Facile synthesis of SnS hollow nanoparticles via laser ablation followed by chemical etching†
Ming-Yan
Sun
,
Jing
Yang
,
Tao
Lin
and
Xi-Wen
Du
*
Tianjin Key Laboratory of Composite and Functional Materials, School of Materials Science and Engineering, Tianjin University, Tianjin 300072, P. R. China. E-mail: xwdu@tju.edu.cn
First published on 27th June 2012
Abstract
SnS hollow nanoparticles (HNPs) were fabricated by a two-step method, where laser ablation of a Sn target was first conducted in ethanol solution of thioacetamide to synthesize Sn@SnS core-shell nanoparticles, and then acid etching was employed to eliminate the Sn cores. The as-obtained SnS HNPs are uniform and nearly spherical with a size of ∼40 nm. Moreover, the shell thickness of the HNPs can be facilely modified by adjusting the concentration of thioacetamide in the first step.
Introduction
As narrow-band-gap semiconductors, tin chalcogenides are optically active in the near-infrared and infrared spectral regions; hence they show a high potential for infrared optics applications.1,2 Moreover, they are expected to be promising substitutes for toxic materials such as PbS, PbSe and PbTe due to their nontoxicity.3 Among several tin chalcogenides, tin sulphide (SnS) possesses a stable orthorhombic layered crystal structure,1,2,4,5 and a direct band gap of 1.3 eV, which is efficient for the absorption of sun light.2–4,6 As a result, SnS has been widely applied in near-infrared detectors,1,3 electrochemical capacitors,7,8 lithium ion batteries,9 photovoltaic solar cells4,10,11 and so on.Hollow nanoparticles (HNPs) possess many unique properties such as low density, high surface-to-volume ratio, and the effect of void space.12 Several approaches have been developed to synthesize hollow nanostructures and, according to how the hollow structure is formed, they are categorized into four classes: the Kirkendall effect, chemical etching, galvanic replacement, and template-mediated approach.13 Among them, chemical etching has been demonstrated as a simple and high yield route for the synthesis of hollow or porous nanomaterials. For example, Zeng and co-workers put forward a selective etching strategy to fabricate hollow oxide nanoparticles by using metal@oxide core-shell nanostructures as the starting material.14 Hyeon et al. synthesized various hollow oxide nanoparticles by heating and etching as-prepared manganese oxide or iron oxide nanocrystals in technical grade trioctylphosphine oxide.15
So far, a variety of SnS nanostructures including nanoparticles,1,2,16 nanobelts,17 nanowires,18 nanosheets5,19 have been prepared via chemical routes. For example, Hickey and co-workers fabricated monodisperse SnS nanoparticles smaller than 10 nm in size by hot injection.2 Liu and co-workers prepared single crystalline SnS nanowires by a solution-based technique;18 Zhu and co-workers synthesized SnS nanosheets by a thioglycolic acid assisted hydrothermal method.19 Hitherto, the synthesis of SnS nanostructures is based on traditional solvothermal or hot injection methods, where the products experience a nucleation and growth process, and finally grow into solid nanostructures. On the other hand, the SnS system is not a good Kirkendall effect system,20 namely the diffusion coefficient of the metal Sn is lower than that of S atoms owing to a larger atomic size of Sn. As a result, when Sn nanoparticles are sulphurized, only Sn@SnS core-shell nanoparticles can be obtained. Up to now, the synthesis of SnS HNPs has never been reported.
Laser ablation in liquid (LAL) is known as a powerful technique to fabricate nanostructures, where a pulsed laser is adopted to irradiate a solid target in a liquid medium.21,22 By employing different laser sources, distinct ablation products (plasma, vapour, and metal micro- or nanosized droplets) can be generated. For example, short-pulse-width lasers like a nanosecond pulsed laser tend to generate the vapour and plasma,21,23 while long-pulse-width lasers like millisecond pulsed laser usually induce nanodroplets.12,24,25 These ablation products will further react with the surrounding liquid to form final nanostructures. Hitherto, the LAL technique has successfully produced a large number of nanostructures with various compositions (metals, alloys, oxides, carbides, hydroxides, etc.) and morphologies (nanoparticles, nanocubes, nanorods, nanocomposites, etc.).26
In the present work, we propose a facile way to synthesize SnS HNPs by combining laser ablation and chemical etching; a similar method has been employed to fabricate oxide hollow nanoparticles.14 Specifically, we chose a millisecond pulsed laser which facilitates obtaining pure hollow nanoparticles. As shown in Scheme 1, first, a long-pulse-width (millisecond) laser is employed to ablate the Sn target in ethanol solution of thioacetamide (TAA), which gives a large amount of hot metal nanodroplets (Scheme 1a). Almost at the same time, the hot metal nanodroplets react with ambient TAA to produce Sn@SnS core-shell nanoparticles (Scheme 1b). Second, the as-prepared core@shell nanoparticles are etched by dilute hydrochloric acid (HCl) to eliminate the Sn cores but preserve the SnS shells, resulting in the formation of SnS HNPs (Scheme 1c). The obtained SnS HNPs are nearly spherical with an average diameter about 40 nm, and the thickness of the SnS shells can be easily tuned by adjusting the concentration of sulphur source. This simple method can be expanded to other material systems to synthesize HNPs.
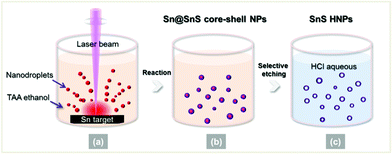 | ||
| Scheme 1 Schematic formation of SnS HNPs by combining laser ablation and chemical etching. | ||
Experimental
Preparation of Sn@SnS core-shell nanoparticles
Sn target (5 mm thick, purity quotient 99.99%) was ablated with a millisecond pulsed Nd:YAG laser (wavelength 1064 nm) in 12 ml of TAA solution in ethanol with a certain concentration. The depth of solution was 5 mm above the target and the whole process was protected by Ar gas at a flow rate of 60 sccm. The laser power density, pulse width, and frequency used in the experiments were 106 W cm−2, 1.5 ms, and 5 Hz, respectively, and the irradiation time was kept at 10 min. After laser ablation, the suspension was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, washed with ethanol three times, and finally dispersed in ethanol.
000 rpm, washed with ethanol three times, and finally dispersed in ethanol.
Preparation of SnS HNPs
To prepare SnS HNPs, 1 ml of dilute HCl (pH value 2.5∼3) was injected into 2 ml ethanol solution of Sn@SnS core-shell nanoparticles. The deep brown solution released bubbles and then turned colourless, indicating that the Sn metal cores were etched out completely.Preparation of porous SnS HNPs
The synthesis procedure for porous SnS HNPs is same with that for Sn@SnS core-shell nanoparticles, except that the laser source was replaced with a nanosecond pulsed laser (wavelength 1064 nm), and the laser power density, voltage, and frequency were set as 108 W cm−2, 460 V, and 5 Hz, respectively.Characterization
The product morphology was observed by an FEI Technai G2 F20 transmission electron microscope (TEM) equipped with a field-emission gun, and the composition was measured by an Oxford INCA energy-dispersive X-ray spectroscopy (EDS) module attached to the TEM. The phase structure was investigated by using a Bruker D8 advance X-ray diffractometer (XRD). UV-vis absorption spectra were detected by using a Hitachi U4100 spectrophotometer.Results and discussion
