Amplified electrochemical microRNA biosensor using a hemin-G-quadruplex complex as the sensing element†
Yunlei
Zhou
b,
Mo
Wang
a,
Xiaomeng
Meng
a,
Huanshun
Yin
*a and
Shiyun
Ai
*a
aCollege of Chemistry and Material Science, Shandong Agricultural University, Taian, 271018, Shandong, China. Tel: +86 538 8247660; Fax: +86 538 8242251
bCollege of Life Science, Beijing Normal University, 100875, Beijing, China. E-mail: yinhs@sdau.edu.cn; ashy@sdau.edu.cn
First published on 13th June 2012
Abstract
A novel signal amplified electrochemical microRNA (miRNA) biosensor was developed here using hemin-G-quadruplex as signal unit. A hairpin structure DNA probe (43 bases) was immobilized on the gold nanoparticles (AuNPs) modified gold electrode surface with the segment at its 3′-end complementary to miRNA-21 (22 bases) and the segment at its 5′-end complementary to capture DNA (21 bases). Upon hybridization with the target miRNA-21, the hairpin structure was unfolded, which was further hybridized with capture DNA loaded on AuNPs. The AuNP contained two kinds of DNA; one was complementary to the hairpin structure DNA probe, while the other was aptamer for hemin. The electrochemical signal of hemin self-assembled in the center of G-quadruplex structure of the aptamer was measured using chronoamperometry. The miRNA-21 target was analyzed with a detection limit of 3.96 pM. Using isolated total RNA from human gastric carcinoma BGC-823 cells, human breast adenocarcinoma MCF-7 cells, human hepatocarcinoma HepG2 cells and human colon cancer HT-29 cells, the assay detected specifically miRNA-21 and over-expression of oncogene miRNA-21 was demonstrated.
1 Introduction
MicroRNAs (miRNAs) belong to a recently discovered class of non-protein-coding small RNA molecules, which regulate gene expression at the post-transcriptional level in plants, animals, and humans1–3 Gene regulation by miRNAs plays an important role in many critical biological processes such as development, differentiation, metabolism, and immunological response.4–6 It has also been reported that miRNAs can regulate multiple genes associated with human cancers, neurological diseases, and viral infections.7 Therefore, miRNAs have been regarded as a kind of useful diagnostic and prognostic markers, candidates for therapeutic intervention, and targets for basic biomedical research. Because mature miRNAs contain only about 17–25 nucleotides, their sequences are highly similar and the expression levels are low, it is crucial to develop a kind of accurate and sensitive method for miRNAs detection.Up to now, many analytical methods have been established to identify and quantify miRNAs, such as northern blotting,8in situ hybridization,9 bioluminescence technology,10 microarray technique,11 reverse transcription polymerase chain reaction (RT-PCR),12 surface enhanced Raman spectroscopy,13 surface plasmon resonance.14 Although these methods can detect miRNAs with good sensitivity, there is still great demand for innovative detection methods because of several reasons. Firstly, many techniques mentioned above require expensive and sophisticated instruments, specialized and expensive reagents, sophisticated readout system, skilled instrument operator, and some involved sophisticated and time-consuming processes, which increase the experimental cost and complexity. Secondly, the specificity of some of the above techniques needs improvement. Thirdly, most of the reported methods require a large amount of miRNAs and relatively pure miRNA samples, which could not directly detect miRNAs in complicated biological samples without a separation and enrichment process. Fourthly, some reported bioanalytical techniques could not be used to quantitatively detect miRNAs. Fifthly, an important aspect, some of the reported methods need miRNAs labeling and enrichment, which increase the operation complexity and decrease the detection accuracy due to the low expression level of miRNAs in plants, animals and humans. Therefore, there remains a need for a new miRNAs detection method that is simple, rapid, free-label and provides the measurement of miRNAs directly from a cellular extract in a sensitive manner.
Electrochemical biosensors have long been viewed as a particularly attractive alternative for bioanalysis because of their high sensitivity and selectivity, low detection limit, low cost, and ease of automatization.15 Recently, several reports have demonstrated the application of direct electrochemical miRNAs detection with sensitivity and selectivity.16–21 For instance, Gao and co-workers reported several examples of this approach in which they used inorganic nanoparticle catalysts as a label to detect miRNA in total RNA from cells.20–22 Pöhlmann and Sprinzl utilized four-component hybridization for sensitive and specific miRNA detection with 2 aM detection limit and diagnostic capabilities in total RNA extracts from human breast adenocarcinoma MCF-7 cells.19 Yang et al. developed another ultrasensitive electrochemical miRNA detection method using Fe–Ru redox pair as a reporter based on a novel nanostructured electrode platform.23 This method showed detection limit of 10 aM and can detect the upregulation of miR-21 and miR-205 in total RNA samples from three human head and neck cancer cell lines.
In this work, we reported a simple and sensitive electrochemistry biosensor for miRNA-21 detection without miRNA-21 labelling, enrichment and PCR amplification. The designed sensing strategy using hemin-G-quadruplex complex as the amplification element was shown in Scheme 1. We first prepared capture DNA S2 (21 bases) and aptamer DNA S3 (21 bases) functionalized gold nanopartilces by thiolated DNA via gold-sulfur affinity, in which capture DNA S2 was complementary to the 3′-end of hairpin DNA probe S1 (43 bases), while aptamer DNA S3 was noncomplementary to probe S1. However, aptamer DNA S3 and hemin can form hemin-G-quadruplex complex. In this tactic, the gold substrate electrode was modified with gold nanopartilces by electrochemical deposition. Then, the 5′-thiol-modified hairpin DNA probe S1 was assembled onto the AuNPs modified gold electrode surface via thiol-Au interaction. After the 22-base segment close to the 5′-end of hairpin DNA probe S1 hybridized with complementary target miRNA, the hairpin structure of the probe was unfolded. Subsequently, the 21-base segment close to the 3′-end of hairpin DNA probe S1 further hybridized with capture DNA S2, which was assembled on the surface of gold nanopartilces with aptamer DNA S3 simultaneously. After the electrode surface was treated with 20 mM Tris–HCl (pH 7.4) containing 40 mM KCl, 200 mM NaCl and 0.06% (v/v) Triton X-100, the aptamer DNA S3 can fold to form G-quadruplex structure. Finally, hemin solution was dripped on the electrode surface to form hemin-G-quadruplex complex. The electrochemical signal of hemin was used to monitor miRNA hybridization event.
 | ||
| Scheme 1 Schematic of label-free approach to electrochemical sensing of the target miRNA using hemin-G-quadruplex complex as the sensing element. | ||
2 Experimental
2.1 Materials and apparatus
PAGE-purified DNA and HPLC-purified miRNA oligonucleotides were obtained from Sangon Biotechnology Co., Ltd. (Shanghai, China) and TaKaRa Biotechnology Co., Ltd. (Dalian, China), respectively. The sequences are as follows: hairpin DNA probe S1, 5′-SH-GAA GTC GAC TCA ACA TCA GTC TGA TAA GCT AGA AGT CGA CTT C-3′; target miRNA-21, 5′-UAG CUU AUC AGA CUG AUG UUG A-3′; single-based mismatch miRNA, 5′-UAG CUU AUC![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) GA CUG AUG UUG A-3′; three-based mismatch miRNA, 5′-U
GA CUG AUG UUG A-3′; three-based mismatch miRNA, 5′-U![[U with combining low line]](https://www.rsc.org/images/entities/char_0055_0332.gif) G CUU AUC GA CUG AU
G CUU AUC GA CUG AU![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) UUG A-3′; non-complementary miRNA, 5′-GUA AGG CAU CUG ACC GAA GGC A-3′; capture DNA S2, 5′-GAA GTC GAC TTC TAG TAT GAT-SH-3′; aptamer DNA S3, 5′-SH-TGA CTG AAT TCT TTG GGT AGG GCG GGT TGG GT-3′). The mismatched bases in the miRNAs are underlined and the italic region identifies the G-quadruplex. All the DNA and miRNA solutions were prepared in TE buffer (pH 8.0) according to the manufacturer's recommendation. Ethidium bromide (EB) was purchased from Sigma (USA). TRIzol reagent was from Invitrogen (USA). Diethylpyrocarbonate (DEPC) was from Solarbio (China). Mercaptopropanoic acid (MPA), tri(2-carboxyethyl) phosphine hydrochloride (TCEP), tris(hydroxymethyl)aminomethane (Tris), disodium ethylenediaminetetraacetic acid (EDTA), sodium citrate and chloroauric acid were purchased from Aladdin (Shanghai, China). All reagents were analytically pure grade.
UUG A-3′; non-complementary miRNA, 5′-GUA AGG CAU CUG ACC GAA GGC A-3′; capture DNA S2, 5′-GAA GTC GAC TTC TAG TAT GAT-SH-3′; aptamer DNA S3, 5′-SH-TGA CTG AAT TCT TTG GGT AGG GCG GGT TGG GT-3′). The mismatched bases in the miRNAs are underlined and the italic region identifies the G-quadruplex. All the DNA and miRNA solutions were prepared in TE buffer (pH 8.0) according to the manufacturer's recommendation. Ethidium bromide (EB) was purchased from Sigma (USA). TRIzol reagent was from Invitrogen (USA). Diethylpyrocarbonate (DEPC) was from Solarbio (China). Mercaptopropanoic acid (MPA), tri(2-carboxyethyl) phosphine hydrochloride (TCEP), tris(hydroxymethyl)aminomethane (Tris), disodium ethylenediaminetetraacetic acid (EDTA), sodium citrate and chloroauric acid were purchased from Aladdin (Shanghai, China). All reagents were analytically pure grade.
The buffer solutions employed in this study are as follows. Oligonucleotide dissolve buffer (TE buffer, pH 8.0): 10 mM Tris–HCl and 1 mM EDTA, probe immobilization buffer: 10 mM Tris–HCl, 1.0 mM EDTA, 1.0 M NaCl, and 1.0 mM TCEP (pH 7.0), miRNA hybridization buffer: 1 × SSC, DNA hybridization buffer: 10 mM Tris–HCl, 1.0 mM EDTA, and 1.0 M NaCl (pH 7.0), electrochemistry determination buffer, 0.1 M PBS (NaH2PO4 and Na2HPO4, pH 7.0). All of the solution and redistilled deionized water used were treated with DEPC and autoclaved to protect from RNase degradation.
Electrochemical experiments were performed with CHI660C electrochemical workstation (CH instruments, Austin, USA) with a conventional three-electrode cell. A bare GCE or modified GCE was used as the working electrode. A saturated calomel electrode (SCE) and a platinum wire were used as the reference electrode and auxiliary electrode, respectively. The RNA integrity was assessed spectrophotometrically at 260 and 280 nm using NanoVue TM equipment (GE-Healthcare, Buckinghamshire, UK). All measurements were carried out at room temperature (25 ± 0.5 °C).
2.2 Preparation of AuNPs modified with two kinds of DNA
Gold nanoparticles were prepared by citrate reduction of HAuCl4 according to a previous report.24 A mixture of 5.0 × 10−9 mol of capture DNA S2 and 2.0 × 10−8 mol of aptamer DNA S3 was activated with acetate buffer (pH 5.2) and 1.5 μL of 10 mM TCEP for 1 h, then added to 1 mL of freshly prepared gold nanoparticles, and shaken gently for 24 h. After that, the DNA-AuNPs conjugates were aged in salts (0.1 M NaCl, 10 mM acetate buffer) for another 24 h. Excess reagents were removed by centrifuging at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min. The red precipitate was washed and centrifuged repeatedly three times. The resulting nanoparticles were dispersed in a buffer solution (pH 8.2) and stored at 4 °C. The obtained dual-DNA functionalized gold nanoparticles were noted as Aptamer-GNP.
000 rpm for 30 min. The red precipitate was washed and centrifuged repeatedly three times. The resulting nanoparticles were dispersed in a buffer solution (pH 8.2) and stored at 4 °C. The obtained dual-DNA functionalized gold nanoparticles were noted as Aptamer-GNP.
2.3 Gold nanoparticle deposition
Au electrode was carefully polished to a mirror-like surface with 0.5 and 0.05 μm alumina slurries, followed by ultrasonication in redistilled deionized water and methanol. Subsequently, the Au electrode was pretreated electrochemically in 0.5 M H2SO4 aqueous solution by potential cycling in a potential range of −0.3 to 1.5 V at a potential scan rate of 100 mV s−1 until the cyclic voltammogram characteristic of a clean Au electrode was obtained. Then, the Au electrode was washed thoroughly with redistilled deionized water and dried under nitrogen gas. The AuNPs electrodeposition on the pretreated planar gold electrode was conducted at −0.2 V in 3 mM HAuCl4 and 0.1 M KNO3 solution under stirring for 300 s. Finally, the electrode was washed with redistilled deionized water and dried at room temperature.2.4 Probe immobilization and hybridization
Gold electrodes coated with AuNPs were immersed into probe immobilization buffer containing 1.0 × 10−7 M hairpin DNA probe for 12 h in humidity conditions. Then the probe modified electrode was further immersed into a blank probe immobilization buffer for 1 h with stirring. After that, the probe DNA modified electrode was immersed into 10 mM Tris–HCl containing 1 mM MCH to further eliminate the non-adsorbed DNA molecules and hold a good orientation of probe DNA for its good recognition ability. The hybridization experiments were carried out by immersing the probe modified electrode into different concentrations of target miRNA (complementary or non-complementary) in a hybridization buffer at 37 °C for 2 h. After that, the hybridized electrode was rinsed three times with hybridization buffer to remove the un-hybridized target miRNA and dried with nitrogen.2.5 Preparation of hemin−G-quadruplex complexes
5 μL Aptamer-GNP was dripped on the hybridized electrode surface and incubated for 2 h to complete the hybridization between the 3′-end probe and capture DNA S2. Then, the electrode was thoroughly rinsed with 10 mM Tris–HCl (pH 7.4) three times. After drying under nitrogen, 5 μL 20 mM Tris–HCl (pH 7.4) containing 40 mM KCl, 200 mM NaCl and 0.06% (v/v) Triton X-100 was dripped on the electrode surface for 1 h at room temperature to allow an appropriate folding structure. Then, the electrode was rinsed with 20 mM Tris–HCl (pH 7.4) three times. Subsequently, hemin solution (40 μM) was dripped on the electrode surface and the electrode was allowed to stand for 2 h at room temperature. Under these conditions, hemin was assembled on the electrode surface based on the G-quardruplex structure of aptamer DNA S3.2.6 Electrochemical determination
Cyclic voltammetry (CV) and chronoamperometry (CA) were performed in 10 mL of 0.1 M PBS (pH 7.0) with a scan rate of 100 mV s−1 for CV and applied potential of −0.4 V for CA. Electrochemical impedance spectroscopy (EIS) was carried out in 5 mM Fe(CN)63−/4− (1:1) containing 0.1 M KCl with the frequency ranging from 10−1 to 105 Hz.
2.7 Total RNA extraction
Human gastric carcinoma BGC-823, human breast adenocarcinoma MCF-7, human hepatocarcinoma HepG2, human colon cancer HT-29 and normal human hepatic L02 cells were obtained from the College of Life Science, Beijing Normal University (China). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM, purchase from GIBICO, USA) containing 10% fetal calf serum (FCS) (Invitrogen, New Zealand) at 37 °C in the presence of 5% CO2. Total RNA from these cells was extracted using TRIzol reagent according to the manufacturer's recommended protocol. RNA concentration was determined by UV-vis spectrophotometry. RNA integrity was checked by agarose gel electrophoresis.3 Results and discussion
3.1 Characterization of the biosensor
Electrochemical impedance spectroscopy (EIS) could provide important information on the impedance changes of the electrode surface during the electrode modification process. Fig. 1 showed the Nyquist plot of impedance for the stepwise modification process with the planar Au electrode. The planar gold electrode witnessed an interfacial electron-transfer resistance value of about 661.5 Ω (curve a). After modifying with electrodeposited AuNPs (curve b), only an almost straight line was observed, which was the characterization of the diffusional limiting step of the electrochemical process, indicating an improvement of the electron transfer ability of Fe(CN)63−/4−. However, a small semi-circle at the high frequency region was observed and the Ret value was 84.5 Ω after hairpin probe S1 immobilization (curve c). The Ret value was further increased to about 175.4 Ω after being hybridized with the complementary target miRNA-21 (curve d). The Ret increased after probe immobilization and hybridization could be well ascribed to the electrostatic repulsion of the negatively charged Fe(CN)63−/4− from the approaching electrode surface by the negatively charged phosphate skeletons of DNA and miRNA. Subsequently, the value of Ret increased to 274.6 Ω when the un-hybridized segment of the probe was further hybridized with capture DNA S2, due to the large amount of DNA linked on the gold nanoparticles (curve e). After forming the hemin-G-quadruplex complex, the Ret value further increased to 390.9 Ω due to the non-conductivity of hemin (curve f), which indicated that the signal amplification element of hemin was successfully immobilized on the electrode surface.![Electrochemical impedance spectra of (a) the bare Au electrode, (b) after electrochemical deposition of AuNPs, (c) after immobilization of the hairpin probe S1, (d) hybridization with target miRNA, (e) hybridization with the capture DNA S2 loaded on the AuNPs and (f) after immobilization of hemin. The data were recorded in the presence of 5.0 mM [Fe(CN)6]3−/4− as redox label with a measuring potential of 0.20 V. The frequency ranged from 10−1 to 105 Hz. Reference electrode: SCE; electrode area: 0.07065 cm2.](/image/article/2012/RA/c2ra20487h/c2ra20487h-f1.gif) | ||
| Fig. 1 Electrochemical impedance spectra of (a) the bare Au electrode, (b) after electrochemical deposition of AuNPs, (c) after immobilization of the hairpin probe S1, (d) hybridization with target miRNA, (e) hybridization with the capture DNA S2 loaded on the AuNPs and (f) after immobilization of hemin. The data were recorded in the presence of 5.0 mM [Fe(CN)6]3−/4− as redox label with a measuring potential of 0.20 V. The frequency ranged from 10−1 to 105 Hz. Reference electrode: SCE; electrode area: 0.07065 cm2. | ||
3.2 Electrochemical behavior of the biosensor
The activation of the miRNA detection system is based on the formation of the hemin-G-quadruplex complex in the presence of target miRNA. To characterize the formation of the hemin-G-quadruplex complex, cyclic voltammetry was carried out. As seen in Fig. 2 (curve b), a reduction peak was observed at −0.374 V in the selected potential range. According to previous reports on the electrochemical behavior of hemin-G-quadruplex complex,25 this reduction peak can be attributed to hemin in the G-quadruplex structure. This peak also indicated the formation of hemin-G-quadruplex complex. A control experiment was also performed, which was treated with the same procedures except for hemin immobilization. There was no redox peak observed in the same potential range (curve a). These results indicated that the peak characterized the formation of hemin-G-quadruplex complex. | ||
| Fig. 2 Cyclic voltammograms of the fabricated biosensor in 0.1 M PBS (pH 7.0) before (a) and after (b) immobilization of hemin. MiRNA-21 hybridization concentration, 5 × 10−9 M. Scan rate: 100 mV s−1; reference electrode: SCE; electrode area: 0.07065 cm2. | ||
3.3 Sensitivity and selectivity for miRNA detection
Fig. 3 showed the amperometric response of hemin after the biosensor hybridized with different concentrations of target miRNA-21. The inset figure showed that the increase of the reduction current is proportional to the logarithm of the concentration of the target miRNA-21 in the range from 5 to 5000 pM. The linear regression equation can be expressed as I = −0.78logc − 0.62 (μA, pM, R = 0.9963). The limit of detection (LOD) was 3.96 pM according to LOD definition given by IUPAC (blank response + 3 × SD). | ||
| Fig. 3 (A) Amperometric responses for the biosensor with a probe hybridized with target miRNA-21 at different concentrations in 0.1 M PBS (pH 7.0). a–h: 0, 5, 10, 50, 100, 500, 1000 and 5000 pM. Insert: calibration curve. Error bars are the relative standard deviation of three independent experiments. Applied potential: −0.15 V (initative potential) and −0.35 V (low potential); electrode area: 0.07065 cm2. | ||
The selectivity of the present biosensor in discriminating perfect complementary miRNA (PCM) from single-base mismatched (SBM), three-base mismatched (TBM) and non-complementary (NC) miRNA sequences was investigated under three concentrations as shown in Fig. 4. Referring to the complementary hybridization at each concentration as 100%, the ratios of hybridization efficiencies were as follows: PCM:SBM:TBM:NC = 100:43.87:25.38:16.13, 100:25.07:14.32:8.26, and 100:26.54:13.18:7.70 for 10, 100 and 1000 pM targets, respectively. It was clear that complete hybridization was not accomplished due to the base mismatch. These results demonstrated that the fabricated biosensor was able to discriminate complementary miRNA-21.
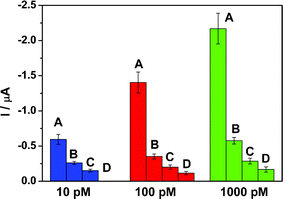 | ||
| Fig. 4 Comparison of the amperometric signal for the biosensor hybridized with a series of target miRNA (10, 100 and 1000 pM). (A) The complementary miRNA, (B) single-base mismatched miRNA, (C) three-base mismatched miRNA and (D) non-complementary miRNA. Error bars are the relative standard deviation of three independent experiments. All data used are background subtracted. The other conditions are the same as in Fig. 3. | ||
Regeneration is one of the significant performances for the biosensor, which is important for the continuous detection of the target. After electrochemical detection, the electrode was immersed in the hybridization buffer at 90 °C with unceasing stirring for 20 min. Then, the electrode was rinsed by hybridization buffer, followed by gradual cooling to room temperature. The resulted electrode was treated with the same process and the same concentration of target miRNA-21. The experimental results demonstrated that the current response to 100 pM target miRNA-21 was about 11.78% to its original response. The signal attenuation might be attributed to the loss of immobilized thiolated hairpin DNA probes on the AuNPs modified gold electrode surface. Considering the instability and low abundance of miRNA-21, we think that the regeneration of the proposed biosensor is acceptable.
Reproducibility is also an extremely important feature of biosensors in the practical applications such as clinical diagnoses. To investigate it, five electrodes were fabricated independently in the same conditions and used to detect 100 pM miRNA-21. The relative standard deviation (RSD) was 12.62%, which was comparable with previous reports20,22,26–30 (Table S1, please see supplemental material).† This result indicated that the proposed biosensor has acceptable reproducibility for miRNA-21 determination.
3.4 Detection of miRNA in cells
It has been identified that the expression level of miRNA-21 upregulated in numerous solid tumor tissues, including stomach, prostate, head and neck, esophagus, glioblastoma, neuroblastoma, breast, lung, liver, colorectal, pancreatic and cervical cancer in comparison to their normal counterparts.31,32 Therefore, miRNA-21 has been recognized as an oncomir.33 We tested the effectiveness of our assay in analysis of real biological samples by measuring the levels of miRNA-21 in total RNA extracted from BGC-823, MCF-7, HepG2 and HT-29 cells. As control, the expression level of miRNA-21 in normal human hepatic L02 cells was also investigated. The integrity of the total RNA was confirmed by agarose gel electrophoresis. The three bands of 28S, 18S and 5S were obtained, indicating the integrity of total RNA. The ratio of A260 and A280 was 1.829, 1.792, 1.842, 1.736 and 1.781 for L02, BGC-823, MCF-7, HepG2 and HT-29 cells, respectively, implying the relatively high purity of total RNA. The express level of miRNA-21 in extracted total RNA was determined using the proposed method in this work. As seen in Fig. 5, the electrochemical response of the biosensor after hybridization with total RNA extracted from BGC-823, MCF-7, HepG2 and HT-29 cells was higher than that of L02 cells, indicating that the higher express level of miRNA-21 in BGC-823, MCF-7, HepG2 and HT-29 cells, which was in accordance with previous reports that the miRNA-21 was over-expressed in human gastric carcinoma,31 human breast adenocarcinoma,34 human hepatocarcinoma35 and human colon cancer cells.31 For testifying these results, we also detected miRNA-21 by RT-PCR according to a previous report.36 The results were similar to that obtained by the proposed method, indicating that this electrochemical method for miRNA detection had good accuracy. These results also demonstrated that this method can be applied to direct determination of miRNA-21 in a sample without separation and enrichment. | ||
| Fig. 5 Relative expression level of miRNA-21 in various cells obtained by electrochemical biosensor and RT-PCR. | ||
4 Conclusions
In summary, we have developed a sensitive and specific electrochemical method for direct detection of miRNA-21 based on a hairpin structure DNA probe and the sensing element of hemin-G-quadruplex complex without miRNA labeling, enrichment or PCR amplification. The developed method was successfully applied for direct, sensitive and selective determination of miRNA-21 in total RNA extracted from human gastric carcinoma BGC-823 cells, human breast adenocarcinoma MCF-7 cells, human hepatocarcinoma HepG2 cells and human colon cancer HT-29 cells. Because this method does not need miRNA labeling and enrichment, it could satisfy the need for a rapid and easy method for early cancer marker detection in clinical diagnostics.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21075078, 21105056) and the Natural Science Foundation of the Shandong province, China (No. ZR2010BM005, ZR2011BQ001).References
- P. P. Medina, M. Nolde and F. J. Slack, Nature, 2010, 467, 86–90 CrossRef CAS.
- M. Yang, Y. Li and R. W. Padgett, Cytokine Growth Factor Rev., 2005, 16, 387–393 CrossRef CAS.
- G. Riddihough, Science, 2005, 309, 1507 CrossRef CAS.
- Y. Zhao, J. Ransom, A. Li, V. Vedantham, M. von Drehle, A. Muth, T. Tsuchihashi, M. McManus, R. Schwartz and D. Srivastava, Cell, 2007, 129, 303–317 CrossRef CAS.
- C. Chen, L. Li, H. Lodish and D. Bartel, Science, 2004, 303, 83–86 CrossRef CAS.
- A. J. Giraldez, R. M. Cinalli, M. E. Glasner, A. J. Enright, J. M. Thomson, S. Baskerville, S. M. Hammond, D. P. Bartel and A. F. Schier, Science, 2005, 308, 833–838 CrossRef CAS.
- T.-Y. Ha, Immune Network, 2011, 11, 135–154 CrossRef.
- E. Varallyay, J. Burgyan and Z. Havelda, Nat. Protoc., 2008, 3, 190–196 CrossRef CAS.
- M. de Planell-Saguer, M. C. Rodicio and Z. Mourelatos, Nat. Protoc., 2010, 5, 1061–1073 CrossRef CAS.
- K. A. Cissell, Y. Rahimi, S. Shrestha, E. A. Hunt and S. K. Deo, Anal. Chem., 2008, 80, 2319–2325 CrossRef CAS.
- R.-Q. Liang, W. Li, Y. Li, C.-y. Tan, J.-X. Li, Y.-X. Jin and K.-C. Ruan, Nucleic Acids Res., 2005, 33, e17 CrossRef.
- J. Li, B. Yao, H. Huang, Z. Wang, C. Sun, Y. Fan, Q. Chang, S. Li, X. Wang and J. Xi, Anal. Chem., 2009, 81, 5446–5451 CrossRef CAS.
- J. D. Driskell, A. G. Seto, L. P. Jones, S. Jokela, R. A. Dluhy, Y. P. Zhao and R. A. Tripp, Biosens. Bioelectron., 2008, 24, 917–922 CrossRef CAS.
- S. Fang, H. J. Lee, A. W. Wark and R. M. Corn, J. Am. Chem. Soc., 2006, 128, 14044–14046 CrossRef CAS.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21, 1192–1199 CrossRef CAS.
- Z. Gao and Y. Yu, Sens. Actuators, B, 2007, 121, 552–559 CrossRef.
- Z. Gao and Y. Yu, Biosens. Bioelectron., 2007, 22, 933–940 CrossRef CAS.
- E. Lusi, M. Passamano, P. Guarascio, A. Scarpa and L. Schiavo, Anal. Chem., 2009, 81, 2819–2822 CrossRef CAS.
- C. Pöhlmann and M. Sprinzl, Anal. Chem., 2010, 82, 4434–4440 CrossRef.
- Y. Peng and Z. Gao, Anal. Chem., 2011, 82, 820–827 CrossRef.
- Z. Gao and Z. Yang, Anal. Chem., 2006, 78, 1470–1477 CrossRef CAS.
- Y. Peng, G. Yi and Z. Gao, Chem. Commun., 2010, 46, 9131–9133 RSC.
- H. Yang, A. Hui, G. Pampalakis, L. Soleymani, F.-F. Liu, E. H. Sargent and S. O. Kelley, Angew. Chem., Int. Ed., 2009, 48, 8461–8464 CrossRef CAS.
- J. Liu and Y. Lu, Nat. Protoc., 2006, 1, 246–252 CrossRef CAS.
- Q. Guo, Y. Bao, X. Yang, K. Wang, Q. Wang and Y. Tan, Talanta, 2010, 83, 500–504 CrossRef CAS.
- Z. Gao and Y. H. Yu, Biosens. Bioelectron., 2007, 22, 933–940 CrossRef CAS.
- G.-J. Zhang, J. H. Chua, R.-E. Chee, A. Agarwal and S. M. Wong, Biosens. Bioelectron., 2009, 24, 2504–2508 CrossRef CAS.
- Z. Gao and Y. Peng, Biosens. Bioelectron., 2011, 26, 3768–3773 CrossRef CAS.
- Y. Fan, X. Chen, A. D. Trigg, C. Tung, J. Kong and Z. Gao, J. Am. Chem. Soc., 2007, 129, 5437–5443 CrossRef CAS.
- Z. Gao and Y. H. Yu, Sens. Actuators, B, 2007, 121, 552–559 CrossRef.
- Z. Lu, M. Liu, V. Stribinskis, C. M. Klinge, K. S. Ramos, N. H. Colburn and Y. Li, Oncogene, 2008, 27, 4373–4379 CrossRef CAS.
- P. P. Medina and F. J. Slack, Cell Cycle, 2008, 7, 2485–2492 CrossRef CAS.
- C. Liu, J. Yu, S. Yu, R. M. Lavker, L. Cai, W. Liu, K. Yang, X. He and S. Chen, J. Hepatol., 2010, 53, 98–107 CrossRef CAS.
- L.-X. Yan, X.-F. Huang, Q. Shao, M.-Y. Huang, L. Deng, Q.-L. Wu, Y.-X. Zeng and J.-Y. Shao, RNA, 2008, 14, 2348–2360 CrossRef CAS.
- C. Liu, J. Yu, S. Yu, R. M. Lavker, L. Cai, W. Liu, K. Yang, X. He and S. Chen, J. Hepatol., 2010, 53, 98–107 CrossRef CAS.
- J. Du, S. Yang, D. An, F. Hu, W. Yuan, C. Zhai and T. Zhu, Cell Res., 2009, 19, 487–496 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20487h/ |
| This journal is © The Royal Society of Chemistry 2012 |