Theoretical study on the possibility of using silicon carbide nanotubes as dehydrogenation catalysts for ammonia–borane
Fenglei
Cao
and
Huai
Sun
*
School of Chemistry and Chemical Engineering and Key Laboratory for Thin Film and Microfabrication of Ministry of Education, Shanghai Jiaotong University, Shanghai, 200240, P. R. China. E-mail: huaisun@sjtu.edu.cn
First published on 23rd July 2012
Abstract
The adsorption and decomposition of ammonia–borane (AB) on the surface of silicon carbide nanotubes (SiCNTs) was investigated using density functional theory. Five adsorption types and four reaction channels were identified. The most favorable reaction channel that generates a H2 molecule is slightly endothermic; furthermore, the energy barrier for the decomposition of the AB molecule is only 9.6 kcal mol−1. The side reactions that generate NH3 or BH3 are highly endothermic; therefore, the generation of side products can be depressed by decreasing the temperature. However, desorption of hydrogen atoms from the surface appears to be a more difficult step. The energy-barrier height for generation of a H2 molecule and its subsequent desorption from the surface is approximately 34.2 kcal mol−1. The migration of hydrogen atoms on the surface of SiCNTs involves lower energy than the desorption process, indicating that the desorption of H2 molecules from the surface may be more complicated.
I. Introduction
Hydrogen energy has been promoted as an efficient alternative to fossil fuels. However, the future application of hydrogen-based energy is limited by the lack of a convenient, cheap and safe storage system. The so-called “hydrogen-storage problem” has attracted much research attention recently.1–8 First synthesized in 1955,9 the physical and chemical properties of ammonia–borane (AB) have been extensively studied. Only in recent decades has AB been suggested as a potential material for hydrogen storage.8,10,11 The AB molecule contains more than 19% of H2 by weight (6.5 wt% and 13.1 wt% for the first and second equivalents of H2, respectively) and 150 g L−1 by volume, superseding the DOE's target.8 However, using AB as a hydrogen-storage material poses serious challenges, such as low release efficiency, high working temperature and unwanted by-products (NH3, BH3, etc.).12–14 In recent years, considerable experimental and theoretical developments have been made in designing new catalysts or additives to solve these problems.2,15–22 Stephan and Erker reported that “frustrated Lewis pairs” (FLPs) reagents can be used to take the protic (N-bound) and hydridic (B-bound) hydrogen atoms from the AB molecule.18 A theoretical study of using N/B-pair FLPs as dehydrogenation catalysts of AB has also been reported by Guo and Zou.19 According to their work, reagents with Lewis pairs can be used as effective catalysts to take the hydrogen atoms off AB molecules and then recombine them into H2 molecules.Here, we report a new approach to catalyze the dehydrogenation of the AB molecule using silicon carbide nanotubes (SiCNTs). SiCNTs, analogs of carbon nanotubes (CNTs), have been synthesized from the reaction of SiO and multiwalled CNTs.23 Due to the polar nature of SiCNTs, SiCNTs are expected to have higher reactivity than CNTs. Theoretical studies have shown that CO,24 HCN,24 NO25 and N2O25 can be chemisorbed on the surface of SiCNTs. Ding et al.26 investigated the cleavage of the N–H bond of ammonia and the O–H bond of H–OX (X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, CH3, or C2H5) on SiCNTs. In previous density functional theory (DFT) calculations, we found that O2 can be dissociated easily on the surface of SiCNTs.27 Due to the significant charge separation between the silicon and carbon atoms,27 the silicon atom (electron deficient) and carbon atom (electron rich) can be viewed as Lewis acid and base, respectively. Therefore, SiCNTs can be considered a nonmetal catalyst with Lewis acid–base pairs. In this work, we found that SiCNTs can indeed take hydrogen atoms from the AB molecule, with a low energy barrier, through interactions between the protic and hydridic hydrogen atoms of AB with the Lewis acid–base pair of the SiCNTs. However, the release of H2 molecules from hydrogenated SiCNTs is a difficult process due to the large energy barrier. Our calculated data also indicate that the generation of by-products that are toxic to the hydrogen-powered fuel cells could be reduced by using SiCNTs.
H, CH3, or C2H5) on SiCNTs. In previous density functional theory (DFT) calculations, we found that O2 can be dissociated easily on the surface of SiCNTs.27 Due to the significant charge separation between the silicon and carbon atoms,27 the silicon atom (electron deficient) and carbon atom (electron rich) can be viewed as Lewis acid and base, respectively. Therefore, SiCNTs can be considered a nonmetal catalyst with Lewis acid–base pairs. In this work, we found that SiCNTs can indeed take hydrogen atoms from the AB molecule, with a low energy barrier, through interactions between the protic and hydridic hydrogen atoms of AB with the Lewis acid–base pair of the SiCNTs. However, the release of H2 molecules from hydrogenated SiCNTs is a difficult process due to the large energy barrier. Our calculated data also indicate that the generation of by-products that are toxic to the hydrogen-powered fuel cells could be reduced by using SiCNTs.
