Excellent catalytic performance of graphite oxide in the selective oxidation of glutaraldehyde by aqueous hydrogen peroxide
Xiaofeng
Chu
,
Quanjing
Zhu
,
Wei-Lin
Dai
* and
Kangnian
Fan
Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Department of Chemistry, Fudan University, Shanghai 200433, P. R. China.. E-mail: wldai@fudan.edu.cn; Fax: 86-21-55665701
First published on 13th June 2012
Abstract
Graphite oxide (GO) was synthesized from natural graphite powder using Hummers' method. A large number of oxygen-containing functional groups (C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) on the surface of graphite oxide were obtained, as identified by Fourier-transform infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS). The GO material shows outstanding catalytic performance and stability in the selective oxidation of glutaraldehyde to glutaric acid with aqueous H2O2. The catalytic activity has a slight increase when the GO is treated with 50% aqueous H2O2 solution and decreases evidently when the GO is treated with KBH4, which could be attributed to the increase in the amount of C–O and CO moieties after oxidation treatment or the decrease in the amount after reduction treatment. The oxygen-containing functional groups on the graphite oxide surface may play an important role in the catalytic process.
O) on the surface of graphite oxide were obtained, as identified by Fourier-transform infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS). The GO material shows outstanding catalytic performance and stability in the selective oxidation of glutaraldehyde to glutaric acid with aqueous H2O2. The catalytic activity has a slight increase when the GO is treated with 50% aqueous H2O2 solution and decreases evidently when the GO is treated with KBH4, which could be attributed to the increase in the amount of C–O and CO moieties after oxidation treatment or the decrease in the amount after reduction treatment. The oxygen-containing functional groups on the graphite oxide surface may play an important role in the catalytic process.
1. Introduction
Graphene, a rapidly rising star on the horizon of materials science and condensed-matter physics,1 is obtained from the reduction of GO. Along with the research on graphene, GO has also received more and more attention. Brodie firstly prepared GO by oxidation of Ceylon graphite with a potassium chlorate and fuming nitric acid mixture in 1859,2 which is time consuming and difficult to operate. In 1958, Hummers and Offeman found a more convenient strong oxidizing method that required less than 2 h for completion at a temperature below 45 °C.3 With the chemical oxidation process, a large number of oxygen-containing functional groups were formed on the surface of graphite. The dominant surface functional groups are epoxides and alcohols located on the basal planes, with carbonyl and carboxyl groups on the edges.4 The amount of functional groups is determined by the oxidation degree. As a novel sp2-hybridized carbon atom layer, modified with lots of oxygen-containing functional groups, GO has gained significant attention in recent years in energy storage,5–8 adsorption,9–11 and catalysis.12–19 Due to the low price of graphite and the convenient preparation method of GO, it has broad industrial perspective.Recently, many researchers have become interested in GO, and a great many of them are in the field of catalysis. In most cases, GO is served as a support to load metal nanoparticles or transition metal oxides.12–15 Although a high activity was obtained, the active catalytic center was the supported species, not the GO. Due to the special structure of GO, some researchers supposed that it can not only be used as a support, but also act as a catalyst directly. Gao et al. reported that reduced graphene oxide was used as a catalyst to reduce nitrobenzene at room temperature while high catalytic activity and stability were exhibited.16 Similarly, Dreyer et al. put forward that graphene oxide was served as a mild and efficient carbon catalyst to catalyze the oxidation of various alcohols and cis-stilbene and the hydration of various alkynes with excellent yields.17 They also found that the unique graphene oxides could activate the small oxygen molecule for catalysis. But until now, the catalytic mechanism of GO or graphene oxide also remains unclear yet. Further studies are still necessary to explore the real mechanism in the GO catalytic process.
Dicarboxylic acids, such as glutaric acid, adipic acid, trimethyladipic acid, and dodecanedioic acid, are essential feed-stocks for the manufacture of polyamides, polyesters, plasticizers, and lubricating oils.20 The current multi-step manufacturing process of glutaric acid is by oxidative cleavage of C–C bonds of mixtures containing the corresponding cyclic alcohols and ketones and their derivatives with nitric acid.21 The severe reaction conditions are necessary and the homogeneous system is impossible for sustainable industrial production with high economic value.
Herein, we report the catalytic behavior of GO in the manufacture of glutaric acid from the oxidation of glutaraldehyde with H2O2. A high yield was obtained under relatively mild conditions. The relationship between the catalytic activity and the GO surface structure was also discussed. Different treatment methods, including oxidation and reduction, were conducted on GO. It is obvious that the catalytic activity is dependent on the functional groups on the GO surface.
2. Materials and methods
2.1 Materials
Potassium permanganate (KMnO4, AP), sodium nitrate (NaNO3, AP), potassium borohydride (KBH4, AP), graphite (CP) and aqueous glutaraldehyde (25%) were all purchased from Sinopharm Chemical Reagent Co., Ltd. 98% concentrated H2SO4 and 50% aqueous H2O2 were of analytical grade.2.2 Catalyst preparation
The Ox-GO was prepared by treating GO with 50% aqueous solution of hydrogen peroxide. Firstly, 250 mg of GO was added into 50 ml of 50% hydrogen peroxide solution and the mixture was then kept at room temperature for 24 h under constant stirring. Finally, the Ox-GO sample was separated by centrifugation, washed with deionized water, and dried in an oven at 50 °C for 24 h. The active carbon was treated by refluxing in concentrated nitric acid for 2 h prior to the catalytic activity test.
2.3 Characterizations
The X-ray diffraction (XRD) patterns were obtained on a Bruker AXS D8 Advance X-ray diffractometer using nickel-filtered Cu-Kα (λ = 0.15418 nm) with an angle (2θ) range 5°∼65°, the tube voltage of 40 kV, the current of 40 mA, and the scanning speed of 4°/min. Transmission electron microscopy (TEM) was used to study the sample morphology. The analyses were performed on a JEOL JEM 2011 TEM operating at 200 kV. The XPS spectra were collected by a Perkin Elmer PHI 5000 C ESCA spectrometer equipped with a hemispherical electron energy analyzer at a pressure lower than 10−9 Torr. The Mg-Kα (hυ = 1253.6 eV) anode was operated at 14 kV and 20 mA. The carbonaceous C 1s line (284.6 eV) was used as the reference to calibrate the binding energies. The FT-IR spectra were recorded on a Nicolet Avatar-360 FT-IR spectrometer. The catalysts were finely ground, dispersed in KBr, and pelletized. The spectral resolution was 0.9 cm−1, and 32 scans were recorded for each spectrum.2.4 Catalytic reaction
The activity test was performed at 90 °C with magnetic stirring in a 25 ml round-bottom flask equipped with a reflux condenser. In a typical experiment, the sample of GO (ca. 0.050 g) and aqueous 50% H2O2 (20.17 mmol) were added into the flask. The mixture was stirred at room temperature for 5 min, and then 25% aqueous glutaraldehyde (10 mmol) was added to the resulting mixture. The mixture was heated at 90 °C for 10 h. The as-obtained product of glutaric acid was gathered and esterified with methanol for GC analysis. The quantitative analysis of the reaction products were performed on GC equipped with a flame ionization detector and an AT. SE-54 capillary column and the identification of different products in the reaction mixture was determined by means of GC-MS on HP 6890GC/5973 MS. The purity of the glutaric acid product was analyzed by melting point test, HPLC and GC (after esterification with methanol) and no other by-products were detected. Therefore, the selectivity is 100% and the yield is the same as the conversion.The recycling procedure is as follows. The reused catalyst was gathered by centrifugation, washed with large amount of deionized water, and dried in an oven at 50 °C for 48 h. The activity test of the resultant catalyst was conducted by the catalytic reaction procedure.
3. Results and discussion
3.1. Characterization of GO
The morphology of the graphite powder and GO sample were characterized by TEM. From the TEM images shown in Fig. 1, it is found that most of the graphite powder exhibits a micrometer-sized layered stack structure. However, the graphite structure was completely delaminated into single or very thin layers during the severe oxidation process. The typical wrinkle morphology of GO23,24 is clearly shown in Fig. 1b. | ||
| Fig. 1 TEM images of (a) graphite and (b) GO. | ||
Fig. 2 shows the X-ray diffraction patterns of GO, Re-GO and their parent graphite powder. A successful oxidation of graphite to GO by Hummers' method was verified by the XRD patterns. The diffraction pattern of graphite gives a narrow peak with very high intensity (2θ = 26.5°, corresponding to the interlayer distance d = 0.336 nm). After the oxidation, there is no reflection at 2θ = 26.5° in the diffractogram of GO and a new diffraction peak appeared at 2θ = 11.4°, indicating the complete oxidation of graphite. After chemical reduction with potassium borohydride in aqueous solution, a significant decrease was observed in the magnified XRD pattern, indicating that the GO structure was partly exfoliated. At the same time, the diffraction peak shown in the graphite powder pattern did not re-emerge. This finding implies that there is still some stacking of graphene layers remaining.
 | ||
| Fig. 2 XRD patterns of GO, Re-GO and their parent graphite powder (the inset is the magnified region from 5° to 18°). | ||
XPS spectra of C 1s for GO, Ox-GO and Re-GO are shown in Fig. 3. The C 1s XPS spectra region can be deconvoluted into three peaks corresponding to three different bonding environments of carbon including C–C (CC) at 284.6 ± 0.1 eV, C–O (hydroxyl and epoxy) at 286.5 ± 0.1 eV and CO (carboxyl or ketone) at 288.0 ± 0.3 eV.25 However, an unexpected peak at 282.5 eV appeared in Fig. 3c. The weak signal is believed to result from the carbide or boride which is probably produced by the reduction treatment.26 As shown in Table 1, in the GO sample, the atomic ratio of O/C is 52%. It is similar to the literature data, which exhibits an O/C ratio of 50% under strong oxidation conditions.27 The result suggests that the graphite was oxidized entirely in the present work.
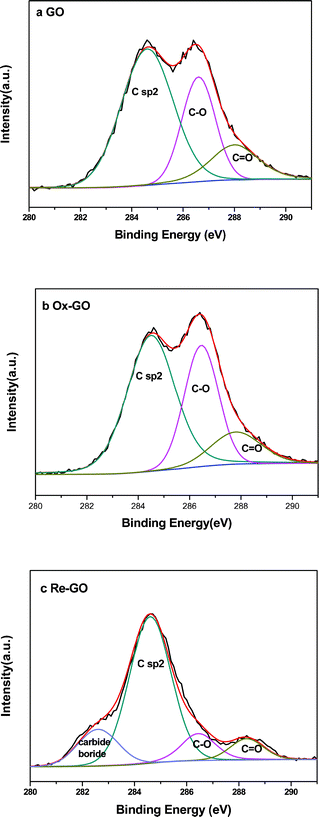 | ||
| Fig. 3 C1s XPS spectra of (a) GO, (b) Ox-GO and (c) Re-GO. | ||
| Sample | Atomic ratio of O/C (%) | C–C (CC) |
C–O (C–O–C,C–OH) | CO |
|---|---|---|---|---|
| a C1s was deconvoluted into three peaks; binding energies and relative area percentages with respect to C–C bonds were included in parentheses. | ||||
| GO | 52 | 284.6 (100) | 286.6 (47.8) | 288.0 (26.1) |
| Re-GO | 39 | 284.6 (100) | 286.5 (16.6) | 288.3 (12.1) |
| Ox-GO | 55 | 284.5 (100) | 286.5 (58.3) | 287.8 (23.4) |
Compared to GO, the content of oxygen significantly reduced from 52% to 39% after the reduction treatment with potassium borohydride. At the same time, we observed that there was also a decrease in the total relative area of the functional groups, including hydroxyl, epoxide, and carboxyl groups. This result confirmed that a large number of oxygen-containing functional groups, including epoxide and carboxyl groups, were reduced during the reducing process with KBH4. On the contrary, the total amount of C–O and CO gave a slight raise when the GO sample was treated with 50% aqueous hydrogen peroxide solution.
In order to further demonstrate the influence of the different treatment methods, FTIR was applied to record the change of functional groups. In Fig. 4, all the three absorption spectra show a broad feature at 3000 to 3800 cm−1 centered at about 3420 cm−1, which corresponds to O–H stretching of the C–OH. The GO sample also exhibits several characteristic features between 800 and 1800 cm−1, including CO(–COOH) stretching vibration at 1720 cm−1, O–H(C–OH) bending at 1618 cm−1, C–O vibration of the C–OH at 1260 cm−1, and epoxy vibration at 1050 and 975 cm−1.26 After reduction treatment, the Re-GO peak intensity of the CO stretching vibration and C–O epoxy vibration reduced obviously, indicating that a considerable part of the oxygenic functional groups of GO sample were efficiently reduced by KBH4. However, a significant amount of oxygenic groups are still remaining. Compared to Re-GO, the FTIR spectrum of Ox-GO shows an opposite mutative trend. The peak intensity of the CO stretching vibration has no significant change, but the epoxy vibration at 1050 and 975 cm−1 increases abruptly. This finding implies that this kind of oxygen-containing group may arise after treatment with hydrogen peroxide, that is, the amount of epoxy groups increases greatly. All these results from FTIR are in line with our conclusion from the XPS.
 | ||
| Fig. 4 IR spectra of GO, Re-GO and Ox-GO. | ||
3.2. Activity test
To investigate the catalytic performance of GO, we used the GO material to catalyze the oxidation of glutaraldehyde with H2O2. The product, glutaric acid, is an important organic chemical raw material to synthesize glutaric anhydride. The yield of glutaric acid is only 15.6% without any catalysts, but to our surprise, GO itself can achieve a very high yield of 94.7%, which was comparable to the best homogeneous tungstic acid catalyst (Table 2), an important and traditional selective oxidation catalyst in the H2O2 system. On the other hand, we use active carbon for comparison. It is found that active carbon showed a much lower yield of glutaric acid (75.6%) compared with GO (Table 2). In addition, active carbon showed certain activity in the decomposition of H2O2 to H2O and O2, which led to the low utilization efficiency of H2O2. These results indicated that GO is indeed a very good catalyst in the oxidation of gluaraldehyde to glutaric acid using hydrogen peroxide as the oxygen donor. It is known that although the traditional tungstic acid is a very active and selective catalyst in the H2O2 system, the recycling ability and the remaining heavy metal content has restricted its wide use in many fine chemicals. Since GO is a new carbon material, its application as an oxidation catalyst can greatly reduce the environmental pollution and the cost in industry.Although there are many studies on the catalytic application of GO in various reactions, little information of the GO catalyzed oxidation reaction with hydrogen peroxide has been reported. Song et al.28 reported that graphene oxide possessed intrinsic peroxidase-like activity that can catalyze the reaction of peroxidase substrate 3,3,5,5-tetramethyl-benzidine in the presence of H2O2. However, there is still no detailed study on the relationship between the GO surface functional groups and the activity.
In order to demonstrate the influence of the different treatment methods of GO on catalytic performance and to explore the oxidative mechanism of hydrogen peroxide with GO, we investigated the catalytic performance of the different treated GO samples. As can be seen from Table 3, when we use the Ox-GO to catalyze the reaction, the yield of glutaric acid increased from 94.7% to 96.3%. On the contrary, when we use the Re-GO as the calalyst, the yield decreased from 94.7% to 83.4%. It is self-evident that the oxidation treatment with hydrogen peroxide for 24 h is in favor of improving the catalytic activity, and an opposite result was given by the reduction treatment with KBH4. Thus, it is supposed that the oxygen-containing functional groups, including C–O and CO groups, on the surface of GO catalysts may be very important for the catalytic oxidation reaction with hydrogen peroxide. Indeed, as shown above in Figs. 3, 4 and Table 1, after treatment with hydrogen peroxide, the amount of CO groups in GO only has a little change, but the relative content of C–O groups shows an evident increase. On the contrary, after treatment with KBH4, most of the oxygen-containing functional groups were reduced, leading to a decrease in the amount of C–O and CO groups, resulting in a low yield of glutaric acid. The above results indicate that the amount of the oxygen-containing functional groups on the surface of the GO samples plays an important role in the catalytic process with H2O2 as the oxidant.
Furthermore, the catalysts have also been used in the oxidation of glutaraldehyde for the investigation of catalytic stability. And it is of significance to mention that GO shows excellent reusability in the reaction (Fig. 5). After four cycles, the catalyst shows nearly the same catalytic activities, suggesting its excellent stability. In addition, a hot filtration test showed that the reaction stopped after the filtration of the catalyst when the reaction was performed for 1 h (50% conversion), indicating that it is a real heterogeneous carbon catalyst. The XRD pattern of GO after four cycles is shown in Fig. 6. It is interesting to find that there is still some stacking of the graphene layers left in GO even after 4 cycles. The oxidations of different substrates to manufacture dicarboxylic acids are currently under way.
 | ||
| Fig. 5 The recycling test of GO in the oxidation of glutaraldehyde to glutaric acid. | ||
 | ||
| Fig. 6 XRD patterns of GO and GO after 4 cycles. | ||
4. Conclusion
GO was demonstrated as a new metal-free catalyst in the oxidation of glutaraldehyde, which shows a very high yield of 94.7%, comparable to the homogeneous tungstic acid catalyst. After treating with H2O2 and KBH4, the amount of the surface functional groups changed obviously. The oxygen-containing functional groups on the GO surface may play an important role in the oxidation reaction. As a new kind of oxidation catalyst, GO shows several advantages compared to traditional transition metal containing counterparts, such as easy to prepare on a large-scale, low-cost, high activity and selectivity, excellent stability and recycling ability that leads to its potential use in industrial plants.Acknowledgements
This work was financially supported by the Major State Basic Resource Development Program (Grant No. 2012CB224804), NNSFC (Project 20973042, 21173052), the Research Fund for the Doctoral Program of Higher Education (20090071110011) and the Natural Science Foundation of Shanghai Science and Technology Committee (08DZ2270500).References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 149, 249 CrossRef.
- W. Hummers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- J. Rourke, P. Pandey, J. Moore, M. Bates, I. Kinloch, R. Young and N. Wilson, Angew. Chem., Int. Ed., 2011, 50, 3173 CrossRef CAS.
- C-P. Tien and H. Teng, J. Power Sources, 2010, 195, 2414 CrossRef CAS.
- J. Xu, K. Wang, S-Z. Zu, B-H. Han and Z. Wei, ACS Nano, 2010, 4, 5019 CrossRef CAS.
- S. Chen, J. Zhu, X. Wu, Q. Han and X. Wang, ACS Nano, 2010, 4, 2822 CrossRef CAS.
- Y. Shao, J. Wang, M. Engelhard, C. Wang and Y. Lin, J. Mater. Chem., 2010, 20, 743 RSC.
- M. Seredych and T. Bandosz, Carbon, 2007, 45, 2130 CrossRef CAS.
- W. H. Slabaugh and B. C. Seiler, J. Phys. Chem., 1962, 66, 396 CrossRef CAS.
- M. Seredych and T. Bandosz, J. Phys. Chem. C, 2007, 111, 15596 CAS.
- Á. Mastalir, Z. Király, M. Benkő and I. Dékány, Catal. Lett., 2008, 124, 34 CrossRef.
- Y. Zu, J. Tang, W. Zhu, M. Zhang, G. Liu, Y. Liu, W. Zhang and M. Jia, Bioresour. Technol., 2011, 102, 8939 CrossRef CAS.
- G. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Muülhaupt, J. Am. Chem. Soc., 2009, 131, 8262 CrossRef CAS.
- J. Bian, M. Xiao, S. J. Wang, Y. X. Lu and Y. Z. Meng, Catal. Commun., 2009, 10, 1529 CrossRef CAS.
- Y. Gao, D. Ma, C. Wang, J. Guan and X. Bao, Chem. Commun., 2011, 47, 2432 RSC.
- D. Dreyer, H-P. Jia and C. Bielawski, Angew. Chem., 2010, 122, 6965 CrossRef.
- H-P. Jia, D. Dreyer and C. Bielawski, Tetrahedron, 2011, 67, 4431 CrossRef CAS.
- B. Machado and P. Serp, Catal. Sci. Technol., 2012, 2, 54 CAS.
- M. Besson, F. Gauthard, B. Horvath and P. Gallezot, J. Phys. Chem. B, 2005, 109, 2461 CrossRef CAS.
- H. Chen, W-L. Dai, X-L. Yang, R. Gao, Y. Cao, H. Li and K. Fan, Appl. Catal., A, 2006, 309, 62 CrossRef CAS.
- Z-L. Hu, M. Aizawa, Z-M Wang, N. Yoshizawa and H. Hatori, Langmuir, 2010, 26, 6681 CrossRef CAS.
- D. Dikin, S. Stankovich, E. Zimney, R. Piner, G. B. Dommett, G. Evmenenko, S. Nguyen and R. Ruoff, Nature, 2007, 448, 457 CrossRef CAS.
- M. Mcallister, J-L. Li, D. Adamson, H. Schniepp, A. Abdala, J. Liu, M. Herrera-Alonso, D. Milius, R. Car, R. Prud'homme and I. Aksay, Chem. Mater., 2007, 19, 4396 CrossRef CAS.
- Z. Lin, Y. Yao, Z. Li, Y. Liu, Z. Li and C-P. Wong, J. Phys. Chem. C, 2010, 114, 14819 CAS.
- J. Ou, J. Wang, S. Liu, B. Mu, J. Ren, H. Wang and S. Yang, Langmuir, 2010, 26, 15830 CrossRef CAS.
- D. Dreyer, R. Ruoff and C. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336 CrossRef CAS.
- Y. Song, K. Qu, C. Zhao, J. Ren and X. Qu, Adv. Mater., 2010, 22, 2206 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |