DOI:
10.1039/C2RA21127K
(Paper)
RSC Adv., 2012,
2, 8055-8060
A new procedure for the synthesis of π-conjugated polymers via ligand-free iron(III)-catalyzed oxidative homocoupling reaction of grignard reagents†
Received
5th June 2012
, Accepted 6th July 2012
First published on 9th July 2012
Abstract
The FeCl3-catalyzed oxidative homocoupling reaction of Grignard reagents was used for the synthesis of π-conjugated polymers in this study. In the presence of an oxidant (1,2-dichloroethane), three polymers were successfully prepared, namely, poly(9,9-dioctylfluorene-2,7-diyl), poly(N-butylcarbazol-3,6-diyl) and poly(2,5-dihexyloxyphenylene-1,4-diyl). In comparison with the polymers obtained by Suzuki coupling, the polymers in this work showed no obvious difference in chemical structures or optical properties. On the basis of the relatively low cost of iron(III)-catalysts and Grignard reagents, this route would be an attractive alternative for the synthesis of π-conjugated polymers.
1. Introduction
Since the conductivity of doped polyacetylenes was revealed,1–3 considerable interest has been aroused to prepare π-conjugated polymers and to explore their chemical and physical properties. After four decades of intensive research, a large number of conjugated polymers, such as polyacetylenes,1–7 polyanilines,8 polythiophenes,9 polyphenylene-ethylenes,10 polyphenylenes,11 and polyfluorenes,12 have been obtained and are widely used as conducting materials,1–6,9b,9c polymer light emitting diodes,9d–9f,12 solar cells,13 field effect transistors,14 biochemical sensors,15 nonlinear optics,16 electrochromic devices17etc.
However, in comparison with the investigations that focused on the properties of π-conjugated polymers, research regarding the synthetic methodology of the polymers has been a little insufficient. Summarily, there have been two main ways to prepare π-conjugated polymers, namely, via a reduction or oxidation process,18 or via a metal-catalyzed coupling method (including Kumada coupling,19 Stille coupling,20 and Suzuki coupling21). Although these well-established coupling routes have played a very important role in the synthesis of many π-conjugated polymers, there still remain some inevitable problems. For instance, during the synthesis of alkyl polyfluorenes via the FeCl3 oxidative route, irregular structures can be generated,18c thus resulting in poor optoelectronic properties. As for the reductive methods, Yamamoto and Ullmann coupling polymerizations are somewhat unsatisfactory because highly toxic and moisture-sensitive Ni(COD)218e is used in the former and a large amount of zinc powder is wasted in the latter.18d Palladium-catalyzed Suzuki coupling polymerization is, undoubtedly, an efficient method for the synthesis of various polyarylenes due to the availability of the monomers and the convenient polymerization route, specifically the compatibility of the functional groups and the designability of the chemical structures used as monomers.21 However, the use of expensive metal catalysts and ligands can cause an increase in the cost of the target polymers.19–21 Accordingly, exploring and developing a cheaper and more convenient procedure for the synthesis of conjugated polymers are urgently required. It is noted that aryl Grignard reagents can be easily converted into the corresponding biaryl compounds with high yields in the presence of FeCl3 and 1,2-dihaloethane.22 Nevertheless, there is little information about the synthesis of conjugated polymers via the oxidative coupling of bis-Grignard reagents.23 Previously, we have reported the synthesis of polyfluorene using a Pd(II)-catalyzed base-free oxidative homocoupling reaction of diboronic esters.24 Our interest is to find cheap and practical methods for the synthesis of π-conjugated polymers. Thus, as part of an extended investigation, in this study we report the synthesis of π-conjugated polymers using the ligand-free iron(III)-catalyzed oxidative homocoupling of Grignard reagents.
2. Experimental
2.1 Materials
Fluorene and carbazole were industrial products, and were recrystallized from ethanol before use. 2,7-Dibromo-9H-fluorene (1),25 2,7-dibromo-9,9-dioctyl-fluorene (2),25 1,4-dibromo-2,5-dihexyloxybenzene (4),26 and 3,6-dibromo-N-butylcarbazole (6)27 were prepared as previously reported. Diisobutylaluminium hydride (DiBALH, 1 M in hexane) and ferric chloride (FeCl3, 0.2 M in 2-methyltetrahydrofuran) were purchased from Aldrich without further purification. Both 1,2-dichloroethane and 1,2-dibromoethane were commercially obtained from Aldrich and distilled over P2O5 before use.)
2.2 Measurement
Proton nuclear magnetic resonance (1H NMR) spectra were recorded with a Varian Mercury 300 spectrometer and carbon nuclear magnetic resonance (13C NMR) spectra were recorded with a Bruker DRX 400 spectrometer. UV-Visible absorption and photoluminescence (PL) spectra were measured with a Hitachi UV2800 spectrometer and a Hitachi F-4500 spectrometer, respectively. Traces of bromine in the polymers were determined on an American Dionex 500 ion chromatograph. A Waters Breeze GPC system was employed to deduce the number-average molecular weight (Mn) and molecular weight distribution (Mw/Mn) based on polystyrene standards with THF as an eluent.
2.3 Synthesis
Synthesis of 9,9-dioctyl-2,7-dibromomagnesiofluorene (3).
Under an argon atmosphere, a mixture of magnesium turnings (5.833 g, 240 mmol) and 120 mL of dry THF in an oven-dried flask was stirred and 0.5 mL of DiBALH (1 M in THF) was added. After being kept at room temperature for 0.5 h, the mixture was heated to 50 °C during a period of 10 min, then a solution of 9,9-dioctyl-2,7-dibromofluorene (2) (13.15 g, 24 mmol) in 60 mL of dry THF was added slowly through a syringe. The mixture was stirred at room temperature for 6 h to give a transparent grayish solution, which was kept carefully and directly used for polymerization.
Synthesis of 1,4-dibromomagnesio-2,5-dihexyloxybenzene (5).
Under an argon atmosphere, a mixture of magnesium turnings (0.972 g, 40 mmol) and 15 mL of dry THF in an oven-dried flask was stirred and 0.08 mL of DiBALH (1 M) was added. The mixture was heated to 50 °C over a period of 10 min, and a solution of 1,4-dibromo-2,5-dihexyloxybenzene (4) (1.744 g, 4 mmol) in 10 mL of dry THF was added slowly through a syringe. The solution was allowed to stir at room temperature for 6 h and the obtained grayish transparent solution was kept carefully and directly used for polymerization.
Synthesis of 3,6-dibromomagnesio-N-butylcarbazole (7).
Under an argon atmosphere, a mixture of magnesium turnings (0.972 g, 40 mmol) and 25 mL of dry THF in an oven-dried flask was stirred and 0.04 mL of DiBALH (1 M in THF) was added. The mixture was heated to 50 °C over a period of 10 min, and a solution of 3,6-dibromo-N-butylcarbazole (6) (1.524 g, 4 mmol) in 10 mL of dry THF was added slowly through a syringe. The solution was allowed to stir for 6 h and the generated grayish mixture was kept carefully and directly used for polymerization.
Under an argon atmosphere, a mixture of FeCl3 (1 mL, 0.2 M in 2-methyltetrahydrofuran), 1,2-dichloroethane (0.396 g, 4 mmol), and 18 mL of THF in a 50 mL oven-dried flask was stirred for 5 min at room temperature. Consequently, the prepared Grignard reagents (2 mmol) were added carefully through a syringe during a period of about 10 min. The mixture was allowed to react for a set time, and then dilute aq. HCl was added to quench the reaction. After removal of THF under vacuum, the residue was extracted with CHCl3 and washed with 10% EDTA twice, and 15 mL of saturated aq. NaCl solution three times. The organic layer was concentrated in vacuo and precipitated in 500 mL of stirring methanol. The obtained solid was redissolved in CHCl3 and reprecipitated in a mixture of methanol and THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v) for poly(9,9-dioctyl-2,7-fluorene), or acetone for poly(N-butylcarbazole) or methanol for poly(2,5-dihexyloxy-1,4-phenylene). Characterization: Poly(9,9-dioctyl-2,7-fluorene) (PFO) 1H NMR (300 MHz, CDCl3δ in ppm): 7.88–7.79 (m, 2H), 7.75–7.60 (m, 4H), 2.22–1.97 (m, 4H), 1.30–1.02 (m, 20H), 0.91–0.75 (m, 10H). 13C NMR (125 MHz, CDCl3, δ in ppm): 151.91, 140.63, 140.12, 126.25, 121.59, 120.03, 55.43, 40.46, 31.86, 30.11, 29.27, 23.90, 22.65, 14.10. Poly(N-butylcarbazol-3,6-diyl) (PBC), 1H NMR (300 MHz, CDCl3δ in ppm): 8.60–8.35 (br, 2H), 7.97–7.74 (br, 2H), 7.57–7.30 (br, 2H), 4.43–4.12 (br, 2H), 1.99–1.72 (br, 2H), 1.04–0.74 (br, 3H). 13C NMR (125 MHz, CDCl3δ in ppm): 140.09, 133.39, 125.58, 123.78, 118.99, 109.00, 43.06, 31.32, 20.66, 13.97. Poly(2,5-dihexyloxy-1,4-phenylene) (PHP), 1H NMR (300 MHz, CDCl3, δ in ppm): 7.10 (br, 2H), 4.01–3.75 (br, 4H), 1.83–1.49 (br, 4H), 1.40–1.12 (m, 12H), 0.98–0.67 (br, 6H). 13C NMR (125 MHz, CDCl3, δ in ppm): 150.90, 127.60, 117.43, 69.64, 31.67, 29.51, 25.78, 22.62, 14.02.
