Synthesis of both enantiomers of ferrocene[1,2-c]1H-quinolin-2-one by diastereoselective deproto-zincation of sugar-derived ferrocene esters†
Aare
Sreeshailam
ab,
Gandrath
Dayaker
ab,
D. Venkata
Ramana
ab,
Floris
Chevallier
a,
Thierry
Roisnel
c,
Shinsuke
Komagawa
de,
Ryo
Takita
de,
Masanobu
Uchiyama
*de,
Palakodety Radha
Krishna
*b and
Florence
Mongin
*a
aChimie et Photonique Moléculaires, UMR 6226 Institut des Sciences Chimiques de Rennes, CNRS-Université de Rennes 1, Bâtiment 10A, Case 1003, Campus Scientifique de Beaulieu, Rennes, 35042, France. E-mail: florence.mongin@univ-rennes1.fr; Fax: +33-2-2323-6955.
bD-211, Discovery Laboratory, Organic and Biomolecular Chemistry Division, CSIR-Indian Institute of Chemical Technology, Hyderabad, 500607, India. E-mail: prkgenius@iict.res.in; Fax: +91-40-27160387.
cCentre de Diffractométrie X, UMR 6226 Institut des Sciences Chimiques de Rennes, CNRS-Université de Rennes 1, Bâtiment 10B, Campus Scientifique de Beaulieu, Rennes, 35042, France
dGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-0033, Japan. E-mail: uchiyama@mol.f.u-tokyo.ac.jp
eAdvanced Elements Chemistry Research Team, RIKEN-ASI, 2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan
First published on 13th June 2012
Abstract
The diastereoselective deproto-metallation of several sugar-derived ferrocene esters using lithium-zinc bases was studied. While bis[(R)-1-phenylethyl]amino as the ligand afforded the diacetone-D-glucose-based (SP)-2-iodoferrocene ester in 91% de after iodination, the RP was synthesized from α-D-glucofuranose using 2,2,6,6-tetramethylpiperidino as the ligand. Both (RP)- and (SP)-ferrocene[1,2-c]1H-quinolin-2-one were reached by subsequent cyclizing couplings, albeit their racemization was noted.
Metallocenes have been intensively studied due to their varied applications in fields such as catalysis, materials science,1 and bioorganometallic chemistry.2 In particular, planar-chiral ferrocenes have attracted the attention of chemists because of their use in homogeneous asymmetric catalysis.3
The presence of a heteroatom-containing substituent on ferrocenes usually directs lithiation onto the adjacent position. With the aim of obtaining enantio-enriched planar-chiral derivatives, reactions of ferrocenes bearing various chiral directing groups have been documented.3b,c We have recently contributed to these studies by identifying sugar-derived esters as suitable groups to induce ferrocene diastereoselective deproto-metallations. With TMPLi (TMP = 2,2,6,6-tetramethylpiperidino) not being efficient enough to achieve this goal, we turned to the synergic (concerning both efficiency and chemoselectivity) lithium-cadmium base (TMP)3CdLi followed by iodination.4 Herein, the use of lithium-zinc combinations as non-toxic basic alternatives to achieve chemo- and diastereoselective syntheses of 2-iodoferrocene esters is described. In order to access the targets such as ferrocene[1,2-c]1H-quinolinones 1 from the latter, coupling-cyclization one-pot sequences are next considered (Scheme 1).
 | ||
| Scheme 1 Retrosynthetic route for 1 | ||
We thus turned our attention to a variety of chiral ferrocene esters, prepared from ferrocenecarboxylic acid and the corresponding secondary alcohols 2a–g depicted in Scheme 2 under classic conditions.5 The metallation reactions were attempted in THF (tetrahydrofuran) at room temperature for 2 h before interception with iodine (Table 1).