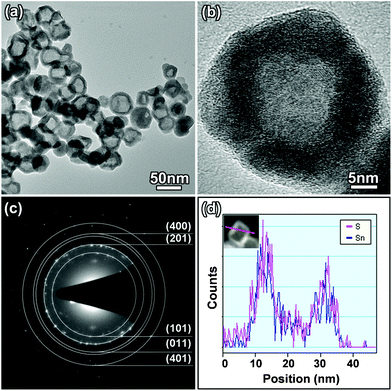
The product of laser ablation of the tin target in TAA solution was characterized, and the results are shown in Fig. 1. As shown in the TEM image in Fig. 1a, core-shell nanoparticles with an average size of 40 nm can be obtained after laser ablation. In the high resolution TEM (HRTEM) image (Fig. 1b), the shell exhibits interplanar distances of 0.284 nm and 0.342 nm, which agree with the (111) and (120) lattice fringes of orthorhombic SnS, respectively. EDS line-scan analysis indicates that the core is mainly composed of the Sn element, while the shell contains both S and Sn elements (Fig. 1c). XRD result indicates that the product comprises of two phases, namely tetragonal Sn (JCPDS card No. 04-0673) and orthorhombic SnS (JCPDS card No. 65-3812) (Fig. 1d). The above characterization results jointly illustrate that the product is Sn@SnS core-shell nanoparticles. | ||
| Fig. 1 (a) TEM images, (b) HRTEM images, (c) EDS line-scan profile and (d) XRD pattern of as-synthesized Sn@SnS core-shell nanoparticles. | ||
In the second step, these core@shell nanoparticles were etched using dilute HCl. As a result, a lot of SnS HNPs were produced. As shown in the TEM image (Fig. 2a), the HNPs are uniform, possessing similar sizes with those of core-shell nanoparticles. The HRTEM image (Fig. 2b) illustrates that the HNPs are of nearly spherical shape, but their crystallinity somewhat deteriorates after chemical etching. The selected area electronic diffraction (SAED) pattern of the HNPs exhibits a set of diffraction rings, which can be indexed as the (101), (201), (011), (401) and (400) planes of orthorhombic phase of SnS (Fig. 2c). The EDS line-scan analysis suggests that the HNPs are composed of Sn and S elements (Fig. 2d). Thus the product is proven to be SnS HNPs.
 | ||
| Fig. 2 (a) TEM image, (b) HRTEM image, (c) SAED pattern and (d) EDS line-scan profile of SnS HNPs. | ||
The shell thickness of Sn@SnS nanoparticles can be tuned by varying TAA concentration during the laser ablation, and the controllable shell thickness is then transferred to SnS HNPs after chemical etching. As shown in Fig. 3, as the TAA concentration in ethanol solution increases from 0.5 to 2 M, SnS shells become thicker, gradually growing from 3.5 to 6.8 nm. At a concentration of 2 M, even a few fully sulphurized SnS nanoparticles could be found in the product (see ESI† Fig. S1).
 | ||
| Fig. 3 Low magnification TEM images and high magnification TEM images of Sn@SnS core-shell nanoparticles prepared in TAA ethanol solution with different concentrations (a1), (a2) 0.1 M; (b1), (b2) 0.5 M; (c1), (c2) 1.0 M; (d1), (d2) 2.0 M. High magnification TEM images of corresponding SnS HNPs after chemical etching (a3) 0.1 M ; (b3) 0.5 M; (c3) 1.0 M; (d3) 2.0 M. The scale bars in (a1), (b1), (c1), (d1) represent 20 nm, while those in (a2), (b2), (c2), (d2), (a3), (b3), (c3), (d3) represent 5 nm. | ||
Next, we discuss the formation mechanism of SnS HNPs. The generation of Sn@SnS nanoparticles by laser ablation is a prerequisite for the subsequent fabrication of HNPs by chemical etching. However, common products by laser ablation are pure metal or pure compound nanoparticles,23,27–29 because ordinary laser ablation techniques adopt short-pulse-width lasers with high power density (108–1010 W cm−2), which heat the metal target rapidly into vapour or plasma state. Subsequently, the vapour or plasma condenses into metal nanoparticles or reacts with ambient medium to form compound nanoparticles.21,30 In the present study, we employed a long-pulse-width laser with a low power density (106 W cm−2), which can generate Sn nanodroplets instead of vapour or plasma, as proven in our previous works.12,24,25,31 The hot nanodroplets can react with TAA molecules in ethanol solution in a controllable way, giving rise to core-shell nanostructures. The reaction extent and then the thickness of SnS shell can be precisely controlled by adjusting the TAA concentration in ethanol, as shown in Fig. 3.
Although we varied the TAA concentration, the products are always Sn@SnS nanoparticles, suggesting that the surface reaction of Sn nanodroplets can not proceed completely under the conditions created by laser ablation. Several factors can be responsible for the incomplete surface reaction. First, although a high TAA concentration facilitates the surface reaction, the TAA concentration in ethanol can not be enhanced unlimitedly, because TAA has a saturated solubility of 2 M. Second, the liquid medium possesses a strong quenching effect, which cools the nanodroplets immediately after their generation, and slows down the diffusion and surface reactions. Third, we believe that the diffusion coefficients of S and Sn atoms in the SnS layer are much lower due to their relatively large atomic sizes. Once an SnS layer forms on the surface of an Sn nanoparticle, it blocks the diffusion of Sn and S atoms. Based on the above, we can conclude that Sn@SnS nanoparticles are a stable product under the conditions created in the present experiment.
As for the chemical etching treatment, the standard reduction potential of the Sn2+/Sn pair (−0.1375 V, vs. SHE) is lower than that of the H+/H pair (0 V), therefore, Sn cores can be etched in acid solution according to the following reaction:
| Sn + 2H+ → Sn2+ + H2↑ | (1) |
We tested different etching reagents, for example, acetic acid, phosphoric acid, tartaric acid, and hydrochloric acid, and found that acetic acid destroyed the SnS shell severely, while leaving the Sn core unchanged. Phosphoric acid and tartaric acid could etch both the SnS shell and the Sn core, producing fragments rather than HNPs, as shown in Fig. S2 in the ESI.† The above results indicate that the SnS shell is sensitive to organic acids, and the selective etching of the Sn core can be realized only by using dilute HCl solutions with a pH value of 2.5∼3. In this case, the SnS shell is stable in acid, and H+ ions can diffuse through the SnS shell or contact directly with an exposed Sn surface due to the discontinuity of the SnS layer. The reaction of H+ ions with Sn cores can eliminate Sn cores completely and leave intact SnS HNPs. Nevertheless, in HCl aqueous solution with higher acidity (pH < 2), the SnS shell becomes unstable and reacts with HCl by the following equation:
| SnS + HCl → H2SnCl4 + H2S↑ | (2) |
As a result, the Sn@SnS nanoparticles are completely damaged, as shown in Fig. S3 in the ESI.† Based on the above, it is concluded that dilute HCl solution is an appropriate reagent for obtaining SnS HNPs.
We also attempted a one-step synthesis of SnS HNPs by laser ablation in liquid, where a nanosecond pulsed laser with a wavelength of 1064 nm (photon energy 1.17 eV) was employed as the light source. The SnS shell with a band gap of 1.3 eV is transparent for the laser beam, while the metal core can absorb the light efficiently. As shown in Fig. 4, both SnS HNPs and HCl solution do not absorb 1064 nm light, while the suspension of Sn@SnS nanoparticles exhibits obvious absorbance at this wavelength. Therefore, a 1064 nm laser can penetrate through the SnS shell and heat the metal core selectively. On the other hand, a nanosecond laser can provide a higher energy flux than a millisecond pulsed laser, heating the metal core to vapour state,12 leaving the laser-transparent shells unchanged and thus producing HNPs.
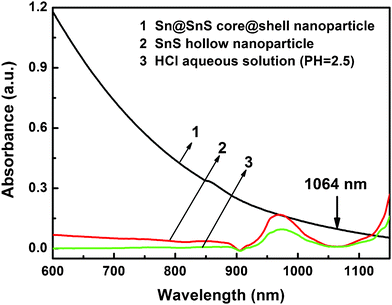 | ||
| Fig. 4 UV-vis absorption spectra of Sn@SnS core-shell nanoparticle, SnS HNPs, and dilute HCl solution. | ||
TEM images shown in Fig. 5 indicate that the product prepared by one-step laser ablation is a mixture of core-shell nanoparticles and porous HNPs which are uniform and partially destroyed. The impurity of the product can be ascribed to the inadequate laser irradiation, and the broken HNPs should arise from the damage effect of Sn vapour which is usually generated under the nanosecond laser pulsed laser with a high power density.21,30 Since the boiling point (2260 °C) of Sn metal is much higher than the melting point (880 °C) of SnS semiconductor, when the core is heated to vapour state by laser beam, the hot Sn vapour can heat SnS to a liquid state, causing a collapse of the shell. The above results illuminate that the two-step process, namely laser ablation plus chemical etching, is a more appropriate method to achieve intact SnS HNPs.
 | ||
| Fig. 5 (a), (b) TEM images of porous SnS HNPs produced by nanosecond pulsed laser ablation. The arrows indicate core@shell nanoparticles and porous HNPs. | ||
Conclusions
SnS HNPs were synthesized by a two-step process, where laser ablation first produced Sn@SnS core-shell nanoparticles, and then chemical etching was conducted to eliminate the Sn core. The shell thickness could be controlled by modifying the concentration of the sulphur source. This special hollow structure of SnS should possess different properties from its solid counterpart, hence it may be widely used in catalysis, sensors, batteries, drug-delivery, etc. In addition, this method can be expanded to synthesize other hollow nanoparticles.Acknowledgements
This work was supported by the Natural Science Foundation of China (Nos. 51102176, 50972102 and 50902103), National High-Tech R&D Program of China (No. 2009AA03Z301), Research Fund for the Doctoral Program of Higher Education of China (No. 20100032120021), and Natural Science Foundation of Tianjin City (Nos. 11JCYBJC02000, 09JCZDJC22600 and 08JCYBJC02900).References
- Y. Xu, N. Al-Salim, C. W. Bumby and R. D. Tilley, J. Am. Chem. Soc., 2009, 131, 15990–15991 CrossRef CAS.
- S. G. Hickey, C. Waurisch, B. Rellinghaus and A. Eychmueller, J. Am. Chem. Soc., 2008, 130, 14978–14980 CrossRef CAS.
- J. Ning, K. Men, G. Xiao, L. Wang, Q. Dai, B. Zou, B. Liu and G. Zou, Nanoscale, 2010, 2, 1699–1703 RSC.
- H. T. Liu, Y. Liu, Z. Wang and P. He, Nanotechnology, 2010, 21(105707), 1–5 Search PubMed.
- J. G. Kang, Y. D. Ko, K. J. Choi, J. G. Park and D. W. Kim, Appl. Phys. A: Mater. Sci. Process., 2011, 103, 505–510 CrossRef CAS.
- E. C. Greyson, J. E. Barton and T. W. Odom, Small, 2006, 2, 368–371 CrossRef CAS.
- Y. Li, H. Q. Xie and J. P. Tu, Mater. Lett., 2009, 63, 1785–1787 CrossRef CAS.
- M. Jayalakshmi, M. M. Rao and B. M. Choudary, Electrochem. Commun., 2004, 6, 1119–1122 CrossRef CAS.
- Y. Li, J. P. Tu, X. H. Huang, H. M. Wu and Y. F. Yuan, Electrochim. Acta, 2006, 52, 1383–1389 CrossRef CAS.
- A. Stavrinadis, J. M. Smith, C. A. Cattley, A. G. Cook, P. S. Grant and A. A. R. Watt, Nanotechnology, 2010, 21(185202), 1–7 Search PubMed.
- Z. J. Wang, S. C. Qu, X. B. Zeng, J. P. Liu, C. S. Zhang, F. R. Tan, L. Jin and Z. G. Wang, J. Alloys Compd., 2009, 482, 203–207 CrossRef CAS.
- K. Y. Niu, J. Yang, S. A. Kulinich, J. Sun and X. W. Du, Langmuir, 2010, 26, 16652–16657 CrossRef CAS.
- K. An and T. Hyeon, Nano Today, 2009, 4, 359–373 CrossRef CAS.
- H. B. Zeng, W. P. Cai, P. S. Liu, X. X. Xu, H. J. Zhou, C. Klingshirn and H. Kalt, ACS Nano, 2008, 2, 1661–1670 CrossRef CAS.
- K. An, S. G. Kwon, M. Park, H. Bin Na, S. I. Baik, J. H. Yu, D. Kim, J. S. Son, Y. W. Kim, I. C. Song, W. K. Moon, H. M. Park and T. Hyeon, Nano Lett., 2008, 8, 4252–4258 CrossRef CAS.
- D. S. Koktysh, J. R. McBride, R. D. Geil, B. W. Schmidt, B. R. Rogers and S. J. Rosenthal, Mater. Sci. Eng., B, 2010, 170, 117–122 CrossRef CAS.
- C. H. An, K. B. Tang, G. Z. Shen, C. R. Wang, Q. Yang, B. Hai and Y. T. Qian, J. Cryst. Growth, 2002, 244, 333–338 CrossRef CAS.
- Y. K. Liu, D. D. Hou and G. H. Wang, Chem. Phys. Lett., 2003, 379, 67–73 CrossRef CAS.
- H. L. Zhu, D. R. Yang, Y. J. Ji, H. Zhang and X. F. Shen, J. Mater. Sci., 2005, 40, 591–595 CrossRef CAS.
- H. J. Fan, U. Gosele and M. Zacharias, Small, 2007, 3, 1660–1671 CrossRef CAS.
- G. W. Yang, Prog. Mater. Sci., 2007, 52, 648–698 CrossRef CAS.
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2009, 11, 3805–3821 RSC.
- R. K. Thareja and S. Shukla, Appl. Surf. Sci., 2007, 253, 8889–8895 CrossRef CAS.
- K. Y. Niu, J. Yang, J. Sun and X. W. Du, Nanotechnology, 2010, 21, 295604 CrossRef , (1–5).
- K. Y. Niu, J. Yang, S. A. Kulinich, J. Sun, H. Li and X. W. Du, J. Am. Chem. Soc., 2010, 132, 9814–9819 CrossRef CAS.
- H. B. Zeng, X. W. Du, S. C. Singh, S. A. Kulinich, S. K. Yang, J. P. He and W. P. Cai, Adv. Funct. Mater., 2012, 22, 1333–1353 CrossRef CAS.
- T. Tsuji, Y. Okazaki, Y. Tsuboi and M. Tsuji, Jpn. J. Appl. Phys., 2007, 46, 1533–1535 CrossRef CAS.
- A. V. Kabashin and M. Meunier, J. Appl. Phys., 2003, 94, 7941–7943 CrossRef CAS.
- P. S. Liu, W. P. Cai and H. B. Zeng, J. Phys. Chem. C, 2008, 112, 3261–3266 CAS.
- T. Sakka, S. Iwanaga, Y. H. Ogata, A. Matsunawa and T. Takemoto, J. Chem. Phys., 2000, 112, 8645–8653 CrossRef CAS.
- F. Lin, J. Yang, S. H. Lu, K. Y. Niu, Y. Liu, J. Sun and X. W. Du, J. Mater. Chem., 2010, 20, 1103–1106 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of fully sulphurized SnS nanoparticles, TEM images of Sn@SnS core-shell nanoparticles after etching by acetic acid, phosphoric acid and tartaric acid, TEM images of Sn@SnS core-shell nanoparticles after etching by high concentration of HCl solution. See DOI: 10.1039/c2ra21112b |
| This journal is © The Royal Society of Chemistry 2012 |