II. Method and model
The all-electron ab initio DFT calculations were carried out using a spin-polarized generalized-gradient approximation with the Perdew–Burke–Ernzerhof (PBE) functional28 and the double numerical basis set including polarization function (DNP) as implemented in the DMol3 package.29 The (8, 0) zigzag single-walled SiCNT was chosen as the basic model for calculations. The periodic boundary condition was used, with a cuboid cell of dimensions 40 × 40 × 10.70 Å. The large separation along x and y directions ensured that the interactions with the image cells could be neglected, and the length along the tube (10.70 Å) equaled twice the periodicity of a zigzag single-walled SiCNT. A larger model with 40 × 40 × 16.05 Å was tested for convergence. In this model, the length along the tube is three times that of the united cell. The Brillouin zone was sampled by 1 × 1 × 2 special k-points using the Monkhorst–Pack scheme30 for structure optimization and energy calculations. Testing using finer grids up to (1 × 1 × 6) k-points indicates the energy data obtained at the (1 × 1 × 2) k-points were converged. The charge transfer between SiCNT and AB molecule was analyzed based on the Hirshfeld population method. The minimum energy pathways (MEP) were searched using the nudged elastic band (NEB) method.31 The geometries of the transition states were searched using the linear/quadratic synchronous transit (LST/QST) method32 and verified using harmonic vibrational frequencies.III. Results and discussion
Based on previous theoretical studies of catalyzed dehydrogenation of AB, the possible reaction pathways of AB dehydrogenation catalyzed by SiCNT are proposed in Scheme 1.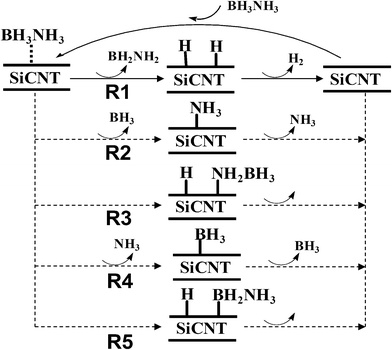 | ||
| Scheme 1 Possible initial reaction pathways of AB dehydrogenation catalyzed by SiCNTs. | ||
The reaction starts with the adsorption of the AB molecule on the surface of the SiCNT. Starting from the adsorbed complex, five reactions are possible: R1, R2, R3, R4 and R5, as shown in Scheme 1. In reaction R1, the Lewis-acid (C) and -base (Si) sites on the surface take the protic (H–N) and hydridic (H–C) hydrogen atoms from AB, yielding hydrogenated SiCNT and NH2BH2. The hydrogen atoms on the surface may interact to form a H2 molecule, which is subsequently desorbed from the surface, yielding a free H2 molecule; thus, the catalytic cycle is completed. In reactions R2 and R4, the B–N bond of AB is broken, forming BH3 and NH3, respectively. In reactions R3 and R5, either the protic or the hydridic hydrogen atom is transferred to the SiCNT. Except R1, the other reaction channels R2–R5 are considered as disadvantageous for the dehydrogenation reactions of AB. Reactions R2 and R4 yield the products BH3 or NH3, which can be toxic to the fuel cell; moreover, reactions R3 and R5 require much greater activation energy to take one hydrogen atom from the AB molecule. In this work, we focus on the factors that may influence the competition between the reaction channel R1 and other reaction channels.
3.1. Adsorption of AB molecule on surface of SiCNT
The optimized structure of the pristine (8, 0) zigzag SiCNT is shown in Fig. 1. The lengths of the C–Si bonds are about 1.80 Å, consistent with the data reported in the literature.27 Charge analysis based on the Hirshfeld method and Mulliken method indicates that the silicon atoms are negatively charged by 0.33 and 0.15 e respectively, and that the carbon atoms are positively charged with the same magnitude. The silicon atom with deficient electrons can be regarded as a Lewis acid, whereas the electron-rich carbon atom plays the role of a Lewis base. The neighboring silicon and carbon atoms form a Lewis acid–base pair. In the optimized free AB molecule, the B–N bond length is 1.66 Å, and the B–H and N–H bond lengths are 1.22 and 1.02 Å, respectively.19 The Hirchfeld (Mulliken) charge distributions on the protic (N–bound) and hydridic (B–bound) hydrogen atoms are 0.16 (0.21) and −0.11 (−1.93) e, respectively.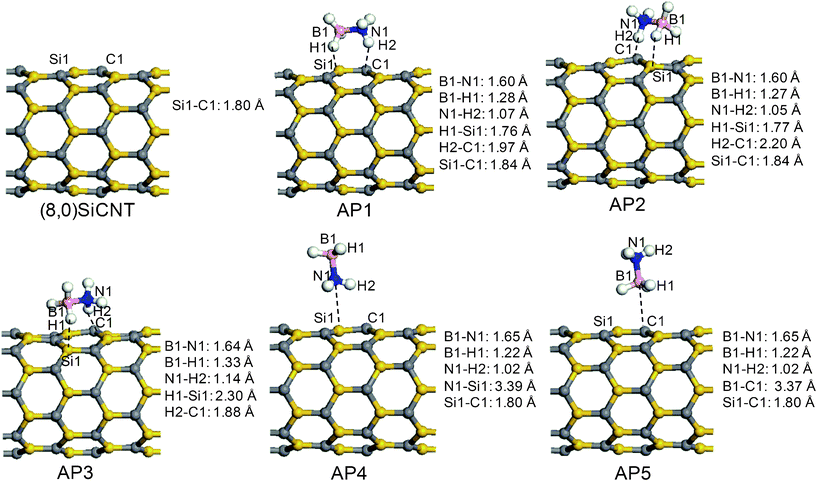 | ||
| Fig. 1 The optimized (8, 0) zigzag SiCNT and various complexes with an adsorbed AB molecule from the PBE/DNP computations. | ||
The AB molecule on the (8, 0) SiCNT yields three chemisorption (AP1, AP2, and AP3 in Fig. 1) and two physisorption (AP4 and AP5 in Fig. 1) stable configurations. In the three chemisorption configurations, the B–N bond of the AB molecule is approximately parallel to the surface; however, it exists in different orientations. The distances of H1⋯Si1 and H2⋯C1 are 1.76 and 1.97 Å, respectively, in AP1, indicating a strong interaction between (1) the protic and hydridic hydrogen atoms and (2) the carbon and silicon atoms. The length of the B1–H1 bond in AP1 is 1.28 Å, longer than that in a free AB molecule by 0.06 Å. Furthermore, the N1–H2 bond is lengthened to 1.07 from 1.02 Å in the free AB molecule, with the shortening of the B–N bond in AP1 from 1.66 to 1.60 Å. Clearly, the AB molecule on the surface of the SiCNT has been activated. In the other two chemisorption-based configurations (AP2 and AP3), the protic and hydridic hydrogen atoms of the AB molecule are attached to the carbon and silicon atoms of the SiCNTs, respectively. The distance of H2⋯C1 in AP2 is 2.20 Å and that of H1⋯Si1 in AP3 is 2.30 Å, which are both longer than that in AP1. Similar to AP1, the AB molecules in AP2 and AP3 have been activated, as deduced by the elongation of the B1–H1 and N1–H2 bonds and the shortening of the B1–N1 bonds, compared with those in the free AB molecule. The adsorption processes to form AP1, AP2 and AP3 are exothermic (−18.4, −17.3 and −18.2 kcal mol−1, respectively). According to the above charge analysis, the interactions are dominated by electrostatic forces.
In the two physisorption configurations, the B–N bond of the AB molecule is approximately vertical to the surface with the NH3 group (AP4) or the BH3 group (AP5) close to the surface, respectively, and the equilibrium distance of AB–SiCNT is larger than 3.0 Å. The AP4 and AP5 configurations are less stable, with low adsorption energies of −4.4 and −4.1 kcal mol−1, respectively. There exists a well-known inaccuracy in van der Waals interactions due to current DFT methods, though this does not affect our whole investigation of chemical nature throughout the paper. The different reaction channels resulting from the different adsorption structures are discussed in the following sections.
3.2. Decomposition of AB molecule on the surface of SiCNT
The energy profiles for the reaction pathways R1, R2, R3 and R4 are shown in Fig. 2. However, we could not identify the reaction pathway of R5 in our calculation. The structures of the various critical points located on the potential surface are shown in Fig. 3–6, along with the values of the most relevant geometrical parameters.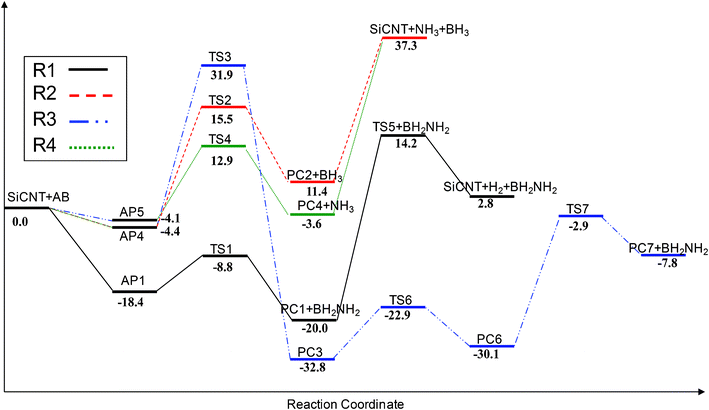 | ||
| Fig. 2 PBE/DNP calculated potential energy profile for the reaction channels 1, 2, 3 and 4. The relative energies are given in kcal mol−1. | ||
 | ||
| Fig. 3 Optimized configurations of the stationary points for reaction channel R1 from the PBE/DNP computations. | ||
 | ||
| Fig. 4 Optimized configurations of stationary points for reaction channel R2 from the PBE/DNP computations. | ||
 | ||
| Fig. 5 Optimized configurations of the stationary points for reaction channel R3 from the PBE/DNP computations. | ||
 | ||
| Fig. 6 Optimized configurations of the stationary points for reaction channel R4 from the PBE/DNP computations. | ||
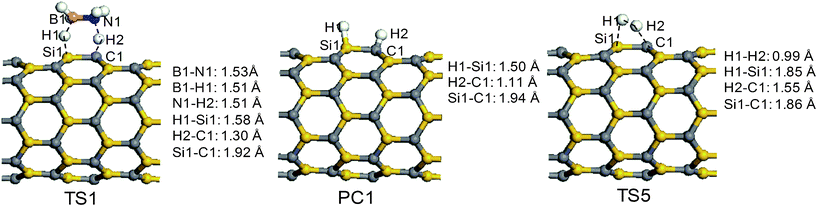
The reaction channel R1 starts from the adsorption structures AP1, AP2, or AP3. Because the three structures are similar, the reaction pathway R1 was studied using the configuration of AP1 only. The optimized structures of the stationary points of R1 are depicted in Fig. 3. Following the configuration of AP1, the transition state TS1 is formed. Compared with AP1, in TS1, the protic (H2) and hydridic (H1) hydrogen atoms are further away from the N1 and B1 atoms and closer to the surface. The B1⋯H1 and N1⋯H2 bond lengths are both extended to 1.51 Å, and the distances of H1⋯Si1 and H2⋯C2 are shortened to 1.58 and 1.30 Å, respectively. Obviously, the protic and hydridic hydrogen atoms are taken by SiCNT in a concerted way. Meanwhile, the B1–N1 bond in the AB molecule is reduced to 1.53 Å and the Si1–C1 bond in the sidewall of the SiCNT is stretched to 1.92 Å. A close examination of the optimized structure shows that the corresponding silicon and carbon atoms of the SiCNT are slightly pulled out of the surface, implying that the local hybridization status of these atoms are transforming from the sp2 to the sp3 mode. We have performed the computations for AP1 and TS1 using a larger model (40 × 40 × 16.05 Å, the length along the tube is three times that of the united cell). The results show that the local structures in the near vicinity around BH3NH3 binding site exhibit very small changes in comparison with that optimized using the small cell model. Especially, the binding energies are slightly changed only by 0.19 kcal mol−1 for AP1 and −1.2 kcal mol−1 for TS1.
To take the hydrogen atoms from the AB molecule, in the case of TS1, an energy-barrier height of approximately 9.6 kcal mol−1, relative to AP1, must be overcome. The reaction is exothermic, with reaction energy of −20.0 kcal mol−1 with reference to SiCNT + AB. Remarkably, the energy-barrier height (9.6 kcal mol−1) is smaller than those obtained by direct thermal decomposition (about 35.0 kcal mol−1)8 or by catalysis using other means (13.4 kcal mol−1).8,19 Because of the smaller barrier and the exothermic nature of the reaction, the reaction can be completed at room temperature. As the B–H and N–H bonds are cleaved, PC1 and BH2NH2 are obtained.
In the intermediate PC1, the H1–Si1 bond length is 1.50 Å, which is slightly longer than the Si–H bond length in silane (1.47 Å). The H2–C1 bond length is 1.11 Å, which is also slightly longer than the C–H bond length in alkanes (approximately 1.07 Å). The Hirshfeld charge distributions on the H1 (Si–bound) and H2 (C–bound) atoms are −0.18 and 0.22 e, indicating that the two hydrogen atoms can be classified as hydridic and protic, respectively. The Si–C bond length (1.94 Å) is significantly longer than that (1.80 Å) found in the pristine SiCNT. The hydrogenated silicon and carbon project out from the surface due to the apparently adopted tetrahedral configuration. To form a H2 molecule, the atoms must collide. In the transition state labeled as TS5 in Fig. 3, the hydrogen atoms are pulled away from the surface and the lengths of the H1⋯Si1 and H2⋯C1 bonds are 1.85 and 1.55 Å, respectively. Meanwhile, the two hydrogen atoms are close to each other, and the distance between them is only 0.99 Å. The transition state, TS5, has a large energy-barrier height, equal to 34.2 kcal mol−1. In our calculations, the hydrogen coverage is only 3.12%. As the number of hydrogen atoms populates the surface, the binding energies of hydrogen atoms on the surface (the C–H and Si–H bonds) would decrease signifcantly33,34, which would decrease the barrier of hydrogen generation. The desorption reaction is endothermic, requiring 22.8 kcal mol−1 relative to PC1.
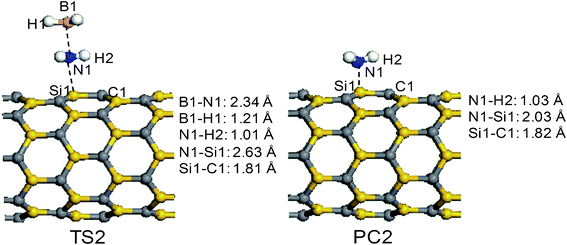
The reaction pathways R2 and R3 start from the adsorption complex AP4. The transition state (TS2) and the product (PC2) of the reaction pathway R2 are displayed in Fig. 4. As Fig. 4 shows, the elongation of the B–N bond of the AB molecule leads to the formation of TS2. In TS2, the length of N1⋯B1 is 2.34 Å, longer than that in a free AB molecule by 0.68 Å, implying that the B–N bond is too close to be dissociated. As the B–N bond elongates, the NH3 group shifts to the Si1 atom on the surface and the distance of N1⋯Si1 becomes 2.63 Å. Interestingly, in the transition state TS2, both NH3 and BH3 groups are planar. The energy-barrier height of TS2, relative to AP4, is calculated to be 19.9 kcal mol−1. The reaction pathway R2 results in the cleavage of the B–N bond and forms borane (BH3) and the product PC2, in which an NH3 molecule is adsorbed above the silicon atom on the surface. As shown in the configuration of PC2, the distance between the nitrogen atom of the ammonia molecule and the silicon atom on the surface is 2.03 Å, which agrees well with the results of previous theoretical investigations.26 The formation of PC2 is endothermic by 11.4 kcal mol−1. In Fig. 2, we can find that the desorption of the NH3 molecule from the surface of SiCNT is an endothermic process with 25.9 kcal mol−1.
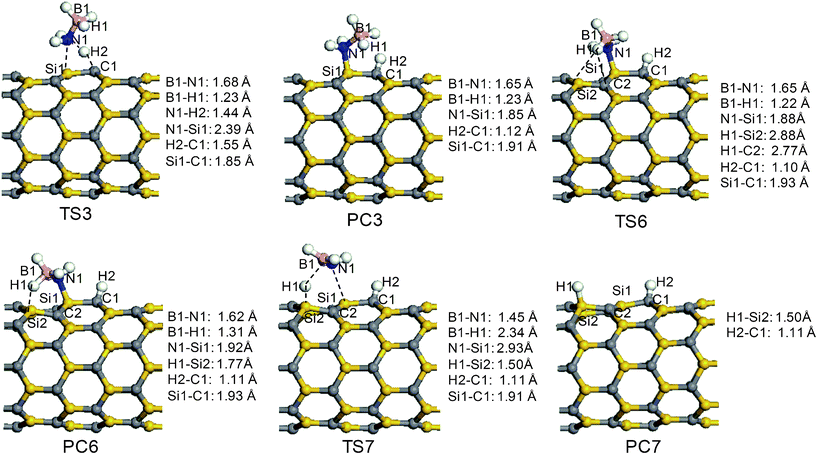
The reaction channel R3 involving TS3 breaks the N–H bond of the AB molecule and the corresponding structures of the stationary points are depicted in Fig. 5. Unlike the elongation of the B1–N1 bond in TS2, the N1–H1 bond is stretched from 1.02 Å to 1.44 Å in TS3, while the distances of H1⋯C1 and N1⋯Si1 are 1.55 Å and 2.39 Å, respectively. As the H2–N1 bond is broken and the H1–C1 and N1–Si1 bonds are formed, product PC3 is obtained. In PC3, the H1–C1 and N1–Si1 bond lengths are 1.12 and 1.85 Å. Although the reaction energy is −32.8 kcal mol−1, channel R3 needs to overcome a large energy-barrier height of 36.3 kcal mol−1, relative to AP4. In fact, product PC3 is an intermediate. The chemically bonded NH2–BH3 group may continue to interact with the surface to release another hydrogen atom and produce NH2BH2. Therefore, the reaction of B-bound H1 being taken by the surface of the SiCNT is considered in this study. Notably, an intermediate product (PC6) exists before the H1 atom is taken up. Product PC6 is formed from PC3 through a transition state TS6. The structure of TS6 is similar to PC3 in geometry, except for the orientation of the –BH3 group. In PC6, the distance of H1⋯Si2 is shortened to 1.77 Å and the B1–H1 and N1–Si bond lengths are extended to 1.31 and 1.92 Å, respectively. The reaction that proceeds from PC3 to PC6 is calculated to have a barrier of 9.9 kcal mol−1 and a binding energy of 2.7 kcal mol−1 relative to PC3. Following the configuration of PC6, the transition state TS7 is formed. In TS7, the H1 atom is further away from the B1 atom and closer to the Si2 atom of the surface, suggesting the formation of the H1–Si2 bond and the cleavage of the B1–H1 bond. Meanwhile, the length of N1⋯Si1 is elongated by 1.01 Å, implying the breaking of the N1–Si1 bond. Finally, a hydrogenation product (PC7) and BH2NH2 is obtained. The transition state TS7 has a large energy-barrier height of 27.2 kcal mol−1 and the reaction is endothermic by 22.3 kcal mol−1. Except that the H1 atom is located on a different site on the surface, PC7 is very similar to PC1. Hence, we speculate that the hydrogen atoms attached to the surface in PC7 may migrate to form a H2 molecule and are thus released from the surface. Therefore, the pathway R3 is another route to generate H2.
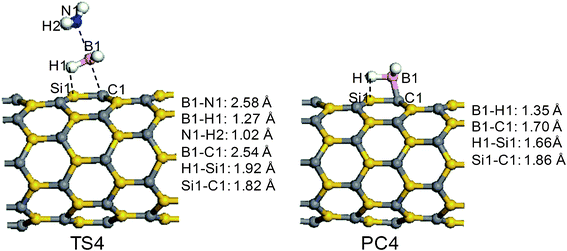
The reaction pathway R4 starts from the adsorption complex AP5. The transition state (TS4) and product (PC4) of the reaction pathway R4 are displayed in Fig. 6. The transition state TS4 is similar to TS2 in that the B1–N1 bond is significantly stretched. However, the distance of B1–N1 is 2.58 Å, 0.24 Å longer than that in TS2. The BH3 group is planar; however, the NH3 group shows a pyramid feature, close to that of a free ammonia molecule. Another important difference is that the boron of the AB molecule is near the carbon atom on the surface in TS4. The hydrogen atom (H1) moves toward the Si1 on the surface, with a distance of 1.98 Å, resulting in an increase in the H1–B1 bond from 1.21 to 1.27 Å. The transition state TS4 has a lower transition-state energy, due to the additional stabilization contributed by the interaction between the H1 and the Si1 atoms. An energy barrier of 17.0 kcal mol−1 has to be overcome before both the decomposition of the AB molecule and the formation of PC4 take place. In contrast to the adsorption of ammonia on the surface in PC2, in PC4, BH3 combines with the surface of the SiCNT, through the formation of a B1–C1 bond with a length of 1.70 Å. Interestingly, a hydrogen bridge links the B1 and Si1 atoms in PC4, and the lengths of the B1–H1 and the H1–Si1 bonds are 1.35 and 1.66 Å, respectively. Therefore, the BH3 on the surface of SiCNT yields a four-membered ring. Due to the unique structure of PC4, the channel R4 is an exothermic process, with an energy change of −3.6 kcal mol−1 relative to SiCNT + AB. Due to the stable structure of BH3 on the surface of the SiCNT, the desorption of BH3 from the surface of the SiCNT is endothermic, with an reaction energy gain of 40.9 kcal mol−1 (Fig. 2).
Based on these results, we can predict the outcomes of the decomposition on the surface of SiCNTs. First, the chemically adsorbed species AP1, AP2 and AP3 are significantly more stable than AP4 and AP5 by 12.0 kcal mol−1. The three possible methods of arranging the AB molecules on the surface with similar adsorption energies also indicates that the adsorption free energies would be much lower for the parallel configurations than for the perpendicular configurations. Therefore, under normal conditions, the AB molecules are predominately adsorbed on the surface in parallel configurations. Note that these configurations lead to the R1 reaction; consequently, the R1 reaction would be predominant. Second, TS1 is a transition state with a much lower transition-state energy than the other transition states. In addition, the reaction leading to the formation of PC1 is exothermic, with the release of a moderate amount of energy. Thus, the R1 channel can occur more easily than other pathways. The reaction R4, which produces NH3 in the first step, is probably the second on the list. Its energy-barrier height (17.0 kcal mol−1) is not very large; moreover, the reaction is exothermic, with the release of −3.6 kcal mol−1. Reaction R2, which produces BH3 in the first step is unlikely to take place under normal conditions, because the energy barrier is very high (19.9 kcal mol−1) and the reaction is endothermic. Although reaction R3, which is the other route to generate the H2 molecule, is the most exothermic, its energy-barrier height is substantially higher than that of the others. Therefore, we conclude that on the surface of SiCNTs, the reaction R1, which abstracts two hydrogen atoms from the AB molecule, is preferred in comparison to the other channels (R2, R3 and R4).
3.3. Migration of hydrogen atoms on the surface of SiCNTs
We calculate the migration of the hydrogen atom from silicon and carbon to adjacent sites in the following section. Based on the configuration PC1, the migration of the hydrogen atom on the sidewall of SiCNT has been studied. Some possible migration sites were considered, shown in the structure of PC1 in Fig. 7, where the two hydrogen atoms are labeled as H1 (Si–bound) and H2 (C–bound). The structures have been optimized with all atoms free to move. The calculated barriers (Ea) and reaction energies (Er), relative to PC1, are tabulated in Table 1.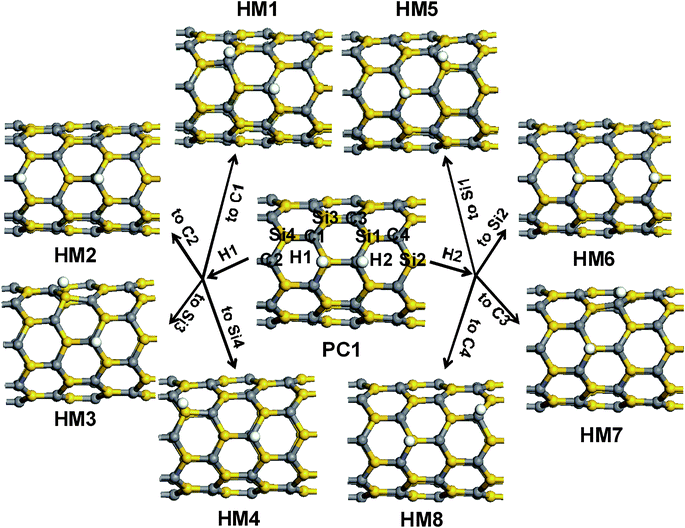 | ||
| Fig. 7 Schematic representations of the possible migration sites of hydrogen, based on the configuration of PC1, and the optimized structures after migration of hydrogen atoms onto the possible sites (HM1–HM8). | ||
| Site | Configuration | E a | E r | |
|---|---|---|---|---|
| H1 to | C1 | HM1 | 29.0 | 21.4 |
| C2 | HM2 | 44.4 | 24.7 | |
| Si3 | HM3 | 42.7 | 5.2 | |
| Si4 | HM4 | 40.1 | 4.7 | |
| H2 to | Si1 | HM5 | 30.8 | 22.4 |
| Si2 | HM6 | 45.8 | 25.8 | |
| C3 | HM7 | 43.3 | 5.1 | |
| C4 | HM8 | 43.0 | 4.6 |
Analyzing the migration process, our considered migration reaction can be categorized into two cases: the first case is that the hydrogen atom migrates along the C–Si bond of the surface of SiCNT, forming HM1 and HM5; and the other is that the hydrogen atom crosses the hexagonal ring of the SiCNT sidewall and moves to a far site, forming the other migration products in Fig. 7. The migration of hydrogen has to overcome energy barriers of approximately 30.0 kcal mol−1 for the first case and greater than 40.0 kcal mol−1 for the other case, indicating that the process involving migration of hydrogen along the C–Si bond is preferred. By comparing similar structures (HM1 and HM5, HM2 and HM6, HM3 and HM7, and HM4 and HM8), we found that the barriers of H1 migration are lower than those of H2 migration, due to the smaller bond energy of the H–Si bond. The results in Table 1 show that all the configurations involving the transfer of the hydrogen atom to the other sites are unfavorable compared to PC1, in terms of thermodynamics. Inspecting the structure of the hydrogenation products (shown in Fig. 7), we can categorize the products into two types, depending on how the hydrogen atoms are attached to the surface of the SiCNTs. In the first type, two hydrogen atoms bond to two carbon or silicon atoms (HM1, HM2, HM5 and HM6) and in the second type, one hydrogen atom bonds to a carbon atom and the other hydrogen atom bonds to a silicon atom (HM3, HM4, HM7 and HM8, as well as PC1). However, the configurations of the first type are unstable structures relative to PC1, with reaction energies greater than 20.0 kcal mol−1, whereas the formation of the second type of configurations is slightly endothermic, needing approximately 5.0 kcal mol−1. This result is similar to the behavior of boron nitride NTs.33,34 This can be explained with a simple molecular orbital description by supposing that the two hydrogen atoms, in turn, bond to the surface of the SiCNTs. In pristine SiCNT, the silicon atoms have partial positive charges and the carbon atoms have partial negative charges. The formation of a bond between the hydrogen atom and the cationic silicon atom (or anionic carbon atom) leads to the creation of a dangling bond on the anionic carbon (or cationic silicon). The formation of a bond between the hydrogen and carbon (or silicon) atoms saturates the dangling bond, which would eliminate the perturbation and reduce the energy of the system. Contrarily, the subsequent functionalization of hydrogen on the silicon (or carbon) atom does not eliminate the dangling bond, but further results in a new dangling bond, which would enhance the perturbation and result in the increase of the energy of the system. Therefore, the stability of the second type is higher than that of the first type. Thus, if many hydrogen atoms are bound on the surface of the SiCNT, the configurations that have the same number of Si–H and C–H bonds are preferred.
Based on the above results, we suggest that on the surface of the SiCNTs, the hydrogen atom migrates along the C–Si bond and that the migration will finally yield a configuration of the second type. For example, for H1 (Si–bound), the migration to C1 is preferred over other migration processes; however, the migration product HM1 is unstable compared with HM3 and HM4. Therefore, H1 will again move along the other C–Si to the Si3 or Si4 site, forming a relatively stable configuration (HM3 or HM4). There is a competition between the migration of hydrogen atoms and the release of H2 molecules. However, the migration of hydrogen on the surface increases the collision of any two hydrogen atoms, which could promote the release of H2 from the surface of the SiCNT. In our case, the hydrogen coverage is only 3.12%. As an increasing number of hydrogen atoms populates the surface, the collision between two hydrogen atoms would considerably increase due to their migration.
IV. Conclusion
In summary, the process of adsorption and decomposition of the AB molecule on the surface of SiCNTs has been investigated using the DFT with periodic boundary conditions. Five adsorption types and four reaction channels were identified. Two of the reaction channels (R1 and R3) release a H2 molecule and the other two generate NH3 (R2) and BH3 (R4). The R1 channel, which is the most direct route for generating H2, occurs more easily than other pathways due to the lower energy barrier and the favorable reaction energy (slightly endothermic). The energy-barrier height for the decomposition of the AB molecule is 9.6 kcal mol−1, significantly lower than that in direct decomposition (∼35.0 kcal mol−1)8 and lower than those obtained using other catalysts (13.4 kcal mol−1).8,19 The other route that generates H2 (namely, R3) is less favorable due to the high energy barrier and the complicated nature of the route. The routes R2 and R4, which generate the by-products NH3 and BH3, are highly endothermic. Therefore, the side reactions can be inhibited by decreasing the temperature.However, the desorption of hydrogen atoms from the surface appears to be a more difficult step. The energy-barrier height for the generation of a H2 molecule and the ensuing desorption from the surface is approximately 32.4 kcal mol−1. We also studied migration of hydrogen on the surface of SiCNTs. The migration along the C–Si bond is preferred and the hydrogen atoms are more likely to be attached to different types of atoms (silicon or carbon) instead of to the same type. The energy-barrier heights of the migration of hydrogen on the surface are lower than that of desorption. This implies that the mechanism of desorption may be more complicated than that studied here. The formation and migration of H2 on the surface is relevant to the surface coverage, which is an objective of future study. In addition, hydrogen is the lightest element and dynamic factors, involving multi-body interactions and free-energy changes of the reactions, may significantly change the behavior related to its desorption from the surface of SiCNTs.35
Acknowledgements
This work was partially funded by the National Science Foundation of China (No. 21073119 and No 21173146) and the National Basic Research Program of China (No. 2007CB209701).References
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS.
- M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048 CrossRef CAS.
- M. Dincă and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766 CrossRef.
- S.-i. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111 CrossRef CAS.
- V. V. Struzhkin, B. Militzer, W. L. Mao, H.-k. Mao and R. J. Hemley, Chem. Rev., 2007, 107, 4133 CrossRef CAS.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656 RSC.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079 CrossRef CAS.
- A. G. Anderson and D. O. Barlow, J. Am. Chem. Soc., 1955, 77, 6048 CrossRef CAS.
- C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279 RSC.
- N. C. Smythe and J. C. Gordon, Eur. J. Inorg. Chem., 2010, 509 CrossRef CAS.
- M. G. Hu, R. A. Geanangel and W. W. Wendlandt, Thermochim. Acta, 1978, 23, 249 CrossRef CAS.
- F. Baitalow, J. Baumann, G. Wolf, K. Jaenicke-Röβler and G. Leitner, Thermochim. Acta, 2002, 391, 159 CrossRef CAS.
- W. J. Shaw, J. C. Linehan, N. K. Szymczak, D. J. Heldebrant, C. Yonker, D. M. Camaioni, R. T. Baker and T. Autrey, Angew. Chem., Int. Ed., 2008, 47, 7493 CrossRef CAS.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS.
- N. Blaquiere, S. Diallo-Garcia, S. I. Gorelsky, D. A. Black and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 14034 CrossRef CAS.
- M. Käβ, A. Friedrich, M. Drees and S. Schneider, Angew. Chem., Int. Ed., 2009, 48, 905 CrossRef.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS.
- Y. Guo, X. He, Z. Li and Z. Zou, Inorg. Chem., 2010, 49, 3419 CrossRef CAS.
- A. Feaver, S. Sepehri, P. Shamberger, A. Stowe, T. Autrey and G. Cao, J. Phys. Chem. B, 2007, 111, 7469 CrossRef CAS.
- D. J. Heldebrant, A. Karkamkar, N. J. Hess, M. Bowden, S. Rassat, F. Zheng, K. Rappe and T. Autrey, Chem. Mater., 2008, 20, 5332 CrossRef CAS.
- Z. Li, G. Zhu, G. Lu, S. Qiu and X. Yao, J. Am. Chem. Soc., 2010, 132, 1490 CrossRef CAS.
- X.-H. Sun, C.-P. Li, W.-K. Wong, N.-B. Wong, C.-S. Lee, S.-T. Lee and B.-K. Teo, J. Am. Chem. Soc., 2002, 124, 14464 CrossRef CAS.
- R. Q. Wu, M. Yang, Y. H. Lu, Y. P. Feng, Z. G. Huang and Q. Y. Wu, J. Phys. Chem. C, 2008, 112, 15985 CAS.
- G. Gao and H. S. Kang, J. Chem. Theory Comput., 2008, 4, 1690 CrossRef CAS.
- J.-x. Zhao, B. Xiao and Y.-h. Ding, J. Phys. Chem. C, 2009, 113, 16736 CAS.
- F. Cao, X. Xu, W. Ren and C. Zhao, J. Phys. Chem. C, 2009, 114, 970 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1991, 94, 7245 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978 CrossRef CAS.
- N. Govind, M. Petersen, G. Fitzgerald, D. King-Smith and J. Andzelm, Comput. Mater. Sci., 2003, 28, 250 CrossRef CAS.
- X. Wu, J. Yang, J. G. Hou and Q. Zhu, J. Chem. Phys., 2004, 121, 8481 CrossRef CAS.
- S. S. Han, S. H. Lee, J. K. Kang and H. M. Lee, Phys. Rev. B, 2005, 72, 113402 CrossRef.
- T. BuCko, L. Benco, O. Dubay, C. Dellago and J. Hafner, J. Chem. Phys., 2009, 131, 214508 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |