheme 4 | ||
Formation of Breslow intermediate 8 provokes an umpolung reactivity of the carbonyl carbon, due to its highly nucleophilic character. In spite of the electrophilic character of the unsaturated ester framework present in 8, the C–C bond formation would not be feasible without a further electrophilic activation of the C4–C5 bond achieved by an intramolecular HB formation between the acidic H1 hydrogen and C5 carbon of Breslow intermediate 8.12
In the present study, an ELF analysis of the intramolecular C–C bond-formation step in Breslow intermediate 12 (see Scheme 5) is performed in order to establish the mechanism of the C–C bond-formation step of the NHC-catalyzed hydroacylation of unactivated C–C double bonds. The results are compared with those recently reported for the intermolecular Stetter reaction.12 Note that the steps associated with the formation of Breslow intermediate 12 and that related to the extrusion of the NHC in intermediate 13 are similar to those in the intermolecular Stetter reaction shown in Scheme 4.
 | ||
| Scheme 5 | ||
Computational methods
DFT calculations were carried out using the B3LYP13 exchange–correlation functionals, together with the standard 6-31G** basis set.14 The optimizations were carried out using the Berny analytical gradient optimization method.15 The stationary points were characterized by frequency calculations and the TS was found to have one and only one imaginary frequency. The intrinsic reaction coordinate (IRC)16 paths were traced in order to check the energy profiles connecting the TS to the two associated minima of the proposed mechanism using the second order González–Schlegel integration method17 Solvent effects were considered by optimizing the gas-phase stationary points using a self-consistent reaction field (SCRF)18 based on the polarizable continuum model (PCM) of Tomasi's group.19 Although the experimental solvent was dioxane, ε = 2.21, in the PCM calculations, benzene was used due to their similar dielectric constants, ε = 2.27. The UFF radii model was used to generate the molecular cavity in the PCM calculations. Values of free energies in benzene were calculated with standard statistical thermodynamics at 120 °C and 1 atm.14The electronic structures of stationary points were analyzed by the natural bond orbital (NBO) method20 and by the topological analysis of the ELF, η(r).21 The ELF study was performed with the TopMod program22 using the corresponding monodeterminantal wavefunctions of the selected structures of the IRC. All calculations were carried out with the Gaussian 03 suite of programs.23
The global electrophilicity index,24ω, is given by the following simple expression ω = (μ2/2η), in terms of the electronic chemical potential μ and the chemical hardness η. Both quantities may be approached in terms of the one electron energies of the frontier molecular orbital HOMO and LUMO, εH and εL, as μ ≈ (εH + εL)/2 and η ≈ (εL − εH), respectively.25 Recently, we introduced an empirical (relative) nucleophilicity index, N,26 based on the HOMO energies obtained within the Kohn–Sham scheme,27 and defined as N = EHOMO(Nu) − EHOMO(TCE). N nucleophilicity refers to tetracyanoethylene (TCE), because it presents the lowest HOMO energy in a large series of molecules already investigated in the context of polar reactions. This choice allows us conveniently to handle a nucleophilicity scale of positive values.26 Local electrophilicity28 and nucleophilicity29 indices, ωk and Nk, were evaluated using the following expressions: ωk = ωf+k and Nk = Nf−k where f+kand f−k are the Fukui functions for nucleophilic and electrophilic attacks, respectively.30
Very recently, we proposed a local reactivity difference index Rk31 able to predict the local electrophilic and/or nucleophilic activation within an organic molecule, and defined as:31
| if (1 < ωk/Nk < 2) or (1 < Nk/ωk < 2) |
| then Rk ≈ (ωk + Nk)/2 ⇒ ambiphilic (Rk = ±n.nn) |
| else Rk ≈ (ωk − Nk) |
| where Rk > 0 ⇒ electrophilic (Rk = +n.nn) |
| and Rk < 0 ⇒ nucleophilic (Rk = −n.nn) |
| if |Rk| < 0.10, then Rk = 0.00 |
In the Rk index, the sign (+, −, ±) indicates the electrophilic or/and nucleophilic character of the centre k, while the magnitude n.nn provides a measure of the local activation.
Results and discussion
a) Analysis of the intramolecular C–C bond-formation step in Breslow intermediate 12
An analysis of the potential energy surface (PES) associated with the C–C bond-formation step in Breslow intermediate 12 indicates that both hydrogen transfer and C–C bond-formation processes take place in an elementary step via a high asynchronous TS3. Note that in the intramolecular Stetter reaction, formation of the Michael adduct 10 takes place through a two-step mechanism via the zwitterionic intermediate 9 (see Scheme 4) Therefore, Breslow intermediate 12, one TS, TS3, and the corresponding intermediate 13, associated with the intramolecular C–C bond-formation step in unactivated C–C double bonds, were located and characterized (see Scheme 5). IRC calculations from TS3 towards the corresponding intermediate 13 end in species 14 in which, while the H1 hydrogen has been completely transferred to the olefinic C5 carbon, the C3–C4 bond formation has not yet started (see Fig. 1).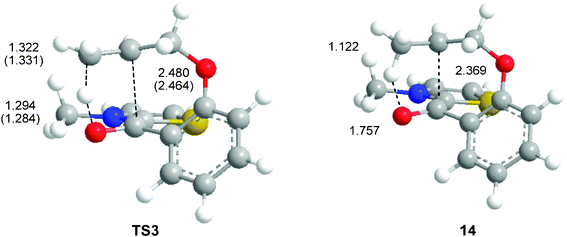 | ||
| Fig. 1 Most relevant gas-phase distances of the transition structure associated with the intramolecular C–C bond formation in Breslow intermediate 12, TS3, and of the IRC structure 14 (benzene distances are in parenthesis). All distances are given in Å. | ||
The free activation energy associated with the intramolecular C–C bond formation in Breslow intermediate 12 is 18.6 kcal mol−1; formation of adduct 13 is exergonic by −5.1 kcal mol−1 (see Table 1). This free activation energy is higher than that associated with the intramolecular Michael addition at intermediate 8, 7.9 kcal mol−1 (see Scheme 4).12
| H | ΔH | S | ΔS | G | ΔG | |
|---|---|---|---|---|---|---|
| 12 | −1145.550982 | 160.5 | −1145.651523 | |||
| TS3 | −1145.530144 | 13.1 | 146.6 | −13.9 | −1145.621950 | 18.6 |
| 13 | −1145.568620 | −11.1 | 145.4 | −15.2 | −1145.659664 | −5.1 |
The geometries of TS3 and species 14 are given in Fig. 1. A comparison of the gas-phase and solvent (benzene) geometries of TS3 indicates that there are no appreciable changes. Along the intramolecular process, the lengths of the O2–H1 breaking and H1–C5 forming bonds at TS3 are 1.294 and 1.322 Å, respectively, while the distance between the C3 and C4 carbons is 2.480 Å. The lengths of the O2–H1 breaking and H1–C5 forming bonds are similar to those found by Glorius in his model for the intramolecular process at the BP86-D/TZVP level; 1.226 and 1.388 Å.10 Considering that the O–H distance is shorter than the C–H one, we can assume that, from a geometrical point of view, at this TS, the hydrogen OH bond is half broken and the newly created CH bond is half formed. On the other hand, the long distance between the C3 and C4 carbons indicates that the C3–C4 bond formation has not started yet.
At species 14, the length of the H1–C5 bond is 1.122 Å while the distance between the O2 oxygen and H1 hydrogen is 1.757 Å. The distance between the C3 and C4 carbons remains at 2.369 Å. These geometrical parameters indicate that at this point of the IRC, while the H1 hydrogen has already been transferred to the terminal olefinic C5 carbon, C3–C4 bond formation has not started yet.
At TS3, the value of the unique imaginary frequency is −1217.9 cm−1. This high value indicates that this TS is mainly associated with the participation of a light hydrogen atom. Note that the value of the unique imaginary frequency of TS1, associated with the Michael-type addition in the Breslow intermediate 8, is −190.5 cm−1. Analysis of the atomic movement at the imaginary frequency of TS3 clearly reveals that it is only associated with the displacement of the H1 hydrogen atom from the hydroxyl O2 oxygen to the terminal olefinic C5 carbon; C3 and C4 carbons do not participate in these vibrations.
In order to obtain a more precise idea of the bond-breaking and bond formation extension at the TS, we performed a bond order (BO) analysis.32 At TS3, the BO values of the O2–H1 breaking and H1–C5 forming bonds are 0.32 and 0.47, respectively, while the BO value between the C3 and C4 carbons is 0.20. These values indicate that while the O2–H1 breaking bond is more advanced than the H1–C5 forming bond, the C3–C4 bond formation is more delayed. At species 14, the BO value of the H1–C5 bond is 0.80, while the BO value between C3 and C4 carbons remains at 0.30.
The polar or non-polar nature of this NHC-catalyzed C–C bond-formation process was evaluated by an NBO analysis of the natural charges at the corresponding stationary points. The natural charges at Breslow intermediate 12, TS3 and species 14 were shared between the Breslow framework and C4–C5 ethene framework, along the C4–C7 single bond. At TS3, the transferring hydrogen H1 atom was considered separately. The sum of the natural charges at the ethene fragment is: −0.02e at 12, −0.23 at TS3 and −0.03e at species 14. Two interesting conclusions can be reached from these values: i) the similar charges found at the ethene frameworks in Breslow intermediate 12 and species 14 indicate that along the first stage of the reaction, one hydrogen atom has been transferred; and ii) the increase of the negative charge in the ethene fragment found at TS3, 0.21e relative to Breslow intermediate 12, indicates that along the hydrogen transfer process a slight amount of electron-density is advanced. Consequently, the NBO analysis corroborates that the C–C bond-formation step in the NHC-catalyzed hydroacylation of unactivated C–C double bonds is associated with a non-polar process that is initialized by a hydrogen transfer process to yield an unstable pseudodiradical species,33 which undergoes a rapid C-to-C diradical coupling process to yield the corresponding intermediate. The barrierless character of the pseudodiradical coupling process justifies the unfeasibility of characterizing species 14 as a stationary point. Note that the sum of the natural charges in the CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Me framework of TS1 and zwitterionic intermediate 9, associated with the intramolecular Michael addition in the Breslow intermediate 8, are 0.44e and 0.66e, respectively, in clear agreement with the strong polar character of this process (see Scheme 4).
CHCO2Me framework of TS1 and zwitterionic intermediate 9, associated with the intramolecular Michael addition in the Breslow intermediate 8, are 0.44e and 0.66e, respectively, in clear agreement with the strong polar character of this process (see Scheme 4).
b) Analysis based on DFT reactivity indices
Analysis of the reactivity indices defined within the conceptual DFT allows for the establishment of the non-polar or polar nature of a reaction. Along a polar reaction, an amount of electron density is transferred from the nucleophile to the electrophile, a behavior that can be easily anticipated by the analysis of the electrophilicity, ω, and nucleophilicity, N, indices of the reagents. The best polar interactions take place between strong electrophiles, ω > 1.7 eV, and strong nucleophiles, N > 3.0 eV, being the first essential requirement. Thus, while a moderate electrophile, 0.80 < ω < 1.50 eV, can react with a strong nucleophile, a strong nucleophile does not react with a marginal electrophile, ω < 0.80 eV, in a polar process. Consequently, in a polar reaction, at least one of the two reagents should be a moderate electrophile, ω > 0.80 eV. The static global properties of the reagents involved in the intramolecular C–C bond-formation steps given in Schemes 4 and 5, namely electronic chemical potential (μ), chemical hardness (η), global electrophilicity (ω), and global nucleophilicity (N), are shown in Table 2.| μ | η | ω | N | |
|---|---|---|---|---|
| Methyl acrylate | −0.1586 | 0.2267 | 1.51 | 1.72 |
| 8 | −0.0975 | 0.0974 | 1.33 | 5.14 |
| Propene | −0.1141 | 0.2687 | 0.66 | 2.36 |
| 12 | −0.0806 | 0.1364 | 0.65 | 5.07 |
Recent studies devoted to intramolecular Diels–Alder reactions34 showed that the analysis of the global electrophilicity and nucleophilicity indices for the reagents is able to predict the polar or non-polar character of these intramolecular reactions. The electrophilicity of Breslow intermediate 8 involved in the Stetter reaction, ω = 1.33 eV, allows for its classification as a moderate electrophile. Note that this value is slightly lower than that for methyl acrylate, ω = 1.51 eV. On the other hand, the nucleophilicity index of 8, N = 5.14 eV, allows for its classification as a strong nucleophile.
On the other hand, the electrophilicity of Breslow intermediate 12, ω = 0.65 eV, allows for its classification as a marginal electrophile. The nucleophilicity index of 12, N = 5.07 eV, allows for its classification as a strong nucleophile. Consequently, just as in propene, ω = 0.66 eV, the marginal electrophilic character of Breslow intermediate 12 does not allow for its participation in a polar reaction. Note that the electrophilicity of Breslow intermediate 8 is twice the electrophilicity of 12.
Recently, we proposed a local reactivity difference index Rk able to predict the local electrophilic and/or nucleophilic activation within an organic molecule.31 Together with the electrophilic and/or nucleophilic behavior of the centre k given by the sign, the magnitude of the Rk index accounts for the extent of the electronic activation. The representation of the significant Rk indices, |Rk| > 0.10 eV, in a molecule constitutes the Rk molecular map of reactivity (RMMR).31 The RMMRs for Breslow intermediates 8 and 12 are given in Fig. 2.
 | ||
| Fig. 2 RMMRs of Breslow intermediates 8 (see Scheme 4) and 12 (see Scheme 5). Rk = +n.nn in red corresponds to electrophilic centers, Rk = −n.nn in blue corresponds to nucleophilic centers, and Rk = ±n.nn in green corresponds to ambiphilic centers. |Rk| values below 0.1 eV are not given. | ||
Breslow intermediate 12 presents nucleophilic activation at atoms belonging to NHC and umpolung formyl groups. The most nucleophilically activated centre of Breslow intermediate 12 is the C3 carbon, RC3 = −1.34 eV, which corresponds to the umpolung carbonyl C3 carbon. As expected, the olefinic C4 and C5 carbons do not show any electrophilic/nucleophilic activation. Consequently, Breslow intermediate 12 cannot participate in an intramolecular polar reaction.
Interestingly, inclusion of an electron-withdrawing methoxycarbonyl group at the terminal olefinic C5 carbon of Breslow intermediate 12 provokes a relevant change in the reactivity of Breslow intermediate 8. As can be seen in the RMMR of 8, the atoms belonging to NHC and formyl groups are the most nucleophilically activated centres. Just as with intermediate 12, the most nucleophilically activated centre of Breslow derivative 8 is also the C3 carbon, RC3 = −1.63 eV. However, all atoms belonging to the α,β-unsaturated ester framework are now electrophilically activated. The most electrophilically activated centre of 8 is the olefinic C4 carbon, RC4 = +0.45 eV, which corresponds to the β-conjugated position of the α,β-unsaturated ester. Consequently, the most favorable intramolecular polar interaction will take place between the C3 and C4 centres of Breslow intermediate 8, allowing for the formation of the new C3–C4 bond via an intramolecular Stetter reaction.
We can conclude that the analysis of the DFT reactivity indices of Breslow intermediates allows for the characterization of the intermolecular reactions studied by Glorius as non-polar processes, unlike the Stetter reaction that has a marked polar character, as a consequence of the electrophilic activation of the C3–C4 bond achieved by the presence of the terminal electron-withdrawing methoxycarbonyl group.
c) ELF bonding analysis of the IRC path of the C–H and C–C bond-formation step in the intramolecular reaction involving Breslow intermediate 12
Recent theoretical studies have shown that the topological analysis of the ELF along the reaction path associated with an organic reaction is a valuable tool for understanding the bonding changes along the reaction path, and thus, to characterize the molecular mechanism.35 Consequently, a topological analysis of the ELF of some relevant points of the IRC path associated with the C–C bond-formation step in the intramolecular reaction involving Breslow intermediate 12 was carried out in order to characterize the nature of these NHC-catalyzed C–C bond-formation processes involving unactivated C–C double bonds. The N populations of the more relevant ELF valence basins of selected structures along the IRC of the one-step mechanism are listed in Table 3, while the most relevant attractor positions and atom numbering are shown in Fig. 3. | ||
| Fig. 3 Most relevant ELF attractors in some selected points of the IRC associated with the C–C bond-formation step in Breslow intermediate 12. | ||
| 12 | TS3 | 14 | 13 | |
|---|---|---|---|---|
| V(H1,O2) | 1.58 | — | — | — |
| V(O2) | 2.33 | 2.99 | 2.92 | 3.21 |
| V′(O2) | 2.49 | 2.84 | 2.84 | 2.86 |
| V(O2,C3) | 1.26 | 1.55 | 1.82 | 1.48 |
| V(C3,C6) | 2.12 | 3.64 | 2.92 | 2.42 |
| V′(C3,C6) | 2.00 | — | — | — |
| V(C3) | — | — | 0.30 | — |
| V(C4) | — | — | 0.55 | — |
| V(C3,C4) | — | — | — | 1.96 |
| V(C4,C5) | 1.74 | 2.77 | 2.17 | 1.88 |
| V′(C4,C5) | 1.77 | — | — | — |
| V(H1) | — | 0.73 | — | — |
| V(C5) | — | 0.87 | — | — |
| V(H1,C5) | — | — | 1.90 | 2.01 |
| V(C7,O8) | 1.16 | 1.15 | 1.15 | 1.30 |
| V(O8) | 2.43 | 2.33 | 2.33 | 2.21 |
| V′(O8) | 2.59 | 2.62 | 2.64 | 2.50 |
| V(C4,C7) | 2.05 | 2.12 | 2.09 | 1.96 |
ELF analysis of Breslow intermediate 12 shows one disynaptic basin V(H1,O2) and one disynaptic basin V(O2,C3), each one integrating 1.58e and 1.26e, respectively, associated with the H1–O2 and O2–C3 σ bonds, and two monosynaptic basins, V(O2) and V′(O2), integrating a total of 4.42e, which are associated with the two lone pairs of the O2 oxygen. The populations of these four valence basins show a strong polarization of electron-density of the single H1–O2 and O2–C3 σ bonds towards the electronegative O2 oxygen. Breslow intermediate 12 also presents two pairs of disynaptic basins, V(C3,C6) and V′(C3,C6) and V(C4,C5) and V′(C4,C5), which integrate a total of 4.12e and 3.51e, respectively, corresponding to the C3–C6 and C4–C5 double bonds present in the Lewis structure of intermediate 12.
At TS3 some relevant changes take place relative to the electronic structure of Breslow intermediate 12. The two disynaptic basins V(C4,C5) and V′(C4,C5) have merged into one disynaptic basin V(C4,C5), integrating 2.77e. Consequently, a strong reduction of electron-density in the C4–C5 double bond region has taken place. Concurrently, a new monosynaptic basin V(C5), integrating to 0.87e, has emerged at the terminal olefinic C5 carbon. These changes account for the formation of a pseudoradical centre at the C5 carbon assisted by the reduction of electron-density in the C4–C5 region. On the other hand, while the disynaptic basin V(H1,O2) associated with the H1–O2 single bond of the hydroxyl group present in 12 has disappeared, a new monosynaptic attractor V(H1), integrating 0.73e, appears. This attractor, which is located at the same position of the H1 hydrogen points to a hydrogen atom structure with some radical character for H1 instead of a cationic centre associated with the participation of hydroxyl hydrogen in a polar process (see Fig. 4). Interestingly, the integration of this monosynaptic basin is closer to the natural charge found at the H1 hydrogen in TS3, 0.38e. Finally, the two disynaptic basins V(C3,C6) and V′(C3,C6) associated with the C3–C6 double bond present in the intermediate 12, have also merged into one disynaptic basin V(C3,C6), integrating 3.64e.
 | ||
| Fig. 4 A schematic representation of disynaptic and monosynaptic basins in some selected points of the IRC associated with the C–C bond-formation step in Breslow intermediate 12, represented by full lines and by ellipses with one dot, respectively. Dotted lines indicate a low basin population while thick lines indicate a large basin population. | ||
For species 14, the ELF analysis supplies relevant information about the mechanism of the NHC-catalyzed C–C bond-formation step in unactivated C–C double bonds. While the monosynaptic basins V(C5) and V(H1) have disappeared, a new disynaptic basin associated with the formation of the H1–C5 σ bond, V(H1,C5), integrating 1.90e, appears. At this point of the reaction path, the H1 hydrogen has been completely transferred from the O2 oxygen to the terminal olefinic C5 carbon. On the other hand, while the electron-density of the disynaptic basin V(C4,C5) is reduced to 2.17e, a new monosynaptic basin emerges at C4, V(C4), with a population of 0.55e. Concurrently, a new monosynaptic basin is created at the C3 carbon, V(C3), integrating 0.30e. These two new monosynaptic basins will permit the formation of the new C3–C4 single bond along the second stage of the reaction pathway. On going from TS3 to species 14, the disynaptic basin V(C3,C6) population is reduced by 0.72e. A part of the loss of electron-density in the C3–C6 region could be involved in the creation of the new monosynaptic basin V(C3).
The ELF analysis of intermediate 13 presents a disynaptic basin V(O2,C3) which integrates 1.48e, and two monosynaptic basins V(O2) and V′(O2), which integrate a total of 6.07e. This high electron-density points to an anionic alcohoxy oxygen. The disynaptic basins V(C3,C4) and V(H1,C5), which integrate 1.96e and 2.01e, respectively, are associated with the new C3–C4 and H1–C5 σ bonds formed along the NHC-catalyzed C–C bond formation. Note that the population of the disynaptic basin V(H1,C5) in species 14 is closer to that in 13, indicating that the H1–C5 σ bond has already been formed at this point of the IRC. Finally, the low electron-density that is present in the disynaptic basin V(C7,O8), between 1.16e in 12 and 1.30 e in 13, and the high electron-density that is present in the monosynaptic basins V(O8) and V′(O8), between a total of 5.02e in 12 and 4.71e in 13 are noteworthy. As with the disynaptic basin V(O2,C3), these integrations point to a large polarization of the C7–O8 σ bond toward the electronegative O8 oxygen. This polarization accounts for the positive charge accumulated in the olefin framework at TS3, 0.12e, obtained by NBO analysis in section a) after sharing the intramolecular process along the O8–C7 single bond.
ELF bonding analysis of Breslow intermediate 12, TS3, species 14 and intermediate 13 asserts that the NHC-catalyzed C–C bond-formation step involving unactivated C–C double bonds takes place via a two-stage one-step mechanism with a non-polar character, which is completely different to that associated with the NHC-catalyzed intramolecular Michael-type addition given in Scheme 4. In the first stage of the reaction, the H1 hydrogen atom is transferred from the hydroxyl O2 oxygen to the terminal olefinic C5 carbon. Once the H1 hydrogen has been completely transferred to species 14, which possesses some pseudodiradical character, the formation of the new C3–C4 σ bond at the second stage of the reaction begins. Note that while at 14 the disynaptic basin V(H1,C5) has been filled, there is no disynaptic basin V(C3,C4) associated with the formation of the new C3–C4 bond unlike the intramolecular Stetter reaction, where the Michael addition takes place in two separated steps (see Scheme 4). Along the first step, the new C3–C4 bond is already formed at TS1, while the H1–O2 bond remains practically unchanged. Along the second step, the H1 hydrogen is transferred from the O2 oxygen to the C5 carbon. This second step starts at TS2, where the H1–O2 bond is broken.
Conclusions
The mechanism of the C–C bond-formation step in the NHC-catalyzed hydroacylation of unactivated C–C double bonds has been studied at the B3LYP/6-31G** level. The C–C bond formation takes place via a two-stage one-step mechanism with a non-polar character. In the first stage a hydrogen atom is transferred from the hydroxyl group of Breslow intermediate 12 to the terminal olefinic carbon atom to yield a pseudodiradical species, which by a barrierless C–C bond formation allows for the formation of the corresponding alcohoxy intermediate 13 in the second stage of the reaction.Analysis of the DFT reactivity indices indicates that in spite of the high nucleophilicity of Breslow intermediate 12, its marginal electrophilic character prevents any polar process. This behavior is supported by an ELF analysis of the more relevant structures along the IRC associated with this one-step process. ELF bonding analysis supports the pseudodiradical character of this step, which is initialized by the hydroxyl hydrogen transferred to the terminal carbon of the ethylene residue (see (a) in Fig. 5). The subsequent barrierless C-to-C pseudodiradical coupling allows for the formation of the corresponding intermediate. The present theoretical study establishes that the mechanism of this C–C bond-formation step is completely different from that involved in the intramolecular Stetter reaction, which is initialized by a nucleophilic attack of the umpolung carbonyl carbon on the β-conjugated position of the unsaturated ester,12 and from that proposed by Glorius in which the initial proton transfer activates electrophilically the unactivated C–C double bond (see (b) in Fig. 5).10
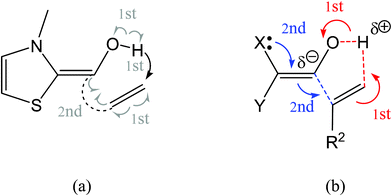 | ||
| Fig. 5 (a) O-to-C hydrogen transfer and C-to-C coupling processes in the C–C bond-formation step in the NHC-catalyzed hydroacylation of unactivated C–C double bonds. (b) Proposed electrophilic activation of unactivated C–C double bonds through a proton transfer.10 | ||
Acknowledgements
This work was supported by research funds provided by the Ministerio de Ciencia e Innovación of the Spanish Government (project CTQ2009-11027/BQU).References
- (a) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691 RSC; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS; (c) N. Marion and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS.
- (a) D. Enders and U. Kallfass, Angew. Chem., Int. Ed., 2002, 41, 1743 CrossRef CAS; (b) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370 CrossRef CAS; (c) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205 CrossRef CAS.
- (a) Y. Hachisu, J. W. Bode and K. Suzuki, J. Am. Chem. Soc., 2003, 125, 8432–8433 CrossRef CAS; (b) H. Takikawa and K. Suzuki, Org. Lett., 2007, 9, 2713 CrossRef CAS; (c) H. Takikawa, Y. Hachisu, J. W. Bode and K. Suzuki, Angew. Chem., Int. Ed., 2006, 45, 3492 CrossRef CAS; (d) D. Enders, O. Niemeier and T. Balensiefer, Angew. Chem., Int. Ed., 2006, 45, 1463 CrossRef CAS; (e) C. Burstein, S. Tschan, X. L. Xie and F. Glorius, Synthesis, 2006, 2418 CAS; (f) W. Schrader, P. P. Handayani, C. Burstein and F. Glorius, Chem. Commun., 2007, 716 RSC; (g) K. Hirano, I. Piel and F. Glorius, Adv. Synth. Catal., 2008, 350, 984 CrossRef CAS; (h) V. Nair, S. Vellalath, M. Poonoth, R. Mohan and E. Suresh, Org. Lett., 2006, 8, 507 CrossRef CAS; (i) A. Chan and K. A. Scheidt, J. Am. Chem. Soc., 2006, 128, 4558 CrossRef CAS.
- (a) S. M. Mennen, J. D. Gipson, Y. R. Kim and S. Miller, J. Am. Chem. Soc., 2005, 127, 1654 CrossRef CAS; (b) G.-Q. Li, L.-X. Dai and S.-L. You, Chem. Commun., 2007, 852 RSC; (c) M. He and J. W. Bode, Org. Lett., 2005, 7, 3131 CrossRef CAS; (d) M. Rommel, T. Fukuzumi and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 17266 CrossRef CAS.
- For excellent reviews of the Stetter reaction see: (a) J. Read de Alaniz and T. Rovis, Synlett, 2009, 1189 CAS; (b) M. Christmann, Angew. Chem., Int. Ed., 2005, 44, 2632 CrossRef CASFor a theoretical investigation of the mechanism, see (c) K. J. Hawkes and B. F. Yates, Eur. J. Org. Chem., 2008, 5563 CrossRef CAS; (d) J. He, S. Tang, J. Liu, Y. Su, X. Pan and X. She, Tetrahedron, 2008, 64, 8797 CrossRef CAS; (e) J. M. Um, D. A. DiRocco, E. L. Noey, T. Rovis and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 11249 CrossRef CAS.
- (a) H. Stetter, Angew. Chem., Int. Ed. Engl., 1976, 15, 639 CrossRef; (b) H. Stetter and H. Kuhlmann, Org. React., 1991, 40, 407 CAS; (c) H. Stetter and M. Schreckenberg, Angew. Chem., Int. Ed. Engl., 1973, 12, 81 CrossRef.
- K. Hirano, A. T. Biju, I. Piel and F. Glorius, J. Am. Chem. Soc., 2009, 131, 14190 CrossRef CAS.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS.
- B. K. Corkey and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 17168 CrossRef CAS.
- I. Piel, M. Steinmetz, K. Hirano, R. Fröhlich, S. Grimme and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 4983 CrossRef CAS.
- (a) L. R. Domingo, M. J. Aurell and M. Arno, Tetrahedron, 2009, 65, 3432 CrossRef CAS; (b) L. R. Domingo, R. J. Zaragozá and M. Arnó, Org. Biomol. Chem., 2010, 8, 4884 RSC; (c) L. R. Domingo, R. J. Zaragozá and M. Arnó, Org. Biomol. Chem., 2011, 9, 6616 RSC.
- L. R. Domingo, R. J. Zaragozá, J. A. Saéz and M. Arnó, Molecules, 2012, 17, 1335 CrossRef CAS.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- (a) H. B. Schlegel, J. Comput. Chem., 1982, 3, 214 CrossRef CAS; (b) H. B. Schlegel, in Modern Electronic Structure Theory, ed. D. R. Yarkony, World Scientific Publishing: Singapore, 1994 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS.
- (a) C. González and H. B.Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef; (b) C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853 CrossRef.
- (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS; (b) B. Y. Simkin and I. Sheikhet, Quantum Chemical and Statistical Theory of Solutions-A Computational ApproachEllis Horwod, London, 1995 Search PubMed.
- (a) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS; (b) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- (a) A. Savin, A. D. Becke, J. Flad, R. Nesper, H. Preuss and H. G. Vonschnering, Angew. Chem., Int. Ed. Engl., 1991, 30, 409 CrossRef; (b) A. Savin, B. Silvi and F. Colonna, Can. J. Chem., 1996, 74, 1088 CrossRef CAS; (c) A. Savin, R. Nesper, S. Wengert and T. F. Fassler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS; (d) B. Silvi, J. Mol. Struct., 2002, 614, 3 CrossRef CAS.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922 CrossRef CAS.
- (a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS; (b) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- (a) L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615 CrossRef CAS; (b) L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168 RSC.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133 CrossRef.
- L. R. Domingo, M. J. Aurell, P. Perez and R. Contreras, J. Phys. Chem. A, 2002, 106, 6871 CrossRef CAS.
- P. Pérez, L. R. Domingo, M. Duque-Noreña and E. Chamorro, THEOCHEM, 2009, 895, 86 CrossRef.
- R. Contreras, P. Fuentealba, M. Galván and P. Pérez, Chem. Phys. Lett., 1999, 304, 405 CrossRef CAS.
- P. K. Chattaraj, S. Duley and L. R. Domingo, Org. Biomol. Chem., 2012, 10, 2855, 10.1039/C2OB06943A.
- K. A. Wiberg, Tetrahedron, 1968, 24, 1083 CrossRef CAS.
- In 1960 Errede et al. studied the high chemical reactivity of p-xylylene, which was attributed to its pseudodiradical character. They defined a pseudodiradical as a diamagnetic compound that behaves chemically as if were a diradical. L. A. Errede, J. M. Hoyt and R. S. Gregorian, J. Am. Chem. Soc., 1960, 82, 5224–5227 CrossRef CAS.
- (a) J. Soto-Delgado, L. R. Domingo and R. Contreras, Org. Biomol. Chem., 2010, 8, 3678 RSC; (b) J. Soto-Delgado, A. Aizman, L. R. Domingo and R. Contreras, Chem. Phys. Lett., 2010, 499, 272 CrossRef CAS.
- (a) S. Berski, J. Andres, B. Silvi and L. R. Domingo, J. Phys. Chem. A, 2003, 107, 6014 CrossRef CAS; (b) V. Polo, J. Andrés, R. Castillo, S. Berski and B. Silvi, Chem.–Eur. J., 2004, 10, 5165 CrossRef CAS; (c) L. R. Domingo, M. T. Picher, P. Arroyo and J. A. Sáez, J. Org. Chem., 2006, 71, 9319 CrossRef CAS; (d) S. Berski, J. Andres, B. Silvi and L. R. Domingo, J. Phys. Chem. A, 2006, 110, 13939 CrossRef CAS; (e) V. Polo, J. Andres, S. Berski, L. R. Domingo and B. Silvi, J. Phys. Chem. A, 2008, 112, 7128 CrossRef CAS; (f) L. R. Domingo, E. Chamorro and P. Pérez, Lett. Org. Chem., 2010, 7, 432 CrossRef CAS; (g) L. R. Domingo and J. A. Sáez, J. Org. Chem., 2011, 76, 373 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |