Exploiting conformationally restricted N,N′-dimethyl-N,N′-diarylureas as biologically active C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C double bond analogues: synthesis and biological evaluation of combretastatin A-4 analogues†
C double bond analogues: synthesis and biological evaluation of combretastatin A-4 analogues†
Timothy J.
Snape
*a,
Katherine
Karakoula
b,
Farzana
Rowther
b and
Tracy
Warr
b
aSchool of Pharmacy and Biomedical Sciences, Maudland Building, University of Central Lancashire, Preston, PR1 2HE, UK. E-mail: tjsnape@uclan.ac.uk; Fax: +44(0)1772 892929; Tel: +44(0)1772 895805
bBrain Tumour UK Neuro-Oncology Research Centre, University of Wolverhampton, Wulfruna Street, Wolverhampton, WV1 1LY, UK
First published on 23rd July 2012
Abstract
The biological efficacy of a number of suitably functionalised conformationally restricted N,N′-dimethyl-N,N′-diarylureas, which occupy a similar 3-dimensional space to combretastatin A-4 (CA4), has been evaluated for their ability to inhibit tubulin polymerisation and inhibit the growth of short term glioblastoma multiforme (GBM) cell cultures and an established GBM cell line (U251MG). The results show that the ureas most like CA4, with regards to benzene ring oxygenation and overall shape, are the most active.
Introduction
It is well known that changes in the N-substitution of benzanilides and diarylureas result in alterations in their conformation, a feature which can be exploited to gain control of the C–N bonds of these compounds, both in solution and solid states. For example, N,N′-dimethylation has been used as a strategy to control C–N bond rotation to enable a switch between the (trans, trans)-conformation and the (cis, cis)-conformation thus placing the aryl rings in close proximity to one another, presumably favoured because of π-stacking.1–10 Consequently, this property has been exploited in diarylureas to prepare compounds that act as fluorescent sensors by changing their conformation upon exposure to the N-alkylating agent, methyl iodide.10 Similarly, the conformational preference of aromatic amides has been applied to the preparation of compounds which modify the bioactivity of synthetic retinoids3 and cytokinins,11 where it has been shown that amides representing retinobenzoic acids are biologically active only when the amide nitrogen is unsubstituted, and the conformation predominantly resembles that of an (E)-stilbene, in contrast to the biologically inactive N-methylated analogues whose conformation resembles the (Z)-isomer.3Another series of compounds that rely on shape in order to maintain function are the family of biologically active stilbenes known as combretastatins (Fig. 1). These compounds possess promising antimitotic activity and exert their effect by interacting with the colchicine binding-site on tubulin.12,13 The most active of the series, combretastatin A-4 (1) (CA4), shown in Fig. 1 along with combretastatin (3) and the eponymous ligand, colchicine (4), is currently being used in multiple clinical trials as a treatment for solid tumours as its phosphate salt (CA4P).14 Despite its simple structure, the synthesis of CA4 analogues has been the subject of an intense amount of investigation including: (i) the modification of the substitution of rings A and B as well as the incorporation of heteroatoms into the rings;15–17 and (ii) the modification of the double bond,18 including its substitution,19 in addition to the preparation of analogues where the olefin is replaced by various other groups.20–28 It is known that the (Z)-isomer of stilbenes can easily isomerise under the action of heat, light, and protic media,13 and thus a number of research groups have attempted to modify the double bond in CA4 in an attempt to stabilise the (Z)-configuration and thus maintain its biological activity under such conditions.18–28 Additional structure–activity studies have found that the hydroxyl group at position-3 in ring B of CA4 (i.e.2) is not essential to activity,18 and that substituting the three methoxy groups in ring A for methyl groups, does not have a deleterious effect on activity, but in fact produces an analogue which is more active than combretastatin A-4 in inhibiting the assembly of tubulin.16
 | ||
| Fig. 1 The structure of combretastatin A-4 (1), a structurally related biologically active analogue lacking the -OH group (2), combretastatin (3) and colchicine (4). | ||
It is believed that the olefinic bond plays more of a constitutive role rather than a binding role; its function acting to position the aromatic rings in the correct relative orientation thus giving the molecule the correct shape to maximise its interaction with the target. As such, the double bond must be in a (Z)-configuration to possess high cytotoxic and antitubulin activity, while the (E)-isomer is significantly less potent, if not inactive altogether.12,13
With the (Z)-configuration of the aromatic rings in CA4 and its analogues essential for biological activity, we postulated that a hybrid structure, combining the aromatic rings of a biologically active combretastatin analogue with the (cis, cis)-conformational preference of N,N′-dimethyl-N,N′-diarylureas, would reveal a novel class of combretastatin analogues for further investigation as anticancer agents.
Results and Discussion
Initial attempts to synthesise a direct diarylurea–combretastatin A-4 hybrid met with problems due to the free hydroxyl group in the B-ring, but since this had been shown not to be important for activity,18 we turned our attention to the hybrid which lacks this hydroxyl group; in the event this proved to be a much simpler synthesis, Scheme 1. The ureas were prepared through a simple union between a substituted aniline (5–8) and an appropriate isocyanate (9 or 10), followed by N-methylation to generate the (cis, cis)-N,N′-dimethyl-N,N′-diarylureas 11–14 (54–74% yield, 2 steps). The (cis, trans)-urea 15, mimicking the inactive (E)-CA4, was prepared from 4-methoxy-N-methylaniline (6) and isocyanate 9 in 81% yield (Scheme 1).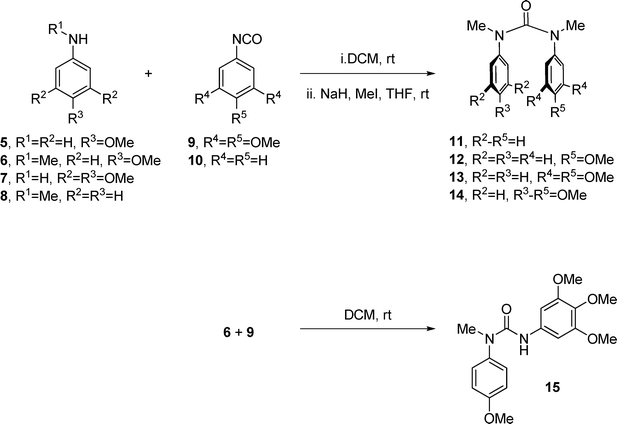 | ||
| Scheme 1 Synthesis of ureas. | ||
As can be seen from the X-ray crystal structure of 14 in Fig. 2,29 the aromatic rings are indeed orientated in close proximity, as anticipated, allowing the urea to adopt a conformation similar to that of CA4, also, comparison of the 1H-NMR spectra of 14 and 15 showed the characteristic up-field shift of the aromatic protons, the consequence of a newly formed close interaction between the two aromatic rings, confirming that the expected conformational change from trans to cis had occurred. Preliminary results from the molecular modelling of three conformationally-templated structures are also shown in Fig. 2,30 which show that a reasonable level of observed overlap (shape similarity 0.798) can be achieved between 14 (shown in pink) and the natural combretastatins (shown in green and pale blue). The positions of the aromatic rings are well aligned, as are the three methoxy groups of the A-rings and the p-methoxy group of the B-rings, which can be attributed to the close alignment of the aryl–aryl linker; carbon–carbon double and single bonds in the combretastatins and NC(O)N in the urea analogue.
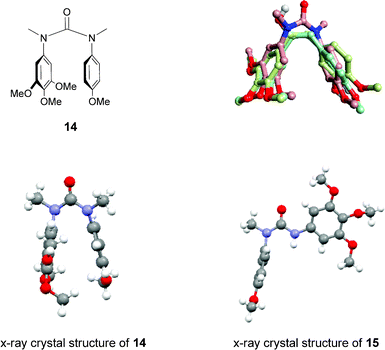 | ||
| Fig. 2 The structure of the urea–combretastatin hybrid, 14 (top left); the X-ray crystal structures of 14 and 15 (bottom, showing the (cis, cis)- and (cis, trans)-conformations);29 and the superposition of combretastatin A-4 (1, pale blue), combretastatin (3, green) and 14 (pink) (top right).30 | ||
Inhibition of tubulin polymerisation (TPI)
The ability of each of the compounds to inhibit tubulin polymerisation in vitro was evaluated at a single dose of 10 μM (Table 1). CA4P, used as a comparison, was the most effective inhibitor of microtubule assembly with ∼50% inhibition over 1 h compared to the DMSO-treated control. The relative TPI activity of the 5 analogues reflects their shape and predicted activity. For example, we expected 14, which is most like CA4P in shape and oxygenation, to be the most active and the activity to drop off as the analogues became less like CA4P with fewer methoxy groups. The results indicate that the impact of the variations of the –OMe (both number and position) in the modified compounds was evident; the 2 analogues bearing the most –OMe groups, and thus most like CA4 (and CA4P) (13 and 14) inhibited tubulin polymerisation by 34% and 31%, respectively, whilst those lacking some or all –OMe groups (11 and 12) had the TPI activity reduced to 23% and 25%, respectively. The analogue which has a completely different shape (15), and is thus more reminiscent of the inactive (E)-isomer of CA4, was essentially inactive here too. This suggests, at least to some extent, that the oxygenation of the phenyl rings in these urea-analogues in combination with a defined shape is important, but that having too much oxygenation (e.g.14) may be detrimental to tubulin binding. Based on the modelling in Fig. 2 it is unclear at present as to why this would be the case.| Compound | % TPIa |
|---|---|
| a The TPI activity is expressed as the percentage of polymerisation relative to the control (100% TPI) at a single dose of 10 μM. Values are representative of 2 experiments. | |
| CA4P | 49 |
| 13 | 34 |
| 14 | 31 |
| 12 | 25 |
| 11 | 23 |
| 15 | 5 |
In vitro cell culture assays
The anti-proliferative and cytotoxic effects of CA4P and the 5 analogues were evaluated in 2 GBM short-term cell cultures (IN1472 and IN1760) and the established GBM cell line U251MG at 10 μM of the test compound (Table 2). There was good correlation between the TPI data and the ability of the compounds to induce growth inhibition, mitotic arrest and apoptosis with CA4P displaying a higher activity than the 5 analogues in all the in vitro assays. Indeed, CA4P was the only compound to induce significant levels of growth inhibition, apoptosis and mitotic arrest at 10 μM in all 3 cell cultures whereas U251MG and IN1760 were resistant to the analogues. For example, against the IN1472 cells, CA4P exhibited ∼83% growth inhibition compared with ∼32%, 26%, 24% and 7% for 13, 14, 12 and 11 respectively—a trend which mirrors that of the TPI assay. Urea 15 showed mild cytotoxicity (15% growth inhibition) in this assay, which is surprising given the lack of activity in the tubulin binding assay, suggesting that an alternative mitotic control mechanism may be involved.| Compound | % Growth Inhibitiona | Apoptotic Indexb | Mitotic Indexc | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IN1472 | IN1760 | U251MG | IN1472 | IN1760 | U251MG | IN1472 | IN1760 | U251MG | |
| a The reduction of proliferation in cell cultures treated with 10 μM of test compounds for 72 h compared to solvent-treated controls. b The induction of apoptosis in cell cultures treated with 10 μM of test compounds for 24 h compared to solvent-treated controls. c The induction of mitotic arrest in cell cultures treated with 10 μM of test compounds for 24 h compared to solvent-treated controls. Results are expressed as the mean ± SD of 3 experiments. d P < 0.05 (treated versus control). | |||||||||
| CA4P | 82.84 ± 0.16d | 29.78 ± 1.04d | 38.49 ± 0.27d | 6.21 ± 1.36d | 6.51 ± 1.21d | 1.51 ± 0.26 | 24.83 ± 2.49d | 4.70 ± 0.29d | 19.97 ± 1.20d |
| 13 | 31.57 ± 0.02d | 0 ± 4.33 | 6.68 ± 0.087 | 1.74 ± 0.239 | 0.22 ± 0.29 | 0.61 ± 0.14 | 6.37 ± 0.06d | 2.10 ± 0.52 | 4.17 ± 0.06 |
| 14 | 25.54 ± 0.29 | 0 ± 2.55 | 9.45 ± 0.17 | 2.90 ± 0.11d | 0.43 ± 0.10 | 0.80 ± 0.08 | 6.40 ± 1.13d | 2.33 ± 0.12 | 4.33 ± 0.35 |
| 12 | 24.19 ± 0.10 | 8.59 ± 1.58 | 6.63 ± 0.084 | 2.78 ± 0.49d | 0.13 ± 0.34 | 0.62 ± 0.08 | 3.37 ± 0.31 | 2.07 ± 0.07 | 3.77 ± 0.41 |
| 11 | 6.91 ± 0.14 | 0 ± 3.23 | 0 ± 0.12 | 2.06 ± 0.02 | 0.21 ± 0.31 | 0.91 ± 0.17 | 2.70 ± 0.21 | 2.13 ± 0.15 | 4.20 ± 0.05 |
| 15 | 15.46 ± 0.17 | 0 ± 2.86 | 9.6 ± 0.32 | 2.19 ± 0.46 | 0.63 ± 0.12 | 0.15 ± 0.26 | 5.73 ± 0.46d | 2.17 ± 0.05 | 3.87 ± 0.11 |
As can be seen from the apoptotic index data, whilst CA4P, 14 and 12 did show a significant induction of apoptosis compared to vehicle controls against the IN1472 cells, the analogues were only half as effective at being able to induce apoptosis compared to CA4P (∼3 vs. ∼6 (arbitrary values)). The analogues were unable to induce apoptosis at all against the IN1760 and U251MG cells whereas CA4P maintained its ability to do so against IN1760, but not U251MG.
In the mitotic index assay against the more sensitive IN1472 cells, 13 and 14 both induced significant levels of mitotic arrest although the microtubule spindle apparatus was still evident in some dividing cells (data not shown). The levels of mitotic arrest were only ∼25% of the levels achieved by CA4P indicating that in these whole cell systems other factors than molecular shape and oxygenation are influential to anticancer activity. That said, 15 also induced significant mitotic arrest in IN1472, further corroborating that additional factors are at play here. Conversely, treatment with 10 μM CA4P completely disrupted the microtubule network leading to the formation of metaphase chromosome clusters and apoptotic DNA fragmentation (data not shown). Urea 13 significantly inhibited cell proliferation by 32% although it was a poor inducer of apoptosis.
Conclusions
In summary, the preference for N,N′-dimethyl-N,N′-diarylureas to exist predominantly in the (cis, cis)-conformation, and thus possess the ability to mimic the (Z)-stereochemistry of stilbenes, has been exploited to prepare combretastatin A-4 analogues, some of which possess the ability to inhibit tubulin polymerisation and glioblastoma cell growth, albeit not as effectively as CA4P. These combined results suggest that further investigation is warranted to optimise biological activity in this area and identify compounds that can match the activity of CA4P. Nevertheless, these compounds present a novel scaffold for tubulin binding inhibitors, and along with the conflicting results of compound 15 in the TPI and mitotic index assays, suggest that they deserve further investigation in this area.Acknowledgements
The authors thank Dr Susan Short, UCL, for providing CA4P, Dr Gino Brunori, GSK, for assistance with the tubulin polymerization assay and Dr Madeleine Helliwell for crystallographic analysis, as well as the Universities of Central Lancashire and Wolverhampton for support. This work was part-funded by Brain Tumour UK and Brain Tumour North West.References
- G. Lepore, S. Migdal, D. E. Blagdon and M. Goodman, J. Org. Chem., 1973, 38, 2590–2594 CrossRef CAS.
- U. Lepore, G. C. Lepore, P. Ganis, G. Germain and M. Goodman, J. Org. Chem., 1976, 41, 2134–2137 CrossRef CAS.
- H. Kagechika, T. Himi, E. Kawachi and K. Shudo, J. Med. Chem., 1989, 32, 2292–2296 CrossRef CAS.
- A. Itai, Y. Toriumi, N. Tomioka, H. Kagechika, I. Azumaya and K. Shudo, Tetrahedron Lett., 1989, 30, 6177–6180 CrossRef CAS.
- I. Azumaya, H. Kagechika, Y. Fujiwara, M. Itoh, K. Yamaguchi and K. Shudo, J. Am. Chem. Soc., 1991, 113, 2833–2838 CrossRef CAS.
- A. Itai, Y. Toriumi, S. Saito, H. Kagechika and K. Shudo, J. Am. Chem. Soc., 1992, 114, 10649–10650 CrossRef CAS.
- K. Yamaguchi, G. Matsumura, H. Kagechika, I. Azumaya, Y. Ito, A. Itai and K. Shudo, J. Am. Chem. Soc., 1991, 113, 5474–5475 CrossRef CAS.
- J. Clayden, L. Vallverdu and M. Helliwell, Org. Biomol. Chem., 2006, 4, 2106–2118 CAS.
- J. Clayden, L. Lemiegre, M. Pickworth and L. Jones, Org. Biomol. Chem., 2008, 6, 2908–2913 CAS.
- T. Hirano, T. Osaki, S. Fujii, D. Komatsu, I. Azumaya, A. Tanatani and H. Kagechika, Tetrahedron Lett., 2009, 50, 488–491 CrossRef CAS.
- T. Okamoto, K. Shudo, S. Takahashi, E. Kawachi and Y. Isogai, Chem. Pharm. Bull., 1981, 29, 3748–3750 CrossRef CAS.
- A. Cirla and J. Mann, Nat. Prod. Rep., 2003, 20, 558–564 RSC.
- G. C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca and A. A. Genazzani, J. Med. Chem., 2006, 49, 3033–3044 CrossRef CAS.
- http://www.oxigene.com/ .
- N. J. Lawrence, L. A. Hepworth, D. Rennison, A. T. McGown and J. A. Hadfield, J. Fluorine Chem., 2003, 123, 101–108 CrossRef CAS.
- K. Gaukroger, J. A. Hadfield, N. J. Lawrence, S. Nolan and A. T. McGown, Org. Biomol. Chem., 2003, 1, 3033–3037 CAS.
- D. Simoni, R. Romagnoli, R. Baruchello, R. Rondanin, G. Grisolia, M. Eleopra, M. Rizzi, M. Tolomeo, G. Giannini, D. Alloatti, M. Castorina, M. Marcellini and C. Pisano, J. Med. Chem., 2008, 51, 6211–6215 CrossRef CAS.
- M. Cushman, D. Nagarathnam, D. Gopal, A. K. Chakraborti, C. M. Lin and E. Hamel, J. Med. Chem., 1991, 34, 2579–2588 CrossRef CAS.
- J. A. Hadfield, K. Gaukroger, N. Hirst, A. P. Weston, N. J. Lawrence and A. T. McGown, Eur. J. Med. Chem., 2005, 40, 529–541 CrossRef CAS.
- R. Shirai, K. Tokuda, Y. Koiso and S. Iwasaki, Bioorg. Med. Chem. Lett., 1994, 4, 699–704 CrossRef CAS.
- M. Medarde, A. C. Ramos, E. Caballero, R. P. L. de Clairac, J. L. Lopez, D. G. Gravalos and A. S. Feliciano, Bioorg. Med. Chem. Lett., 1999, 9, 2303–2308 CrossRef CAS.
- S. L. Gwaltney, H. M. Imade, K. J. Barr, Q. Li, L. Gehrke, R. B. Credo, R. B. Warner, J. Y. Lee, P. Kovar, J. Y. Wang, M. A. Nukkala, N. A. Zielinski, D. Frost, S. C. Ng and H. L. Sham, Bioorg. Med. Chem. Lett., 2001, 11, 871–874 CrossRef CAS.
- N. J. Lawrence, D. Rennison, A. T. McGown and J. A. Hadfield, Bioorg. Med. Chem. Lett., 2003, 13, 3759–3763 CrossRef CAS.
- N. J. Lawrence, E. S. M. Armitage, B. Greedy, D. Cook, S. Ducki and A. T. McGown, Tetrahedron Lett., 2006, 47, 1637–1640 CrossRef CAS.
- Q. Zhang, Y. Peng, X. I. Wang, S. M. Keenan, S. Arora and W. J. Welsh, J. Med. Chem., 2007, 50, 749–754 CrossRef CAS.
- D. J. Kerr, E. Hamel, M. K. Jung and B. L. Flynn, Bioorg. Med. Chem., 2007, 15, 3290–3298 CrossRef CAS.
- R. Romagnoli, P. G. Baraldi, M. D. Carrion, C. L. Cara, D. Preti, F. Fruttarolo, M. G. Pavani, M. A. Tabrizi, M. Tolomeo, S. Grimaudo, A. Di Cristina, J. Balzarini, J. A. Hadfield, A. Brancale and E. Hamel, J. Med. Chem., 2007, 50, 2273–2277 CrossRef CAS.
- J.-P. Liou, Z.-Y. Wu, C.-C. Kuo, C.-Y. Chang, P.-Y. Lu, C.-M. Chen, H.-P. Hsieh and J.-Y. Chang, J. Med. Chem., 2008, 51, 4351–4355 CrossRef CAS.
- X-ray data for 14 and 15. CCDC 744677 and 744673 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/cgi-bin/catreq.cgi.
- Structures were aligned using FieldTemplater™ from Cresset BioMolecular Discovery Limited: http://www.cresset-bmd.com/.
Footnote |
| † Electronic supplementary information (ESI) available: biological assays, experimental data for the synthesised compounds and 1H and 13C NMR spectra. CCDC reference numbers 744673 and 744677. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21096g |
| This journal is © The Royal Society of Chemistry 2012 |