Fluorescent labeling techniques in biomolecules: a flashback
Harekrushna
Sahoo
*
Department of Biophysics, Technische Universität Dresden, Biotechnologisches Zentrum (BIOTEC), Tatzberg 47-51, 01307, Dresden. E-mail: harekrushna.sahoo@biotec.tu-dresden.de; Fax: +49-351-46340342; Tel: +49-351-46340326
First published on 20th April 2012
Abstract
Recent advancements in analytical methods relating organic chemistry, molecular biology, spectroscopy, engineering, and chemical biology have provided powerful tools to explore different selective fluorescent labeling techniques for analyzing molecular events in cells. New fluorescent labeling techniques have been developed and improvised depending on requirements like non-specificity, selectivity, small size, and of course, high yield. Along with synthesized small fluorophores that are used mostly for chemical labeling, fluorescent proteins such as green fluorescent protein (GFP) and its variants are used to label living cells genetically, both in vivo and in vitro. Fluorescent labeling has been extended over a large area of interdisciplinary research to visualize the functions and conditions of molecules using lower concentrations of the fluorescent material. Considering the wide applications of fluorescent techniques in various fields, the basic principles of different fluorescent labeling techniques relevant to chemistry and biology are discussed in this review.
 Harekrushna Sahoo | Harekrushna Sahoo received his masters degree in physical and nuclear chemistry from Utkal University (India) in 2000. He finished his PhD at Jacobs University Bremen (Germany) in 2007 on peptide conformation and dynamics using fluorescence-based techniques with Prof. Werner M Nau. He started working on protein folding using fluorescence as the primary technique with Prof. Lila M Gierasch at University of Massachusetts-Amherst (USA) in 2007. In 2010, he moved to Dresden (Germany) to work on extra-cellular matrices in the group of Prof. Petra Schwille. His focus is to study the chemical aspects of biological processes. |
Introduction
Tracking biomolecules (proteins, antibodies, amino acids, and peptides) often requires labeling with a reporter or sensor. At present, several labeling methods such as isotope markers,1 radioactive tracers,2 colorimetric biosensors,3 photoswitchable biomaterials,4 photochromic compounds,5,6 electrochemical sensors,7 and fluorescent labels8,9 are available for this purpose. Among the labeling methods, fluorescent labeling has the upper hand due to its non-destructive nature and the high sensitivity of the fluorescence technique, as well as meeting the requirements of small measurement volume and low concentration of the fluorescent material. Fluorescent labeling is generally accomplished by using a reactive derivative of the fluorophore that selectively binds to a functional group contained in the target biomolecule. In order to select a good fluorescent probe and labeling strategy, there are a few requirements that should be considered, i.e., (i) the fluorescent probes should be small in size and chemically stable, with minimal interference on the folding and biological functions of the target protein and (ii) the labeling reaction should be highly efficient and adaptable to the target molecule, preferably establishing a covalent linkage between the synthetic probe and a specific residue in the target molecule.An efficient method of specific fluorescent labeling at the point of interest in the target molecules is in high demand.10–12 Fluorescent labeling with synthetic organic molecules in the ultraviolet and visible regions have been established with several applications.13–17 Synthetic fluorescent probes offer a wide range of properties and functionalities, since they can be derivatized with different reactive chemical groups to meet the requirement of the labeling technique. Despite having a wide array of excitation and emission wavelengths with better photostability and a higher fluorescence quantum yield, synthetic organic fluorophore molecules are not used in cell imaging studies because of the difficulties in establishing a specific linkage between the target proteins and their toxic effects inside the cell. The presence of specific enzymes is also advantageous in labeling biomolecules with small synthetic fluorophores selectively. Despite having the selective labeling as an advantage, the low yield obtained in enzymatic labeling is a big concern. There are a few other labeling aspects to be considered, such as the synthesized small fluorophore molecules being conjugated to antibodies, which are then bound to the target proteins.18,19 The small fluorophore molecules that are conjugated to antibodies must be cell permeabilized and fixed to allow non-specific binding with the intracellular proteins, but high background labeling could arise as a result of the low specificity. In order to overcome the difficulties with specificity in live cells, labeling using a new class of proteins (i.e., fluorescent proteins), which are genetically tagged to the target proteins, has been introduced to the fluorophore-labeling world. The fusion of fluorescent proteins with target proteins has extracted a wealth of information on many biological networks over the last decade.20
Efficient and selective fluorescent labeling on biomolecules has been used to exploit their dynamics, kinetics and photophysical properties. These aspects are reviewed in several recent publications.18,21–25 Considering the in vitro labeling selectivity in a protein, cysteine is the most frequently used amino acid for chemical attachment of fluorophores due to its relatively low abundance and high nucleophilicity compared to other amino acid side chains. Specific and non-interfering dual fluorescent labeling in a peptide or protein molecule allows conformational investigations in terms of intramolecular distances.13 Site-specific fluorescent labeling, one of the most efficient labeling techniques, is developed as a tool to measure the intra and intermolecular protein dynamics.26–29 Some of the widely used fluorescent labeling methods are cited in Chart 1.
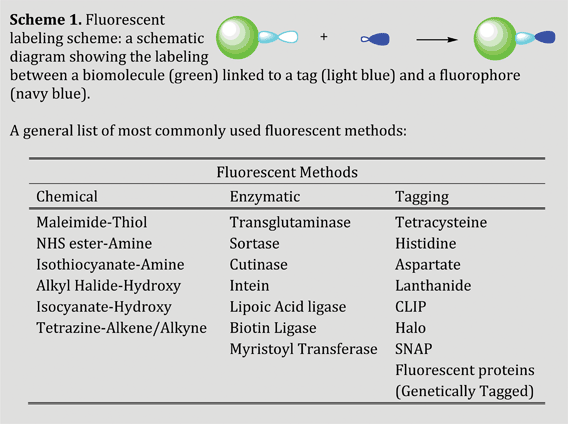 | ||
| Chart 1 General fluorescent labeling scheme and commonly used labeling techniques. | ||
With the advent of fluorescent labeling techniques, several cellular functions have been explored. For example, fluorescence resonance energy transfer (FRET) and fluorescence correlation spectroscopy (FCS) explore the activity of signal transduction by visualizing protein binding or folding.30 Protein dynamics and trafficking by fluorescence recovery after photobleaching (FRAP)19,31 and protein turnover have been explored by employing molecular tags that specifically bind to particular membrane-permeable dyes.32 Fluorescent labeling methods along with advanced fluorescence microscopy have enabled researchers to investigate the protein distribution, translocation and interactions in both in vivo and in vitro systems.33 In this way, the protein can be visualized in real time for the elucidation of its function in a complex biological network with specific and efficient fluorescent labeling. Also, protein–protein interactions, which are of fundamental importance for intra- and inter-cellular communications, are investigated with different fluorescence-based techniques.34
The main aim of this review article is to provide an insight into the state of the art techniques in the field of fluorescent labeling that are intensively used in labeling biomolecules. The fluorescent labeling methods that are systematically discussed here include chemical, enzymatic, peptide/protein tag (can be chemical or genetic), and genetic labeling techniques. The different methods are compared, including a discussion of their advantages and disadvantages.
Labeling overview
Mostly, the followed fluorescent labeling techniques generally adopted (as mentioned in Chart 1) allow specific labeling with functional groups attached to an amino acid with high selectivity and specificity. The fluorophores are designed with a reacting moiety, which may be bound covalently or non-covalently to the target biomolecules. Fluorescent molecule attachment to the biomolecules can be achieved chemically (chemical modification through non-covalent or covalent binding) or biologically (genetic incorporation of unnatural amino acids, fusion of biomolecules with a peptide tag having a fluorescent probe, or enzyme catalysis). Besides the above-mentioned chemical and biological labeling methods, a new type of labeling is widely employed (i.e., tag-labeling) which can be carried out both chemically and biologically.1. Chemical labeling
Despite the existence of several fluorescent-labeling techniques, chemical labeling is of great interest as it permits novel types of experiments by targeting chemicals with a wider range of functionality. From the chemical labeling point of view, covalent attachment of the chemical probes with reactive moieties and specific amino acids has the advantage of greater irreversibility compared to the non-covalent binding. Under certain conditions, the reactive groups non-specifically react with the amino acids at the C–H and N–H bonds. As a result of their nature and strategy, the chemical labeling methods are very robust, easy to handle and provide maximum efficiency with a wide range of fluorophores that can be coupled covalently to the target molecule. Chemical labeling methods are more suitable for in vitro studies rather than in vivo. The most commonly used chemical labeling methods are discussed below.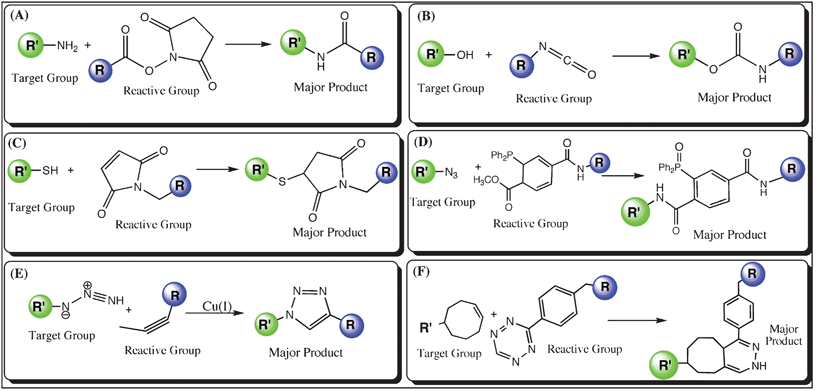 | ||
| Fig. 1 Schematic diagram of chemical labeling techniques (R′ in green represents the biomolecule to be labeled with a target moiety, while R is the fluorophore linked with a reactive group); (A) Amine labeling: an NHS (N-hydroxysuccinimide)-ester group couples to the –NH2 moiety, (B) Hydroxy labeling: a fluorophore is modified with an isocyanate group, which couples to the –OH moiety, (C) Thiol labeling: coupling between a maleimide modified fluorophore and the –SH moiety, (D) Azide labeling: Staudinger ligation of azide moiety with phosphine group, (E) Azide labeling: copper(I)-catalyzed cycloaddition of a fluorophore with the azide group linked to a biomolecule, and (F) Tetrazine labeling: A biomolecule labeled with tetrazine undergoes Diels–Alder cycloaddition with a dienophile linked to a fluorophore. | ||
The reactive groups, which are used to conjugate to the amine group, can also be coupled to hydroxy (–OH) groups in the target molecule under experimental conditions (isocyanate; see Fig. 1(B)) with a few exceptions.37 Basically, these reactions can be carried out in the presence of organic solvents (i.e. dimethyl sulfoxide, DMSO), which can absorb the liberated water molecule and avoid hydrolysis. Labeling reactions involving the hydroxyl group can be termed as hydroxy labeling, which is efficient at alkaline pH values (e.g., ∼ pH 8.5). The hydroxyl group can be temporarily activated for coupling with a secondary functional group (e.g. secondary amine) as well as allowing a direct and stable linkage upon reacting with the reactive groups.24–30 Like amine labeling, hydroxy labeling is also not very specific because of the existence of multiple hydroxyl groups (e.g. in the side chains of serine, threonine and tyrosine).
2. Enzymatic labeling
Enzyme catalyzed labeling techniques have several distinct advantages over their non-enzymatic counterparts. For example, enzymatic reactions are generally fast, efficient and more selective, allowing labeling both in vivo and in vitro with a high signal-to-noise ratio, including their multiple types of signal output, signal amplification and the wide selection of enzyme-labeled products, especially antibodies. Certain enzymes have properties that enable them to function as highly sensitive probes with versatility for the detection of proteins in tissues, whole cells or lysates. Besides having the ability to target selectively and permeability across the membrane, enzymatic labeling has a few disadvantages due to its large size or use of a large polypeptide tag. As a consequence, there is a chance of alternation in the function of the target molecule, as it needs to be fused with the target protein molecule. One more disadvantage is its low yield compared to chemical labeling.There are two different types of enzymatic labeling, In one case, the employed enzyme acts as a catalyst (e.g., Transglutaminase) and in the other case, the enzyme undergoes self-modification prior to the fluorescent labeling of the target molecule (e.g., Cutinase). The advantage of protein labeling with self-modifying enzymes is that the attachment of the chemical probes to the enzyme tag is directly coupled to enzyme catalysis, and thus the corresponding labeling reaction is of high efficiency and specificity. Following are some classic examples of the enzymes that are used for fluorescent labeling.
2.1.1. Transglutaminase. Transglutaminases (TGases) are popularly known as “Nature's biological glues” for their presence in various cell types and their wide substrate pool for protein cross-linking, including glutathione S-transferases, actin, myosin, β-tubulin, etc.52 TGase catalyzes the fluorescent labeling process in the cell upon forming an isopeptide bond between Gln and Lys residues.52 The mechanism of TGase catalysis involves an acyl group transfer from the γ-carboxamide group of a Gln (glutamine) residue of a protein to the ε-amino group of a Lys (lysine) residue linked to the fluorophore. The acyl transfer alternatively establishes a ε-(γ-glutamyl)lysine isopeptide linkage between the protein and the fluorophore (Fig. 2(A)). Microbial TGase (MTGase, ∼38 kDa) demonstrates high specificity for Gln-containing proteins as the acyl donors compared to the Guinea pig liver TGase (gpTGase, ∼80 kDa). It is found that cadaverine (with a primary amine at the terminus) can be readily used as the substrate for the modification of short peptide tags (TGase-tag: –Pro–Lys–Pro–Gln–Gln–Phe–Met–, 6-7 amino acids) with an embedded Gln residue in the target molecule both in vitro and in vivo.53,54 TGase has a wide substrate pool in the cell, which restricts the selective labeling of the peptide tag with Gln or Lys and therefore causes background labeling with other cellular proteins, which is a major drawback.
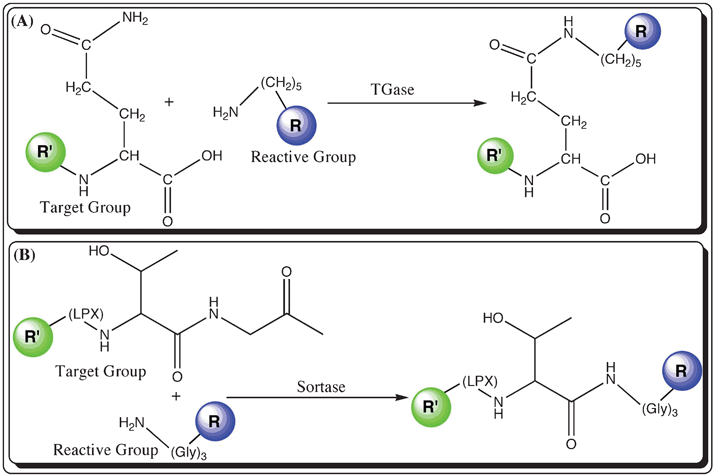 | ||
| Fig. 2 Schemes showing labeling techniques mediated by the catalysis of post-translational modified enzymes (R′ in green represents the biomolecule with the target group, whereas R in blue is a fluorophore modified with a reactive group); (A) Transglutaminase (TGase): R′ presented with a side chain amide group (e.g., Arginine) and R (blue) represents the fluorophore attached to cadaverine with the reactive primary amine group and (B) Sortase: fluorescent labeling of a R′ with the peptide tag, –L–P–X–T–G– (where ‘X’ is any amino acid) and a R is attached to a tri-glycine motif with a primary amine. | ||
2.1.2. Sortase. Sortase, a prokaryotic enzyme with transpeptidase activity, is used to label proteins with fluorophores linked to an amine group. The basic function of sortase is to anchor important cell surface proteins to peptidoglycans as a part of the cell wall of gram-positive bacteria.55,56 During the fluorescent labeling process, sortase acts as a catalyst rather than modifying itself. Staphylococcus aureus sortase (or SrtA, ∼28 kDa) catalyzes the hydrolysis of the peptide bond between Thr and Gly in a soratse recognition motif (sortase-tag: –Lys–Pro–X–Thr–Gly–, where ‘X’ is any amino acid) in the cell surface protein, and furthermore forms a new peptide bond between the carboxyl group of the exposed Thr and the amino terminal of the penta- or tri-glycine (Fig. 2(B)).55 Currently, sortase-mediated fluorescent labeling has been demonstrated with fluorophores conjugated to peptides with an oligo-Gly peptide. Sortase-mediated fluorescent labeling allows site-specific labeling of the biomolecule, but only at the C-terminus of the protein.57,58 Dual labeling can be carried out using sortase as a catalyst through protein splicing.59 The labeling of the amine group to the carboxy group of hydrolyzed threonine in the sortase tag has the advantage in terms of its specificity and efficiency, where as its ability to hydrolyze the peptide bond serves as a disadvantage.
2.2.1. Cutinase. Cutinase (∼22 kDa) is a serine esterase with the native activity of cutin (a polyester polymer composed of 16-hydroxy palmitic acid and 18-hydroxy stearic acid) hydrolysis. Cutinase is also used to hydrolyze fatty acid esters and triacyl glycerol.60 In fluorescent labeling process, an alkyl phosphonate linked to a fluorophore forms a covalent adduct with cutinase on conjugation with the Ser residue in the enzyme active site.61 Mechanistically, in the first step the target molecule is fused to cutinase and in the second step the fused cutinase with a serine at the active site is coupled to p-nitrophenyl phosphonate linked to a fluorophore (Fig. 3(A)).62 Cutinase-mediated labeling has been used to label proteins, which are immobilized on a self-assembled monolayer.63 Despite having the advantage of its negligible nonspecific in vivo labeling, the size of cutinase limits the technique’s applicability to small proteins. In addition, there is a chance of alteration in the target protein’s activity and conformation upon fusion with cutinase.
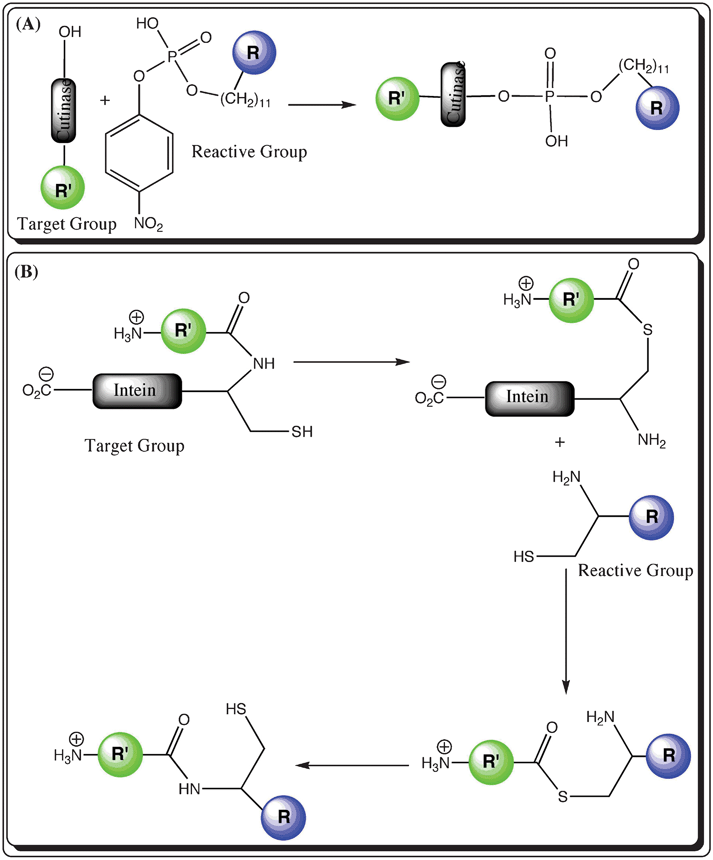 | ||
| Fig. 3 Schematic representation of self-modified enzyme-based labeling techniques (R′ in green represents the biomolecule with the target group, whereas R in blue is a fluorophore modified with a reactive group; (A) Cutinase: a fluorophore is modified with a phosphate group, which is transferred to the selective site with a serine amino acid in the cutinase tag fused with the target biomolecule and (B) Intein: the biomolecule is fused to an intein with a free cysteine group and a fluorophore is conjugated to a cysteine amino acid residue as well as a free amine group. The various steps display the exchange between the amine and thiol groups during the labeling reaction. | ||
2.2.2. Intein. Inteins are intercepting protein domains that are expressed in frame with flanking N- and C-terminal polypeptides (exteins) and are engineered to carry out selective labeling in the protein segments.64 Intein is considered as a self-modified enzyme for protein splicing as it is self-excised from the precursor protein fusion to afford ligated exteins.65 Intein-mediated labeling proceeds through multiple steps: (i) an intein is employed to create a protein possessing a C-terminal thioester that can be ligated to a protein or peptide with an amino-terminal cysteine via a native peptide bond66 and (ii) the conjugation of the target protein with a cysteine-modified fluorophore in a reaction known as native chemical ligation (Fig. 3(B)).67 In this strategy, the target protein is first expressed as a fusion with a C-terminal intein tag followed by the formation of a protein–intein conjugation linked with a thioester bond, which subsequently undergoes ester exchange with the cysteine-modified fluorophore to release intein and afford target protein conjugated to the fluorescent probe with a thioester linkage. The intermediate gives rise to a native peptide bond between the C-terminus of the target protein and the cysteine-conjugated fluorescent probe upon spontaneous rearrangement (as shown in Fig. 3(B)). The site specificity and the lack of non-specific labeling are the foremost advantages, while the inability to label the loops inside the protein is among the major disadvantages.
2.2.3. Haloalkane dehalogenase. Haloalkane dehalogenase (∼36 kDa) belongs to the family of hydrolases and catalyzes the hydrolysis of haloalkanes via a covalent enzyme–substrate intermediate that undergoes the hydrolytic process.68 The basic function of the haloalkane dehalogenase enzyme is to remove halides from aliphatic hydrocarbons through hydrolysis and thereafter afford aliphatic alcohols.69 The enzymatic catalysis starts with the nucleophilic displacement of the terminal chloride in the substrate by the Asp106 (Asp: aspartate) in the enzyme, giving rise to a covalent alkyl–enzyme intermediate. Therefore, chemical probes of diverse structures have been linked to alkyl chloride to serve as the substrates for the target protein fused with a Halo-tag (∼33 kDa). The Halo-tag has also been applied to protein labeling with quantum dots functionalized with aliphatic chlorides.70,71 The Halo-tag is widely used in the labeling of proteins inside mammalian cells due to the membrane permeability of the alkyl halide substrates.72 Despite having advantages like membrane permeability, the large size of the Halo-tag is regarded as an inconvenience during the protein expression of the target fusion protein and is thus expected to interfere with the native function of the target proteins.
2.3.1. Biotin protein ligase. Biotin protein ligase (BPL) plays an important role in attaching biotin to a specific Lys side chain covalently through an amide linkage in biotin carboxyl carrier protein (BCCP, ∼17 kDa).73 The attachment mechanism occurs through two steps. In the first step, the biotin prosthetic group in BCCP undergoes N-carboxylation giving rise to N-carboxybiotin. Then in the second step the carboxylated biotin moves to the active site of the carboxyl transferase for the delivery of the carboxyl group to acetyl-CoA to afford malonyl-CoA. Thus, the biotin conjugated to lysine in BCCP functions as a swinging arm for the transfer of an activated carboxyl group between the enzyme active sites. As biotin has a strong affinity towards streptavidin, fluorophores can be attached to a biotin-modified protein through biotin–streptavidin binding. Recently, to overcome the large size of BCCP, BCCPΔ100 (69 residues, ∼7 kDa) has been used to label the cell-surface proteins at the termini in both in vivo and in vitro systems.74
The biotin ligase technique is also used to ligate biotin to a 15-amino-acid acceptor peptide (AP, ∼2 kDa). The enzyme accepts a ketone isostere of biotin and ligates to AP fused with cell surface proteins and then to the ketone group specifically conjugated to hydrazide- or hydroxylamine-functionalized fluorophores.75 The drawback of fluorescent labeling mediated by biotin ligase is its selectivity towards the N- and C-termini, which restricts labeling in the loops inside the target protein.
2.3.2. Lipoic acid ligase. Lipoic acid ligase (LAL; ∼37 kDa) catalyzes the modification of the lysine residue in a lipoyl domain molecule, which is of 80 residues in length, by conjugating lipoic acid in the presence of ATP.76 In order to make the lipoyl domain short during the expression of the fused target protein, a 22-residue peptide (Lipoic Acid Peptide, LAP; ∼2.5 kDa) – an efficient substrate of E.coli LAL, is used instead of the lipoyl domain.40 It is also observed that LAL transfers an azide conjugated carboxylic acid to LAP. The azide moiety further undergoes azide-alkyne cycloaddition with an alkyne group conjugated to a fluorophore (as shown in Fig. 4). The advantage of having a swinging arm is that it does not interfere much with the conformation or the activity of the protein. The limitation towards labeling in the middle of the protein is the major disadvantage.
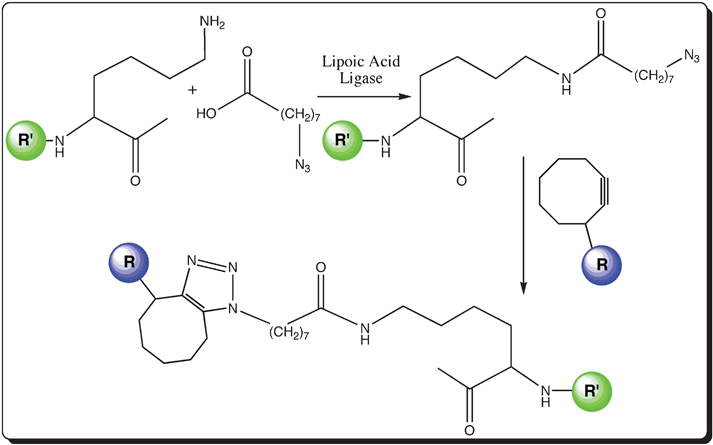 | ||
| Fig. 4 Schematic representation of the enzymatic-mediated labeling of a protein with a swinging arm; Lipoic acid ligase labeling: attachment of an azide-conjugated carboxylic acid moiety to create a swinging arm in the target biomolecule (R′ in green) and subsequent fluorescent labeling with a fluorophore (R in blue) linked to a cyclic alkyne group. | ||
2.3.3. Phosphopantetheinyl transferase. Phosphopantetheinyl transferases (PPTases) catalyzed protein labeling has recently been found to be a useful method for site specific protein labeling with small fluorophore molecules of diverse structures.77,78 For this purpose, chemical probes conjugated to coenzyme A (CoA) are used as the substrate and the substrate molecule is covalently attached to a specific serine residue in the target protein or fused protein with a short peptide tag (11 residues, ∼1.2 kDa).79 The basic function of PPTases is to transfer the phosphopantetheinyl group from CoA to a conserved serine residue of acyl carrier protein (ACP) or peptidyl carrier protein (PCP) domains as a part of fatty acid synthases, nonribosomal peptide synthetases and polyketide synthases.80,81 In general, the ACP and PCP carrier protein domains include 80 to 100 residues and the phosphopantetheinyl group acts as a swinging arm, which can be used to anchor fatty acid, polyketide and nonribosomal peptide chains.82 Besides the advantage of using a short peptide tag or ACP/PCP domains for site-specific protein labeling, the leading disadvantage is its inability towards position-independent labeling.
3. Tag labeling
Protein labeling through the incorporation of a short tag has been developed recently as a useful labeling technique. The use of the smaller tag, which has less potential to disrupt protein folding or function, holds an advantage over other techniques along with its labeling specificity. Here, an attempt is made to explain the tag-mediated protein labeling technique, which includes both external and internal tags with respect to the target protein molecule.Protein labeling involving transition metal linkage with specific residue(s) in a specific tag has a number of remarkable features, such as, (i) simplicity of the labeling procedure, (ii) high selectivity, (iii) application to various site-specific labeling (N-termini, C-termini or internal sites), and (iv) labeling efficiency. All these advantages make this technique an attractive and powerful protein labeling technique.
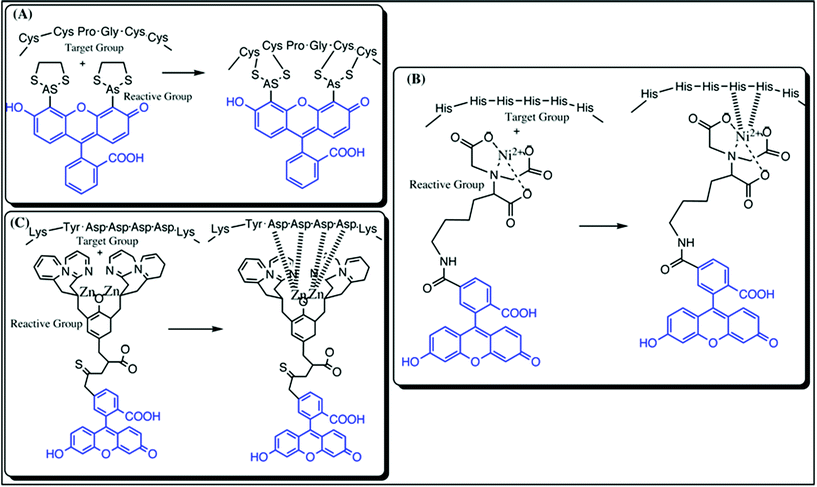 | ||
| Fig. 5 Representation of various types of tag labeling techniques; (A) Tetracysteine/Biarsenic tag: the left panel represents the unbound tetracysteine motif and the biarsenical motif coupled to fluorescein (blue) and the right panel shows the covalent linkage between the thiol groups of cysteine and Arsenic(III); (B) Histidine tag: the left panel represents the unbound histidine tag motif and metal complex with NTA linked to fluorescein (blue) and the right panel represents the covalent linkage between the histidine residues and the Ni2-complex with NTA conjugated to a fluorophore and (C) FLAG-tag: the left panel represents the unbound FLAG tag motif and Zn2NTA complex linked to fluorescein (blue) and the right panel shows the covalent linkage between the aspartate residues and the Zn2-complex with NTA conjugated to fluorescein. | ||
4. Genetic labeling
Genetically-mediated fluorescent labeling in living cells has been developed by using protein domains, small peptides, or single amino acids. Several existing methods are described below to label proteins inside the cells, which are broadly useful to study the localization, movement, interactions, and microenvironments of proteins in vivo.![Schematic representation of labeling techniques using genetic modifications: (A) SNAP-tag: attachment of benzylguanidine conjugated to a fluorophore (R, blue) to the SNAP tag fused to the target protein (R′, green); (B) CLIP-tag: attachment of benzylcytosine conjugated to a fluorophore (R, blue) to the SNAP tag fused to the target protein (R′, green); (C) Fluorescent protein: Green fluorescent proteins and a few examples of its variants, such as GFP (excitation and emission: 488 and 508 nm), YFP (excitation and emission: 514 and 530 nm) and RFP (excitation and emission: 554 and 585 nm). The blue color represents the chromophoric compound in the protein; (D) Unnatural amino acids: a few examples of widely used fluorescent non-natural amino acids, such as microenvironment sensitive (3-(4-nitrobenzo[c]1,2,5 oxadiazol-7-ylamino)-2-aminopropanoic acid (1), amino acids carrying 7-methoxycoumarine (4) and b-anthraniloyl (5)), 2-amino-3-(anthracen-5-yl)propanoic acid (2), which is less sensitive towards polar and nonpolar solvents and a hydrophobic labeled dye (2-amino-3-(pyren-2-yl)propanoic acid (3)). Moieties in blue indicate the fluorescent groups with a longer excitation wavelength than 320 nm, which thus remove the interference from the intrinsic fluorophores in the protein, and (E) tRNA: Reductive cleavage of dihydrouridine in the presence of NaBH4 and subsequent amination to form an amine group at the target site. In the second step, a fluorophore (R in blue) modified with a dihydrazide moiety is coupled to the primary amine group of the target site.](/image/article/2012/RA/c2ra20389h/c2ra20389h-f6.gif) | ||
| Fig. 6 Schematic representation of labeling techniques using genetic modifications: (A) SNAP-tag: attachment of benzylguanidine conjugated to a fluorophore (R, blue) to the SNAP tag fused to the target protein (R′, green); (B) CLIP-tag: attachment of benzylcytosine conjugated to a fluorophore (R, blue) to the SNAP tag fused to the target protein (R′, green); (C) Fluorescent protein: Green fluorescent proteins and a few examples of its variants, such as GFP (excitation and emission: 488 and 508 nm), YFP (excitation and emission: 514 and 530 nm) and RFP (excitation and emission: 554 and 585 nm). The blue color represents the chromophoric compound in the protein; (D) Unnatural amino acids: a few examples of widely used fluorescent non-natural amino acids, such as microenvironment sensitive (3-(4-nitrobenzo[c]1,2,5 oxadiazol-7-ylamino)-2-aminopropanoic acid (1), amino acids carrying 7-methoxycoumarine (4) and b-anthraniloyl (5)), 2-amino-3-(anthracen-5-yl)propanoic acid (2), which is less sensitive towards polar and nonpolar solvents and a hydrophobic labeled dye (2-amino-3-(pyren-2-yl)propanoic acid (3)). Moieties in blue indicate the fluorescent groups with a longer excitation wavelength than 320 nm, which thus remove the interference from the intrinsic fluorophores in the protein, and (E) tRNA: Reductive cleavage of dihydrouridine in the presence of NaBH4 and subsequent amination to form an amine group at the target site. In the second step, a fluorophore (R in blue) modified with a dihydrazide moiety is coupled to the primary amine group of the target site. | ||
Incorporation of unnatural amino acids permits the direct or indirect introduction of fluorophores. Direct fluorophore introduction is restricted to derivatives of intrinsic protein fluorophores and is not compatible with single molecule fluorescence spectroscopy. On the other hand, indirect incorporation is performed by using unnatural amino acids with non-proteinogenic reactive groups, such as the ketone carbonyl group that allows selective, site-specific labeling in a postsynthetic fashion. The indirect incorporation is demonstrated by the site-specific labeling of a single-ketone protein with fluorescein hydrazide.108 Despite the power of the approach, it has not been used widely for preparing labeled proteins mainly due to the very low yields.
Conclusion
Though a number of existing fluorescent labeling methods are presented in this review, new protein labeling techniques in vitro and in vivo are still in demand and are undergoing development. The present review is only an attempt to discuss most of the widely used protein labeling techniques with an internal or external fluorophore. Table 1 summarizes the advantages and disadvantages of the fluorescent labeling techniques that are mentioned in this review. With the current progress in synthetic chemistry and molecular biology technologies along with genetic engineering, protein labeling with a fluorophore plays a key role in understanding the dynamics and biological functions of biomolecules with more accuracy. Fluorescent labeling methods are not only confined to the use of small synthetic fluorophores and fluorescent proteins, but also extend over a wide range of wavelengths using semiconductor nanocrystals (or quantum dots, QDs). The labeling of biomolecules with quantum dots, taking account of the improvements on their disadvantages of cytotoxicity, specificity and aggregation, has continued to inspire a great deal of research in recent years.| Labeling Techniques | Advantages | Disadvantages | Applications (in vitro or in vivo) | |
|---|---|---|---|---|
| CHEMICAL | Amine | Robust and efficient | Limited to in vitro and pH sensitive | in vitro 36 |
| Hydroxy | Robust and efficient | Limited to in vitro and pH sensitive | in vitro 37 | |
| Thiol | Powerful, efficient and selective | Limited to in vitro and pH sensitive | in vitro 38 | |
| Azide | Efficient and selective | Incorporation of azide | Both44 | |
| Tetrazine | Efficient, rapid and selective | Incorporation of reactive groups | Both51 | |
| ENZYMATIC | TGase | Not position specific | Non-specific (in vivo) and low yield | Both53,54 |
| Sortase | Efficient labeling | Hydrolyzes proteins and extracellular | Both57,58 | |
| Cutinase | Selective | Site-specific, large size and extracellular | Both62,63 | |
| Intein | Selective & Efficient | Site-specific (C-terminus) | in vitro 64 | |
| Haloalkane Dehalogenase | Membrane permeability | Large size of the enzyme and incorporation of halide | Both71 | |
| BPL | Selective and efficient | Site-specific and extracellular | Both74 | |
| LAL | Swinging-arm | Large size of LAL tag | in vitro 76 | |
| PPTase | Short recognition motif | Site-specific | in vitro 78 | |
| TAG | FlAsH | Selective and efficient | Non-specific (cysteines other than the tag) | Both83 |
| Histidine | Selective and efficient | Extracellular only | in vitro 93 | |
| FLAG | Selective and efficient | Extracellular only | in vitro 95,98 | |
| GENETIC | SNAP-tag | Selective and efficient | Large size of the tag | Both21 |
| CLIP-tag | Selective and efficient | Large size of the tag | Both99 | |
| Fluorescent Protein | Self-fluorescent and biocompatible | Large size and labeling at the termini | Both100 | |
| Unnatural Amino Acid | Efficient and specific | Limited to UV-visible range | Both108 | |
| tRNA | Efficient and specific | Multiple steps and low yield | Both112 |
Acknowledgements
The author is thankful to Prof. Anadi Charan Dash (retd., Utkal University, India), Dr Monalisa Mishra (Max-Planck Institute for Molecular Cell Biology and Genetics, Germany), Dr Joanna Swain (Adnexus R&D Company, USA), Dr Sujit Kumar Dora (Kanazawa University, Japan), and Dr Jiang Hong (Shanghai University, China) for critically reading the manuscript and offering valuable suggestions. Financial support by the German Research Foundation (DFG) within Transregio SFB 67 is gratefully acknowledged.References
- S. E. Ong and M. Mann, Nat. Protoc., 2007, 1, 2650–2660 CrossRef
.
- A. Varki, Faseb J., 1991, 5, 226–235 CAS
- H. X. Li and L. Rothberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14036–14039 CrossRef CAS
- J. R. Kumita, O. S. Smart and G. A. Woolley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3803–3808 CrossRef CAS
- N. Soh, K. Yoshida, H. Nakajima, K. Nakano, T. Imato, T. Fukaminato and M. Irie, Chem. Commun., 2007, 5206–5208 RSC
- F. Di Benedetto, E. Mele, A. Camposeo, A. Athanassiou, R. Cingolani and D. Pisignano, Adv. Mater., 2008, 20, 314–318 CrossRef CAS
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5985 CrossRef CAS
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS
- M. Zimmer, Chem. Rev., 2002, 102, 759–781 CrossRef CAS
- N. Johnsson and K. Johnsson, ACS Chem. Biol., 2007, 2, 31–38 CrossRef CAS
- T. L. Foley and M. D. Burkart, Curr. Opin. Chem. Biol., 2007, 11, 12–19 CrossRef CAS
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS
- H. Sahoo, J. Photochem. Photobiol., C, 2011, 12, 20–30 CrossRef CAS
- H. Sahoo, A. Hennig, M. Florea, D. Roth, T. Enderle and W. M. Nau, J. Am. Chem. Soc., 2007, 129, 15927–15934 CrossRef CAS
- H. Sahoo and W. M. Nau, ChemBioChem, 2007, 8, 567–573 CrossRef CAS
- H. Sahoo, D. Roccatano, A. Hennig and W. M. Nau, J. Am. Chem. Soc., 2007, 129, 9762–9772 CrossRef CAS
- H. Sahoo, D. Roccatano, M. Zacharias and W. M. Nau, J. Am. Chem. Soc., 2006, 128, 8118–8119 CrossRef CAS
- B. T. Kurien and R. H. Scofield, Methods Mol. Biol., 2009, 536, 557–571 CAS
- T. Miyashita, Methods Mol. Biol., 2004, 261, 399–410 CAS
- Y. Wang, J. Y.-J. Shyy and S. Chien, Annu. Rev. Biomed. Eng., 2008, 10, 1–38 CrossRef CAS
- D. Maurel, L. Comps-Agrar, C. Brock, M.-L. Rives, E. Bourrier, M. A. Ayoub, H. Bazin, N. Tinel, T. Durroux, l. Prézeau, E. Trinquet and J.-P. Pin, Nat. Methods, 2008, 5, 561–567 CrossRef CAS
- J. Yin, P. D. Straight, S. M. McLaoughlin, Z. Zhou, A. J. Lin, D. E. Go, R. Kolter and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15815–15820 CrossRef CAS
- J. Evans and D. T. Yue, Neuron, 2003, 38, 145–147 CrossRef CAS
- D. L. Pan, H. O. Qin and B. S. Cooperman, RNA, 2009, 15, 346–354 CrossRef CAS
- A. R. Katritzky and T. Narindoshvili, Org. Biomol. Chem., 2009, 7, 627–634 CAS
- M. Margittai, J. Widengren, E. Schweinberger, G. F. Schröder, S. Felekyan, E. Haustein, M. König, D. Fasshauer, H. Grubmüller, R. Jahn and C. A. M. Seidel, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15516–15521 CrossRef CAS
- C. Eggeling, P. Kask, D. Winkler and S. Jäger, Biophys. J., 2005, 89, 605–618 CrossRef CAS
- B. K. Müller, E. Zaychikov, C. Bräuchle and D. C. Lamb, Biophys. J., 2005, 89, 3508–3522 CrossRef
- H. Sahoo and P. Schwille, ChemPhysChem, 2011, 12, 532–541 CrossRef CAS
- N. Mochizuki, S. Yamashita, K. Kurokawa, Y. Ohba, T. Nagai, A. Miyawaki and M. Matsuda, Nature, 2001, 411, 1065–1068 CrossRef CAS
- N. Zuleger, D. A. Kelly, A. C. Richardson, A. R. W. Kerr, M. W. Goldberg, A. B. Goryachev and E. C. Schirmer, J. Cell Biol., 2011, 193, 109–123 CrossRef CAS
- G. Gaietta, T. J. Deerinck, S. R. Adams, J. Bouwer, O. Tour, D. W. Laird, G. E. Sosinsky, R. Y. Tsien and M. H. Ellisman, Science, 2002, 296, 503–507 CrossRef CAS
- M. Fernandez-Suarez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef CAS
- C. Bustamante, W. Cheng and Y. X. Mejia, Cell, 2011, 145, 160–160 CrossRef CAS
-
G. T. Hermanson, in Bioconjugate Techniques, Academic Press, Inc., 2008, 1202 Search PubMed
- A. J. Lomant and G. Fairbanks, J. Mol. Biol., 1976, 104, 243–261 CrossRef CAS
- L. Bing, J. M. Davidson and S. A. Guelcher, Biomaterials, 2009, 30, 3486–3494 CrossRef
- M. D. Partis, D. G. Griffiths, G. C. Roberts and R. B. Beechey, J. Protein Chem., 1983, 2, 263–277 CrossRef CAS
- P. M. E. Gramlich, C. T. Wirges, A. Manetto and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 8350–8358 CrossRef CAS
- M. Fernandez-Suarez, H. Baruah, L. Martinez-Hernandez, K. T. Xie, J. M. Baskin, C. R. Bertozzi and A. Y. Ting, Nat. Biotechnol., 2007, 25, 1483–1487 CrossRef CAS
- A. E. Speers, G. C. Adam and B. F. Cravatt, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS
- J. M. Baskin and C. R. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211–1219 CAS
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS
- S. T. Laughlin and C. R. Bertozzi, Nat. Protoc., 2007, 2, 2930–2944 CrossRef CAS
- A. E. Speers, G. C. Adams and B. F. Cravatt, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS
- H.-S. Han, N. K. Devaraj, J. Lee, S. A. Hilderbrand, R. Weissleder and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 7838–7839 CrossRef CAS
- N. K. Devaraj and R. Weissleder, Accounts of Chemical Research, 2011, 44, 816–827 CAS
- M. Griffin, R. Casadio and C. M. Bergamini, Biochem. J., 2002, 368, 377–396 CrossRef CAS
- C. W. Lin and A. Y. Ting, J. Am. Chem. Soc., 2006, 128, 4542–4543 CrossRef CAS
- Y. Sugimura, M. Hosono, F. Wada, T. Yoshimura, M. Maki and K. Hitomi, J. Biol. Chem., 2006, 281, 17699–17706 CrossRef CAS
- A. M. Perry, H. Ton-That, S. K. Mazmanian and O. Schneewind, J. Biol. Chem., 2002, 277, 16241–16248 CrossRef CAS
- S. K. Mazmanian, G. Liu, T. T. Hung and O. Schneewind, Science, 1999, 285, 760–763 CrossRef CAS
- H. Y. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS
- T. Tanaka, T. Yamamoto, S. Tsukiji and T. Nagamune, ChemBioChem, 2008, 9, 802–807 CrossRef CAS
- M. W. Popp and H. L. Ploegh, Angew. Chem., Int. Ed., 2011, 50, 5024–5032 CrossRef CAS
- M. L. M. Mannesse, R. C. Cox, B. C. Koops, H. M. Verheij, G. H. Dehaas, M. R. Egmond, H. Vanderhijden and J. Devlieg, Biochemistry, 1995, 34, 6400–6407 CrossRef CAS
- M. L. M. Mannesse, J. W. P. Boots, R. Dijkman, A. J. Slotboom, H. Vanderhijden, M. R. Egmond, H. M. Verheij and G. H. Dehaas, Biochim. Biophys. Acta-Lipids, 1995, 1259, 56–64 Search PubMed
- R. Bonasio, C. V. Carman, E. Kim, P. T. Sage, K. R. Love, T. R. Mempel, T. A. Springer and U. H. von Andrian, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14753–14758 CrossRef CAS
- C. D. Hodneland, Y. S. Lee, D. H. Min and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5048–5052 CrossRef CAS
- M. Q. Xu, M. W. Southworth, F. B. Mersha, L. J. Hornstra and F. B. Perler, Cell, 1993, 75, 1371–1377 CrossRef CAS
- C. J. Noren, J. M. Wang and F. B. Perler, Angew. Chem., Int. Ed., 2000, 39, 450–466 CrossRef CAS
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef CAS
- P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS
- F. Pries, J. Kingma, G. H. Krooshof, C. M. Jeronimusstratingh, A. P. Bruins and D. B. Janssen, J. Biol. Chem., 1995, 270, 10405–10411 CrossRef CAS
- K. H. G. Verschueren, F. Seljee, H. J. Rozeboom, K. H. Kalk and B. W. Dijkstra, Nature, 1993, 363, 693–698 CrossRef CAS
- H. Q. Yao, Y. Zhang, F. Xiao, Z. Y. Xia and J. H. Rao, Angew. Chem., Int. Ed., 2007, 46, 4346–4349 CrossRef CAS
- Y. Zhang, M. K. So, A. M. Loening, H. Q. Yao, S. S. Gambhir and J. H. Rao, Angew. Chem., Int. Ed., 2006, 45, 4936–4940 CrossRef CAS
- G. V. Los and K. V. Wood, Methods Mol. Biol., 2007, 356, 195–208 CAS
- P. Reche and R. N. Perham, EMBO J., 1999, 18, 2673–2682 CrossRef CAS
- S. Sueda, S. Yoneda and H. Hayashi, ChemBioChem, 2011, 12, 1367–1375 CrossRef CAS
- I. Chen, M. Howarth, W. Lin and A. Y. Ting, Nat. Methods, 2005, 2, 99–104 CrossRef CAS
- X. Zhao, J. R. Miller, Y. F. Jiang, M. A. Marletta and J. E. Cronan, Chem. Biol., 2003, 10, 1293–1302 CrossRef CAS
- M. Sunbul, K. Zhang and J. Yin, Methods Enzymol., 2009, 458, 255–275 CAS
- J. Yin, A. J. Lin, D. E. Golan and C. T. Walsh, Nat. Protoc., 2006, 1, 280–285 CrossRef CAS
- Y. Zou and J. Yin, J. Am. Chem. Soc., 2009, 131, 7548–7549 CrossRef CAS
- R. S. Flugel, Y. Hwangbo, R. H. Lambalot, J. E. Cronan and C. T. Walsh, J. Biol. Chem., 2000, 275, 959–968 CrossRef CAS
- R. H. Lambalot, A. M. Gehring, R. S. Flugel, P. Zuber, M. LaCelle, M. A. Marahiel, R. Reid, C. Khosla and C. T. Walsh, Chem. Biol., 1996, 3, 923–936 CrossRef CAS
- A. C. Mercer and M. D. Burkart, Nat. Prod. Rep., 2007, 24, 750–773 RSC
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS
- B. Liu, C. T. Archer, L. Burdine, T. G. Gillette and T. Kodadek, J. Am. Chem. Soc., 2007, 129, 12348–12349 CrossRef CAS
- X. Y. Zhang and A. C. Bishop, J. Am. Chem. Soc., 2007, 129, 3812–3813 CrossRef CAS
- A. C. Hearps, M. J. Pryor, H. V. Kuusisto, S. M. Rawlinson, S. C. Piller and D. A. Jans, J. Fluoresc., 2007, 17, 593–597 CrossRef CAS
- B. A. Griffin, S. R. Adam, J. Jones and R. Y. Tsien, Methods Enzymol., 2000, 327, 565–578 CAS
- T. Machleidt, M. Robers and G. Hanson, Methods Mol. Biol., 2007, 356, 209–220 CAS
- C. C. Spagnuolo, R. J. Vermeij and E. A. Jares-Erijman, J. Am. Chem. Soc., 2006, 128, 12040–12041 CrossRef CAS
- H. S. Cao, Y. J. Xiong, T. Wang, B. W. Chen, T. C. Squier and M. U. Mayer, J. Am. Chem. Soc., 2007, 129, 8672–8673 CrossRef CAS
- A. N. Kapanidis, Y. W. Ebright and R. H. Ebright, J. Am. Chem. Soc., 2001, 123, 12123–12125 CrossRef CAS
- E. G. Guignet, R. Hovius and H. Vogel, Nat. Biotechnol., 2004, 22, 440–444 CrossRef CAS
- S. Lata, M. Gavutis, R. Tampe and J. Piehler, J. Am. Chem. Soc., 2006, 128, 2365–2372 CrossRef CAS
- A. Ojida, K. Honda, D. Shinmi, S. Kiyonaka, Y. Mori and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 10452–10459 CrossRef CAS
- K. Honda, E. Nakata, A. Ojida and I. Hamachi, Chem. Commun., 2006, 4024–4026 RSC
- K. Honda, S. H. Fujishima, A. Ojida and I. Hamachi, ChemBioChem, 2007, 8, 1370–1372 CrossRef CAS
- K. J. T. Venken, J. Kasprowicz, S. Kuenen, J. Yan, B. A. Hassan and P. Verstreken, Nucleic Acids Res., 2008, 36, e114, DOI:10.1093/nar/gkn486
- A. Gautier, E. Nakata, G. Lukinavicius, T. K. T and K. Johnsson, J. Am. Chem. Soc., 2009, 131, 17954–17962 CrossRef CAS
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS
- D. A. Zacharias, J. D. Violin, A. C. Newton and R. Y. Tsien, Science, 2002, 296, 913–916 CrossRef CAS
- A. Dragulescu-Andrasi and J. H. Rao, ChemBioChem, 2007, 8, 1099–1101 CrossRef CAS
- K. L. Kiick, R. Weberskirch and D. A. Tirrell, FEBS Lett., 2001, 502, 25–30 CrossRef CAS
- I. Chen and A. Y. Ting, Curr. Opin. Biotechnol., 2005, 16, 35–40 CrossRef CAS
- D. Mendel, V. W. Cornish and P. G. Schultz, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 435–462 CrossRef CAS
- V. W. Cornish, K. M. Hahn and P. G. Schultz, J. Am. Chem. Soc., 1996, 118, 8150–8151 CrossRef CAS
- V. W. Cornish, D. R. Benson, C. A. Altenbach, K. Hideg, W. L. Hubbell and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2910–2914 CrossRef CAS
- J. Fei, P. Kosuri, D. D. MacDougall and R. L. Gonzalez, Mol. Cell, 2008, 30, 348–359 CrossRef CAS
- J. B. Munro, R. B. Altman, N. O'Connor and S. C. Blanchard, Mol. Cell, 2007, 25, 505–517 CrossRef CAS
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS
- L. Wang, A. Brock and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 1836–1837 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2012 |