| Entry | 3 | 4, yield (%), dea (%) | 5, yield (%), eeb (%) |
|---|---|---|---|
a Wherever possible, determined from the integration of the 1H NMR spectrum of the crude mixture.
b Determined by HPLC analysis on a chiral stationary phase (AS-H column, eluent: hexane–isopropanol 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1 mL min−1, λ = 252 nm).
c Absolute configuration assigned on the basis of reported data.8
d Estimated yield, due to the presence of starting material.
e Reduction performed on a fraction.
f Using ZnCl2·TMEDA (1.5 equiv) and TMPLi (4.5 equiv).
g Both diastereomers separated by column chromatography purification over silica gel. :1, 1 mL min−1, λ = 252 nm).
c Absolute configuration assigned on the basis of reported data.8
d Estimated yield, due to the presence of starting material.
e Reduction performed on a fraction.
f Using ZnCl2·TMEDA (1.5 equiv) and TMPLi (4.5 equiv).
g Both diastereomers separated by column chromatography purification over silica gel.
|
|||
| 1 | 3a | 4a, 86, 20 | 61, 22 (R)c |
| 2 | 3b | 4b, 50d, 17 | (S)c,e |
| 3 | 3c | 4c, 86, 54 | (S)c,e |
| 4 | 3d | 4d, 87, 56 | 96, 57 (S)c |
| 5 | 3e | 4e, 64d | 86, 32 (S)c |
| 6f | 3f | 4f, 70 | 91, 11 (S)c |
| 7g | 3g | R P- 4g, 35 and SP-4g, 12d | 81, 96 (R) and 88, 92 (S)c |
From the α-D-xylofuranose derivative 3a, using the lithium-zinc base prepared in situ from ZnCl2·TMEDA (1 equiv, TMEDA = N,N,N',N'-tetramethylethylenediamine) and TMPLi (3 equiv)6 afforded 4a in 86% yield but with a moderate 20% de. Subsequent reduction to 2-iodoferrocenemethanol (5) using DIBAL-H,7 analysis by HPLC using a chiral stationary phase, and comparison with the literature showed the predominant formation of 4a as the RP diastereomer (Table 1, entry 1). When the α-D-mannofuranose derivative 3b was treated similarly, the metallation proved incomplete, affording 4b in 50% yield and a low 17% de in favor of the SP diastereomer (Table 1, entry 2). Under the conditions used to functionalize 3a and 3b, the iodo derivatives 4c and 4d were synthesized from the inexpensive α-D-glucofuranose derivatives 3c and 3d in 86 and 87% yield and 54 and 56% de, respectively (major SP diastereomer, Table 1, entries 3 and 4). Involving the α-D-allofuranose derivative 3e in the reaction, a good conversion was observed but with a disappointing 32% de in favor of the SP diastereomer (Table 1, entry 5). In order to evaluate more metal-coordinating groups, the α-D-xylofuranose derivative 3f was tested using 1.5 equiv of base, giving 4f in 70% yield and a low de (Table 1, entry 6). With the α-D-glucofuranose derivative 3g, both diastereomers were separated and the major one, isolated in 35% yield, proved to be the RP (Table 1, entry 7).
A thorough study was then undertaken in order to evaluate the parameters responsible for the diastereoselectivity observed from the promising 3c (Table 2). In order to check the importance of the structural composition of the base, (TMP)2BuZnLi6b was tested; a similar yield and de were obtained (Table 2, entry 2). Using hexane containing TMEDA (5 equiv)9 instead of THF still led to 4c but in lower yield and de (Table 2, entry 3). The presence of TMEDA10 in THF, due to the zinc source used, did not change the diastereoselectivity, as shown by employing ZnCl2 instead of ZnCl2·TMEDA (Table 2, entries 4 and 5). The addition of TMPLi (1 or 2 equiv) to a mixture of (TMP)2Zn and 3c was attempted, and led to 89 and 87% yields, and 64 or 72% de, respectively (Table 2, entries 6 and 7), as if TMPLi (or aggregates containing LiCl)6c was superior to mixed ferrocenyl-TMP zincates when performing the diastereoselective reaction. Similarly, the addition of TMPLi (3 equiv) to a mixture of ZnCl2·TMEDA and 3c led to an improved de (Table 2, entry 8). The sequential addition of (TMP)2Zn and TMPLi (2 equiv) was also attempted using 1,2-dimethoxyethane, 1,4-dioxane, and dimethoxymethane as solvents, but the reaction only proceeded with the latter, affording 4c in 32% yield and 51% de (Table 2, entry 9). Mixing the reagents in different orders at −30 or −50 °C before warming to rt did not change the diastereoselectivity significantly (Table 2, entries 10–13).

| Entry | R | R'(n) | Solvent, conditions | Yield (%), dea (%) |
|---|---|---|---|---|
|
a Determined from the integration of the 1H NMR spectrum of the crude mixture.
b Determined after reduction using DIBAL-H by HPLC analysis on a chiral stationary phase (AS-H column, eluent: hexane–isopropanol 9:1, 1 mL min−1, λ = 252 nm).
c In hexane containing TMEDA (5 equiv).
d Base prepared from ZnCl2 instead of ZnCl2·TMEDA.
e TMEDA slowly added at −30 °C.
f Sequential addition of RLi and, 10 min later, R'Li.
g Substrate mixed with ZnCl2·TMEDA before reaction (no RLi used).
h Using dimethoxymethane as solvent.
i Containing TMEDA (1 equiv).
j Base transferred to the substrate.
k Using (R)-PEAH.
l Using (S)-PEAH.
|
||||
| 1 | TMP | TMP(1) | THF, rt, 2 h | 86, 54 (Sb) |
| 2 | TMP | Bu(1) | THF, rt, 2 h | 89, 55 (Sb) |
| 3 | TMP | TMP(1) | c, rt, 2 h | 50, 42 (Sb) |
| 4d | TMP | TMP(1) | THF, rt, 2 h | 84, 56 (Sb) |
| 5de | TMP | TMP(1) | THF, −30 °C to rt, 2 h | 46, 53 (Sb) |
| 6f | TMP | TMP(1) | THF, rt, 2 h | 89, 64 (Sb) |
| 7f | TMP | TMP(2) | THF, rt, 2 h | 87, 72 (Sb) |
| 8 | g | TMP(3) | THF, −30 °C to rt, 2 h | 70, 68 (Sb) |
| 9df | TMP | TMP(2) | h, rt, 2 h | 32, 51 (Sb) |
| 10f | TMP | TMP(2) | THF, −30 °C to rt, 2 h | 51, 52 (Sb) |
| 11 | TMP | TMP(2) | THF, −30 °C to rt, 2 h | 64, 56 (Sb) |
| 12d | TMP | TMP (2) | THF,i −30 °C to rt, 2 h | 30, 56 (Sb) |
| 13j | TMP | TMP(2) | THF, −50 °C to rt, 2 h | 71, 56 (Sb) |
| 14 | PEAk | PEAk(1) | THF, rt, 2 h | 67, 79 (Sb) |
| 15 | PEAl | PEAl(1) | THF, rt, 2 h | 24, 10 (Sb) |
| 16f | PEAk | PEAk(2) | THF, rt, 2 h | 85, 91 (Sb) |
Double asymmetric induction11 using commercial (R)- and (S)-bis[1-phenylethyl]amine (PEAH) as the ligand source instead of TMPH was next attempted. Thus, when the substrate was reacted with a base obtained from ZnCl2·TMEDA (1 equiv) and (R)- or (S)-PEALi (3 equiv), 4c was obtained in 67 and 24% yield, and 79 and 10% de, respectively (Table 2, entries 14 and 15). With the sequential addition of the zinc diamide (1 equiv) and lithium amide (2 equiv) to the substrate being more efficient in the case of TMPH (Table 2, entry 7), we applied a similar protocol using (R)-PEAH; under these conditions, both a good yield and de were obtained (Table 2, entry 16).
When applied to the ester 3g, the sequential addition of (TMP)2Zn (either prepared from ZnCl2·TMEDA or ZnCl2) and TMPLi (2 equiv) afforded RP-4g in 51 and 57% yield, respectively.
In order to access the quinolinones 1, the iodides SP-4c and RP-4g were reacted with 2-aminophenylboronic acid. Using catalytic Pd(dba)2 (dba = dibenzylidene acetone) and triphenylphosphine, dioxane as solvent, and CsF in order to avoid the use of basic reagents,12 led to the carboxamides RP-1 and SP-1 through the Suzuki cross-coupling and subsequent cyclization of the amino group with the ester function (Scheme 3).
 | ||
| Scheme 3 Cyclizing Suzuki couplings giving 1. | ||
The crystals were analysed by X-ray diffraction and a structure referring to the non-chiral space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) was obtained (Fig. 1). This result is not sufficient to claim racemization,13 but led us to check this possibility using HPLC analysis on the chiral phases. The interconversion (RP-1vs.SP-1) was noted in solution;14 without solvent, it could be stopped at temperatures below −20 °C.
was obtained (Fig. 1). This result is not sufficient to claim racemization,13 but led us to check this possibility using HPLC analysis on the chiral phases. The interconversion (RP-1vs.SP-1) was noted in solution;14 without solvent, it could be stopped at temperatures below −20 °C.
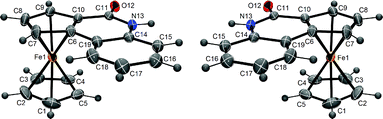 | ||
| Fig. 1 ORTEP diagrams (30% probability) of RP-1 and SP-1. | ||
Lithiation experiments on aryl carboxamides showed that the orientation of the functional group has an impact on the efficiency of the ortho-metallations, which increases with the coplanarity of the oxygen and activated hydrogen within the ring.15 A rationalization of the diastereoselectivity observed using 3c was thus attempted by identifying more stable conformers. Geometrical (local stabilization) optimization was performed by changing the dihedral angle between the upper plane of the ferrocenyl moiety and the ester carbonyl group (B3LYP/6-31G(d), structure of the ferrocenyl group fixed). The dihedral angle was fixed with 30° intervals from 0° to 330°. The two most stable structures, with dihedral angles of 0° (to give the major diastereomer) and 180° (to give the minor diastereomer), were identified and calculated in greater detail (M06/LanL2DZ(Fe)&6-31G(d)) at around 0° and 180°. The conformation with a dihedral angle of −6° proved to be 4.6 kcal mol−1 lower in energy that of 190° (Fig. 2, left).16 These calculated results are in accordance with the observed diastereoselectivity in the deproto-metallation of 3c using the lithium-zinc combination (Scheme 4). It is interesting to note that the structure obtained by the X-ray diffraction of the suitable crystals of 3c corresponds to the most stable conformer (Fig. 2, right).
 | ||
| Fig. 2 The calculated most stable conformer (M06/LanL2DZ(Fe)&6-31G(d)) and ORTEP diagram (30% probability) of 3c. | ||
 | ||
| Scheme 4 The observed diastereoselectivity in the deproto-metallation of 3c. | ||
Conclusions
Sugar-based ferrocene esters were deproto-metallated by using mixed lithium-zinc amido-based bases, with diastereoselectivities up to 91% under the “double asymmetric induction” protocol.Acknowledgements
The authors gratefully acknowledge the Institut Universitaire de France, Région Bretagne (financial support for A.S. and G.D.), University of Rennes 1 and CNRS (financial support for G.D. and D.V.R.). This research has also been performed as part of the Indo-French “Joint Laboratory for Sustainable Chemistry at Interfaces”. The calculations were performed by using the RIKEN Integrated Cluster of Clusters (RICC) facility.References
- N. J. Long, Angew. Chem., Int. Ed. Engl., 1995, 34, 21–38 CrossRef CAS.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5985 CrossRef CAS.
- (a) A. Togni and T. Hayashi, Ferrocene, VCH Verlagsgesellshaft, Weinheim, 1995 Search PubMed; (b) R. C. J. Atkinson, V. C. Gibson and N. J. Long, Chem. Soc. Rev., 2004, 33, 313–328 RSC; (c) W.-P. Deng, V. Snieckus and C. Metallinos, Chiral Ferrocenes in Asymmetric Catalysis: Synthesis and Applications, Wiley-VCH, Weinheim, 2010 Search PubMed.
- (a) A. Sreeshailam, G. Dayaker, F. Chevallier, T. Roisnel, P. R. Krishna and F. Mongin, Eur. J. Org. Chem., 2011, 3715–3718 CrossRef CAS; (b) G. Dayaker, A. Sreeshailam, F. Chevallier, T. Roisnel, P. R. Krishna and F. Mongin, Chem. Commun., 2010, 46, 2862–2864 RSC.
- A. Rauf and H. Parveen, Eur. J. Lipid Sci. Technol., 2004, 106, 97–100 CrossRef CAS.
- (a) J. M. L'Helgoual'ch, A. Seggio, F. Chevallier, M. Yonehara, E. Jeanneau, M. Uchiyama and F. Mongin, J. Org. Chem., 2008, 73, 177–183 CrossRef CAS; (b) K. Snégaroff, S. Komagawa, F. Chevallier, P. C. Gros, S. Golhen, T. Roisnel, M. Uchiyama and F. Mongin, Chem. Eur. J., 2010, 16, 8191–8201 Search PubMed; (c) P. García-Álvarez, R. E. Mulvey and J. A. Parkinson, Angew. Chem., Int. Ed., 2011, 50, 9668–9671 CrossRef.
- T. E. Pickett, F. X. Roca and C. J. Richards, J. Org. Chem., 2003, 68, 2592–2599 CrossRef CAS.
- A. Patti, D. Lambusta, M. Piattelli and G. Nicolosi, Tetrahedron: Asymmetry, 1998, 9, 3073–3080 CrossRef CAS.
- A. Seggio, M.-I. Lannou, F. Chevallier, D. Nobuto, M. Uchiyama, S. Golhen, T. Roisnel and F. Mongin, Chem.–Eur. J., 2007, 13, 9982–9989 CrossRef CAS.
- R. R. Fraser and T. S. Mansour, Tetrahedron Lett., 1986, 27, 331–334 CrossRef CAS.
- C. Metallinos and V. Snieckus, Org. Lett., 2002, 4, 1935–1938 CrossRef CAS.
- S. W. Wright, D. L. Hageman and L. D. McClure, J. Org. Chem., 1994, 59, 6095–6097 CrossRef CAS.
- J.-P. Djukic, A. Hijazi, H. D. Flack and G. Bernardinelli, Chem. Soc. Rev., 2008, 37, 406–425 RSC.
- Concerning previously reported rt racemization of ferrocenes, see for example: M. Tsukazaki, M. Tinkl, A. Roglans, B. J. Chapell, N. J. Taylor and V. Snieckus, J. Am. Chem. Soc., 1996, 118, 685–686 CrossRef CAS.
- (a) P. Beak and A. I. Meyers, Acc. Chem. Res., 1986, 19, 356–363 CrossRef CAS; (b) P. Beak, S. T. Kerrick and D. J. Gallagher, J. Am. Chem. Soc., 1993, 115, 10628–10636 CrossRef CAS; (c) M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206–2225 CrossRef CAS.
- The calculations using the SCRF method (PCM, solvent = THF) gave similar results. For details, see the ESI.†.
Footnote |
| † Electronic Supplementary Information (ESI) available: procedures, X-ray diffraction analysis and CIF files of 1 (CCDC 872139) and 3c (CCDC 798863), NMR spectra of 1, geometrical optimization of 3c and computational details. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21045b/ |
| This journal is © The Royal Society of Chemistry 2012 |