:2, v/v) for poly(9,9-dioctyl-2,7-fluorene), or acetone for poly(N-butylcarbazole) or methanol for poly(2,5-dihexyloxy-1,4-phenylene). Characterization: Poly(9,9-dioctyl-2,7-fluorene) (PFO) 1H NMR (300 MHz, CDCl3δ in ppm): 7.88–7.79 (m, 2H), 7.75–7.60 (m, 4H), 2.22–1.97 (m, 4H), 1.30–1.02 (m, 20H), 0.91–0.75 (m, 10H). 13C NMR (125 MHz, CDCl3, δ in ppm): 151.91, 140.63, 140.12, 126.25, 121.59, 120.03, 55.43, 40.46, 31.86, 30.11, 29.27, 23.90, 22.65, 14.10. Poly(N-butylcarbazol-3,6-diyl) (PBC), 1H NMR (300 MHz, CDCl3δ in ppm): 8.60–8.35 (br, 2H), 7.97–7.74 (br, 2H), 7.57–7.30 (br, 2H), 4.43–4.12 (br, 2H), 1.99–1.72 (br, 2H), 1.04–0.74 (br, 3H). 13C NMR (125 MHz, CDCl3δ in ppm): 140.09, 133.39, 125.58, 123.78, 118.99, 109.00, 43.06, 31.32, 20.66, 13.97. Poly(2,5-dihexyloxy-1,4-phenylene) (PHP), 1H NMR (300 MHz, CDCl3, δ in ppm): 7.10 (br, 2H), 4.01–3.75 (br, 4H), 1.83–1.49 (br, 4H), 1.40–1.12 (m, 12H), 0.98–0.67 (br, 6H). 13C NMR (125 MHz, CDCl3, δ in ppm): 150.90, 127.60, 117.43, 69.64, 31.67, 29.51, 25.78, 22.62, 14.02.
3. Results and discussion
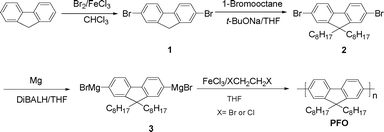
On the basis of the wide applications of fluorene-based polymers,12 our initial efforts were occupied with the synthesis of poly(9,9-dioctylfluorene) via the homopolymerization of 9,9-dioctyl-2,7-dibromomagnesiofluorene (3, Fig. 1). To realize the polymerization, high yields of bis-Grignard reagents were required. Usually, there were two ways for the preparation of Grignard reagents: one was the metal-halogen exchange between halogenated aryl compounds and isopropyl magnesium chloride;28 and the other was direct formation via the reaction of halogenated aryl compounds with magnesium.29 Considering that magnesium was cheaper and readily available, we chose the directly synthetic procedure, e.g., 3 was prepared from the reaction of magnesium with dibromoalkylfluorene (2). It was noted that the directly synthetic procedure needed an “activated magnesium”,30 which was obtained in many cases by pretreating magnesium with I2 or dibromoethane.29 We found that diisobutylaluminum hydride (DiBALH) was more effective for the improvement of the reactivity of magnesium. When a catalytic amount of DiBALH was used, 2 quickly disappeared during a period of 1 h, and the HPLC results showed that 3 was formed in a high yield after the reaction was carried out at room temperature for 6 h.
 |
| | Fig. 1 The procedure for the synthesis of polyfluorene. | |
Subsequently, we investigated the oxidative homopolymerization of the obtained 9,9-dioctyl-2,7-dibromomagnesiofluorene (3). There had been a lot of oxidants used for the Fe(III) catalyzed homocoupling of Grignard reagents.22,31 Initially, we chose dibromoethane as an oxidant, and poly(9,9-dioctylfluorene-2,7-diyl) (PFO, Fig. 1) was obtained with a number average molecular weight (Mn) of 5700 after being extracted with acetone for 24 h in a Soxhlet extractor. However, further optimization of the polymerization conditions, including changing the reaction temperature from −25 °C to refluxing and adding a ten-fold amount of 1,2-dibromoethane, did not enhance Mn. Such results may be attributed to the fact that 1,2-dibromoethane was apt to be decomposed and the generated HBr may inevitably quench the Grignard reagent, resulting in a suppression of chain growth. Interestingly, when 1,2-dichloroethane (Fig. 1) was used as an oxidant, PFO was obtained with a Mn of more than 7300 after being extracted with acetone, and the yield was more satisfactory (Table 1). It was noted that the reaction gave the polymer with a wide polydispersity, as can be seen in Table 1. Removing the fractions with low molecular weight by precipitation of the obtained polymer in a mixture solvent of THF and methanol (2:1, v/v) gave a polymer fraction with a Mn of 11960 and a PDI of 1.34. This result is superior to that previously reported by our group via the Pd(II)-catalyzed oxidative homopolymerization of diboronic esters,24 suggesting that Fe(III) catalyzed oxidative homocoupling can give a high molecular weight of PFO which can meet the requirements of optoelectronic devices.
| Processing method |
M
n
a
|
Polydispersity |
Yield (%) |
|
GPC curves of the samples were shown in supplementary information.
Precipitated in methanol.
Extracted with acetone for 24 h in a Soxhlet extractor.
Reprecipitated in a mixture of methanol and THF (1:2, v/v).
|
| Ab |
4870 |
2.19 |
98 |
| Bc |
7300 |
1.59 |
75 |
| Cd |
11960 |
1.34 |
35 |
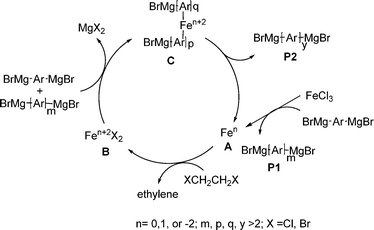
To investigate the reason why Fe(III) catalyzed oxidative homocoupling gave such a wide PDI, we then looked into the relationship between the molecular weight and reaction time, and the results are summarized in Table 2. Surprisingly, the largest polymer fraction had come up to 10300 during a very short period (one minute). Although the largest polymer fraction was near to 12000 when the reaction was run for only 5 min, a lot of oligomers with a molecular weight of ∼3000 were observed. Further prolongation of the reaction time did not increase the molecular weight of PFO. Assuming that the impurities in the Grignard reagent (3) may influence chain growth, we then put 3 into dilute aq. HCl, and the product (9,9-dioctylfluorene) in the organic layer displayed a HPLC purity of 98.7%. This result implied that the wide PDI of PFO can not be attributed to the impurities in monomer 3, and may be due to the special mechanism of the iron-catalyzed homocoupling of bis-Grignard reagents,22 as shown in Fig. 2. Initially, the oxidation of a bis-Grignard reagent by FeCl3 generated an oligomer P1, accompanying the formation of a low valent iron A. The oxidative addition of A to the dihaloethane and the subsequent β-halogen elimination transforms A to the iron species B. Consequently, the transmetalation of magnesium to iron afforded a species C. Finally, the polymer intermediate P2 was obtained via the reductive elimination of C, accompanying the regeneration of the active species A. In the catalytic cycle, 1,2-dihaloethane (1,2-dichloroethane or 1,2-dibromoethane) played a role to oxidize the iron species A (Fen) to the iron species B (Fen+2), undergoing a reductive elimination step.22 Because the iron species B and C displayed complicated oxidation states,22 including Fe1+, Fe2+ and Fe3+, the progress of B to C to P2 may produce the polymer with a wide PDI.
Table 2 The correlation between the largest fraction of PFO and the reaction time using 1,2-dichloroethane as oxidant. All reactions were carried out at room temperaturea
| Entry |
Time (t) |
M
n
|
Polydispersity |
|
GPC curves of the samples are shown in the ESI†.
|
| 1 |
1 min |
10300 |
1.27 |
| 2 |
2 min |
10990 |
1.30 |
| 3 |
3 min |
11070 |
1.29 |
| 4 |
5 min |
11810 |
1.29 |
The chemical structure of the obtained PFO was confirmed by 1H NMR and 13C NMR (See ESI†). In comparison with that of precursor 2, the 1H NMR spectrum of PFO (Fig. 3) showed no extra signal in the area of 1.6–2.2 ppm, suggesting that no cross-coupling product between 3 and the oxidant (1,2-dichloroethane) was produced.
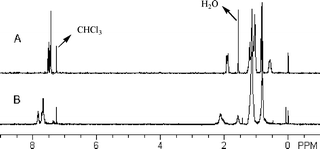 |
| | Fig. 3 A comparison of the 1H NMR spectrum of precursor 2 (A) with that of the obtained PFO (B). | |
To further confirm the chemical structure of PFO, the UV-vis and PL spectra of the PFO prepared from traditional Suzuki coupling were compared with those of the PFO obtained in this work (Fig. 4). As can be seen from Fig. 4, the two samples showed no obvious difference, indicating that Fe(III) catalyzed oxidative coupling gave a polymer with the same chemical structure as that of the polymer prepared by Suzuki coupling.
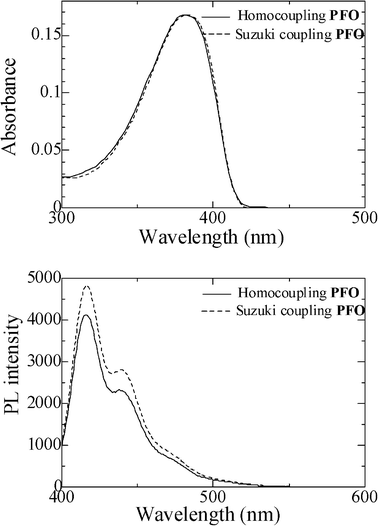 |
| | Fig. 4 UV-vis (top) and PL (bottom) spectra of PFO obtained using different coupling routes. The concentration of all samples in THF was 4 × 10−6 M for UV-vis and 4 × 10−8 M for PL, respectively. | |
Considering that poly(alkyloxyphenylene)s32 and alkyl substituted polycarbazoles33 had emerged as a very attractive class of excellent photoelectric materials, we then tried to synthesize poly(2,5-dihexyloxyphenylene-1,4-diyl) (PHP) and poly(N-butyl-carbazol-3,6-diyl) (PBC) by Fe(III) catalyzed oxidative coupling, and the synthetic route is shown in Fig. 5. The data of the obtained polymers are summarized in Table 3. The chemical structures of PHP and PBC were verified by 1H NMR and 13C NMR (see experimental section and ESI†).
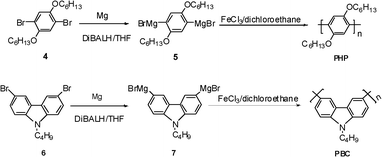 |
| | Fig. 5 The synthetic route of poly(alkyloxyphenylene) and poly(N-alkylcarbazole). | |
Table 3 Synthesis of PHP and PBCa
| Polymer |
M
n
|
Polydispersity |
|
In the presence of 2 eq. 1,2-dichloroethane and 10% FeCl3. All the reactions were carried out at room temperature for 18 h.
|
| PHP |
2800 |
1.27 |
| PBC |
3700 |
1.24 |
Compared to the UV-vis spectra of the corresponding precursors, those of PHP and PBC showed red-shifts, suggesting that the polymers had the expanded π-conjugated system (Fig. 6). It is noteworthy that PHP and PBC each displayed a lower molecular weight in comparison with PFO. The ion chromatograph (Table 4) also indicated that the Br content in PHP and PBC was much higher than that in PFO, suggesting that the formation of monomers 5 and 7 (Fig. 5) was more difficult than that of monomer 3 (Fig. 1). PHP and PBC seem to be considered as the oligomers, however, casting a toluene solution of PHP or PBC on glass plates gave a smooth film. The lower Mn may also be attributed to the special process of the iron-catalyzed homocoupling of bis-Grignard reagents (Fig. 2),22 as well as being potentially due to steric hindrance resulting from the long side chains in PHP34 and the twisted and zigzag-shape structure in PBC.35 Further, more in-depth details will soon be investigated.
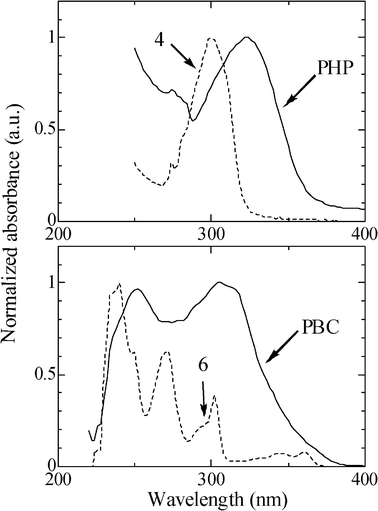 |
| | Fig. 6 UV-Vis spectra of PHP and PBC in THF. For comparison, the spectra of the corresponding precursors 4 and 6 are also given. | |
Table 4 The chemical analysis of the polymers
| Polymers |
Terminal Br content (%)a |
|
Measured by ion chromatography.
|
| PHP |
0.436 |
| PBC |
0.404 |
| PFO |
0.013 |
4. Conclusions
Three π-conjugated polymers, poly(9,9-dioctylfluorene-2,7-diyl) (PFO), poly(N-butylcarbazol-3,6-diyl) (PBC) and poly(2,5-dihexyloxyphenylene-1,4-diyl) (PHP) have been successfully synthesized via Fe(III)-catalyzed oxidative homocoupling of Grignard reagents. The advantages of this route include a short reaction period and low cost, making it an attractive alternative for the synthesis of π-conjugated polymers.
Acknowledgements
Financial support from Ministry of Science and Technology of China (2010BAK67B12 and 2011ZX02703) are gratefully acknowledged.
References
- T. Ito, H. Shirakawa and S. Ikeda, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 11 CrossRef CAS.
- H. Shirakawa, Rev. Mod. Phys., 2001, 73, 713 CrossRef CAS.
- H. Shirakawa and E. J. Louis, J. Chem. Soc., Chem. Commun., 1977, 578 RSC.
- G. Natta, I. Pasquon and A. Zambelli, J. Polym. Sci., 1961, 156, 387 CrossRef.
- C. K. Chiang, M. A. Druy and S. C. Gau, J. Am. Chem. Soc., 1978, 100, 1013 CrossRef CAS.
- B. M. Quillan, G. B. Street and T. C. Clarke, J. Electron. Mater., 1982, 11, 471 CrossRef.
- H. Shirakawa, T. Otaka and G. Piao, Synth. Met., 2001, 117, 1 CrossRef CAS.
-
(a) C. F. Liu, Polym. J., 1993, 25, 363 CrossRef CAS;
(b) Y. K. Kang, Synth. Met., 1992, 52, 319 CrossRef CAS.
-
(a) T. Yamamoto, K. Sanechika and A. Yamamoto, J. Polym. Sci., Polym. Lett. Ed., 1980, 18, 9 CrossRef CAS;
(b) K. T. Jen, G. G. Miller and R. L. Elsenbaumer, J. Chem. Soc., Chem. Commun., 1986, 1346 RSC;
(c) S. Jin and G. Xue, Macromolecules, 1997, 30, 5753 CrossRef CAS;
(d) K. Yoshino, Y. Manda and M. Onoda, Solid State Commun., 1989, 70, 111 CrossRef CAS;
(e) X. J. Wang, M. R. Andersson and M. E. Thompson, Thin Solid Films, 2004, 468, 226 CrossRef CAS;
(f) M. R. Andersson, M. Berggren and G. Gustafsson, Synth. Met., 1995, 71, 2183 CrossRef CAS;
(g) M. Leclerc and G. J. Daoust, J. Chem. Soc., Chem. Commun., 1990, 273 RSC;
(h) T. A. Chen and R. D. Rieke, J. Am. Chem. Soc., 1992, 114, 10087 CrossRef CAS;
(i) S. Hotta, S. Rughooputh, A. J. Heeger and F. Wudl, Macromolecules, 1987, 30, 212 CrossRef;
(j) R. D. McCullough, Adv. Mater., 1998, 10, 93 CrossRef CAS.
-
(a) J. H. Burroughes, D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burn and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS;
(b) D. Braun and A. Heerger, Appl. Phys. Lett., 1991, 58, 1982 CrossRef CAS.
-
(a) G. Grem, G. Leditzky, B. Ullrich and G. Leising, Adv. Mater., 1992, 4, 36 CrossRef CAS;
(b) V. Scherf and K. Muellen, Macromolecules, 1992, 25, 3546 CrossRef;
(c) J. Huber, K. Mullen, J. Salbeck, H. Schenk, U. Scherf, T. Stehlin and R. Stern, Acta Polym., 1994, 45, 244 CrossRef CAS.
-
(a) O. Yutaka, U. Masao and M. Keiro, Jpn. J. Appl. Phys., 1991, 30, 1941 CrossRef;
(b) G. Klaerner and R. D. Miller, Macromolecules, 1998, 31, 2007 CrossRef CAS;
(c) W. L. Yu, J. Pei and W. Huang, Chem. Commun., 1999, 1837 RSC;
(d) S. Setagesh, A. C. Grimsdale and T. Weil, J. Am. Chem. Soc., 2001, 123, 946 CrossRef;
(e) S. L. Mcfarlane, D. G. Piercey and L. S. Coumont, Macromolecules, 2009, 42, 591 CrossRef CAS;
(f) J. Pei, W. L. Yu and W. Huang, Chem. Commun., 2000, 1631 RSC;
(g) W. Y. Wong, L. Liu and D. M. Cui, Macromolecules, 2005, 38, 4970 CrossRef CAS;
(h) C. S. Wu and Y. Chen, Macromolecules, 2009, 42, 3729 CrossRef CAS.
-
(a) J. Hou, Z. Tan, Y. Yan, Y. He, C. Yang and Y. Li, J. Am. Chem. Soc., 2006, 128, 4911 CrossRef CAS;
(b) J. Hou, L. Huo, C. He, C. Yang and Y. Li, Macromolecules, 2006, 39, 594 CrossRef CAS;
(c) J. Hou, C. Yang, C. He and Y. Li, Chem. Commun., 2006, 871 RSC;
(d) E. Zhou, Z. Tan, C. Yang and Y. Li, Macromol. Rapid Commun., 2006, 27, 793 CrossRef CAS.
- X. Zhan, Z. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. Marder, J. Am. Chem. Soc., 2007, 129, 7246 CrossRef CAS.
-
(a) F. He, Y. Tang, S. Wang, Y. Li and D. Zhu, J. Am. Chem. Soc., 2005, 127, 12343 CrossRef CAS;
(b) F. He, Y. Tang, M. Yu, F. Feng, L. An, H. Sun, S. Wang, Y. Li, D. Zhu and G. Bazan, J. Am. Chem. Soc., 2006, 128, 6764 CrossRef CAS;
(c) Y. Tang, F. Feng, F. He, S. Wang, Y. Li and D. Zhu, J. Am. Chem. Soc., 2006, 128, 14972 CrossRef CAS;
(d) X. Feng, L. Liu, S. Wang and D. Zhu, Chem. Soc. Rev., 2010, 39, 2411 RSC;
(e) X. Duan, L. Liu, X. Feng and S. Wang, Adv. Mater., 2010, 12, 1602 CrossRef.
- S. Ellinger, K. R. Graham, P. Shi, R. T. Farley, T. Steckler, R. Brookins, P. Taranekar, J. Mei, L. Padilha, T. Ensley, H. Hu, S. Webster, D. Hagan, E. V. Stryland, K. Schanze and J. R. Reynolds, Chem. Mater., 2011, 23, 3805 CrossRef CAS.
-
(a) F. B. Koyuncu, E. Sefer, S. Koyuncu and E. Ozdemir, Polymer, 2011, 52, 5772 CrossRef CAS;
(b) B. Kim, J. Kim and E. Kim, Macromolecules, 2011, 44, 8791 CrossRef CAS.
-
(a) A. A. Sergei and M. K. Valerii, Macromol. Chem. Phys., 2000, 201, 809 CrossRef;
(b) P. Kovacic and J. J. Oziomek, J. Org. Chem., 1964, 29, 100 CrossRef CAS;
(c) Y. Ito, T. Shimada, J. Ha, M. Vacha and H. Sato, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4338 CrossRef CAS;
(d) Q. Pei and Y. Yang, J. Am. Chem. Soc., 1996, 118, 7416 CrossRef CAS;
(e) T. Yamamoto, A. Morita, Y. Miyazaki, T. Maruyama, H. Wakayama, Z. H. Zhou, Y. Nakamura, T. Kanbara, A. Sasaki and K. Kubota, Macromolecules, 1992, 25, 1214 CrossRef CAS.
-
(a) R. D. McCullough and R. D. Lowe, J. Chem. Soc., Chem. Commun., 1992, 1, 70 RSC;
(b) R. D. McCullough, Synth. Met., 1995, 69, 279 CrossRef CAS;
(c) A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169 CrossRef CAS;
(d) R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542 CrossRef CAS;
(e) R. S. Loewe, S. M. Khersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250 CrossRef CAS;
(f) E. E. Sheina, J. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526 CrossRef CAS.
-
(a) A. Fazio, B. Gabriele, G. Salerno and S. Destri, Tetrahedron, 1999, 55, 485 CrossRef CAS;
(b) R. D. McCullough, P. C. Ewbank and R. S. Loewe, J. Am. Chem. Soc., 1997, 119, 633 CrossRef CAS.
-
(a) S. Guillerez and G. Bidan, Synth. Met., 1998, 93, 123 CrossRef CAS;
(b) C. Kowitz and G. Wegner, Tetrahedron, 1997, 53, 15553 CrossRef CAS;
(c) T. I. Wallow and B. M. Novak, J. Am. Chem. Soc., 1991, 113, 7411 CrossRef CAS;
(d) W. Yang, Q. Hou, C. Liu, Y. Niu, J. Huang, R. Yang and Y. Cao, J. Mater. Chem., 2003, 13, 1351 RSC;
(e) M. Ranger, D. Rondeau and M. Leclerc, Macromolecules, 1997, 30, 7686 CrossRef CAS;
(f) M. Ranger and M. Leclerc, Chem. Commun., 1997, 1597 RSC;
(g) C. F. Shu, R. Dodda and F. Wu, Macromolecules, 2003, 36, 6698 CrossRef CAS;
(h) Q. Peng, Z. Y. Lu, Y. Huang, M. G. Xie, S. H. Han, J. B. Peng and Y. Cao, Macromolecules, 2004, 37, 260 CrossRef CAS;
(i) Q. Hou, Q. Zhou, Y. Zhang, W. Yang, R. Yang and Y. Cao, Macromolecules, 2004, 37, 6299 CrossRef CAS;
(j) C. H. Chou, S. L. Hsu, K. Dinakaran, M. Y. Chiu and K. H. Wei, Macromolecules, 2005, 38, 745 CrossRef CAS.
-
(a) T. Nagano and T. Hayashi, Org. Lett., 2005, 7, 491 CrossRef CAS;
(b) G. Cahiez, C. Chaboche, F. M. Betzer and M. Ahr, Org. Lett., 2005, 7, 1943 CrossRef CAS.
- M. S. Maji, T. Pfeifer and A. Studer, Chem.–Eur. J., 2010, 16, 5872 CAS.
- C. Yuan, J. Hong, Y. Liu, H. Lai and Q. Fang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4098 CrossRef CAS.
-
(a) M. Ghaemy and M. Barghamadi, J. Appl. Polym. Sci., 2009, 114, 3464 CrossRef CAS;
(b) D. Zeng, J. Chen, Z. Chen, W. Zhu, J. He, F. Yu, H. Huang, H. Wu, C. Liu, S. Ren, J. Du, J. Sun, E. Xu, A. Cao and Q. Fang, Macromol. Rapid Commun., 2007, 28, 772 CrossRef CAS.
- Q. Fang and T. Yamamoto, Polymer, 2003, 44, 2947 CrossRef CAS.
-
(a) K. Smith, D. M. James, A. G. Mistry, M. Bye and D. J. Fauikner, Tetrahedron, 1992, 48, 7479 CrossRef CAS;
(b) T. Zhao, Z. Liu, Y. Song, W. Xu, D. Zhang and D. Zhu, J. Org. Chem., 2006, 71, 7422 CrossRef CAS.
-
(a) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302 CrossRef CAS;
(b) A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333 CrossRef CAS;
(c) F. Kopp, A. Krasovskiy and P. Knochel, Chem. Commun., 2004, 2288 RSC;
(d) H. Ren, A. Krasovskiy and P. Knochel, Org. Lett., 2004, 6, 4215 CrossRef CAS;
(e) H. Ren, A. Krasovskiy and P. Knochel, Chem. Commun., 2005, 543 RSC;
(f) A. Krasovskiy, B. F. Straub and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 159 CrossRef CAS.
-
(a) R. D. Rieke and H. Xiong, J. Org. Chem., 1991, 56, 3109 CrossRef CAS;
(b) A. M. Caporusso, L. A. Aronica, R. Geri and M. Gori, J. Organomet. Chem., 2002, 648, 109 CrossRef CAS.
- F. M. Piller, P. Appukkuttan, A. Gavryushin, M. Helm and P. Knochel, Angew. Chem., Int. Ed., 2008, 47, 6802 CrossRef CAS.
-
(a) G. Cahiez, A. Moyeux, J. Buendia and C. Duplais, J. Am. Chem. Soc., 2007, 129, 13788 CrossRef CAS;
(b) M. S. Maji and A. Studer, Synthesis, 2009, 14, 2467 Search PubMed.
-
(a) R. Miyakoshi, K. Shimono, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2006, 128, 16012 CrossRef CAS;
(b) E. L. Lanni and A. J. McNeil, J. Am. Chem. Soc., 2009, 131, 16573 CrossRef CAS.
- M. C. Stefan, A. E. Javier, I. Osaka and R. D. McCullough, Macromolecules, 2009, 42, 30 CrossRef CAS.
- K. Ono, A. Adachi, K. Okita, M. Goto and Y. Yamashita, Chem. Lett., 1998, 545 CrossRef CAS.
- H. Lai, J. Hong, P. Liu, C. Yuan, Y. Li and Q. Fang, RSC Adv., 2012, 2, 2427 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c2ra21127k |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.