N-Doped carbon nanorods as ultrasensitive electrochemical sensors for the determination of dopamine†
Dingsheng
Yuan
*,
Xiaoli
Yuan
,
Shuangli
Zhou
,
Wujun
Zou
and
Tianxiang
Zhou
E-mail: tydsh@jnu.edu.cn; Fax: +8620 8522 1697; Tel: +8620 8522 0597
First published on 28th June 2012
Abstract
Nitrogen-doped carbon nanorods (N-CNRs) are prepared by a direct carbonization method using polyaniline nanorods as the carbon precursor. The electrochemical behavior of the N-CNRs-Nafion modified electrode is evaluated in connection with dopamine and ascorbic acid by cyclic voltammetry and differential pulse voltammetry. Experimental results indicate that the N-CNRs modified electrode has improved current response and fast electron transfer kinetics. The linear response for the selective determination of dopamine in the presence of ascorbic acid is obtained in the range of 0.008 μM to 15.0 μM with a detection limit of 8.9 × 10−9 M (S/N = 3) by differential pulse voltammetry under optimum conditions. The N-CNRs-Nafion modified electrode exhibits a wide linear range, very low detection limit and anti-interference ability. Meanwhile, a kinetic reaction process and a reaction mechanism for the N-CNRs are also proposed.
1. Introduction
The development of electrochemical sensors has been widely studied as an inexpensive method to detect sensitively and selectively a large number of electroactive species, including hydrogen peroxide,1 hydroquinone,2 nicotinamide-adenine dinucleotide (NADH),3 serotonin,4 glucose,5 dopamine (DA)6 and ascorbic acid (AA),7 and so on. Among them, dopamine an important neurotransmitter and attracts considerable researches in the clinical field because of its involvement in motor and cognitive function. It has been found that some serious diseases such as schizophrenia and Parkinson’s disease are associated with low levels of DA in vivo.8,9 Quantitative determination of DA is therefore important and has attracted much attention among neuroscientists and chemists. However, there are some problems with the electrochemical methods due to the nature of the oxidative reaction, involving selectivity and sensitivity as two major problems in dopamine detection. The interference of other neurochemical compounds such as AA and uric acid (UA), results in an overlap of their voltammetric response at bare glassy carbon electrodes (GCEs) because their oxidation potential is close to that of DA. Moreover, the basal concentration of DA and AA changes in a broad range from 1.0 × 10−7 to 1.0 × 10−3 M, and the concentration of AA is usually one thousand times higher than that of DA in the extracellular fluid of the caudate nucleus. Therefore, both the sensitivity and the selectivity are of equal importance for practical applications. In order to improve the selectivity of the electrodes, considerable efforts have been devoted to modify the electrode surface. The key is to choose suitable materials.Some carbon-based materials, such as boron-doped diamond, grapheme, carbon nanotubes (CNTs) and ordered mesoporous carbon (OMC), show high electrocatalytic activity for biosensing applications.10–13 These special carbon materials provide perfect results for simultaneous or selective determination of compounds. CNTs, OMC and functionalized carbon have been intensively studied in the oxidation of DA, UA and AA. Song et al.11 used porous-carbon-modified glassy carbon electrodes in the presence of AA for selective determination of dopamine, based on both cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements. Layer-by-layer assembled CNTs-modified GCE was used for the selective determination of dopamine by Zhang.10 Furthermore, DA and AA were simultaneously detected at a glassy carbon modified electrode containing multiwall carbon nanotubes by Alothman.14 Primary CNTs are chemically inert and therefore must be treated with strong acids to remove the end caps and shorten the length of the CNTs. Acid treatment also adds oxide groups and primarily carboxylic acids to the tube ends and defect sites. Further chemical reaction can be performed at these oxide groups to functionalize with groups such as amides, thiols, or the others.7 However, such procedures are time-consuming and tedious. Therefore, it is necessary to develop new carbon materials to replace those above.
Herein, we present the electrochemical properties of nitrogen-doped carbon nanorods. The electrocatalytic behavior of the N-CNRs-modified GCE towards the oxidation of DA and AA has been investigated for the first time. Notably, the unique porous structure and the characteristic features of nitrogen-doping are helpful for quick mass transfer of the electrolyte and selective determination in N-CNRs. The N-CNRs modified GCE shows faster electron transfer kinetics compared to bare GCE, and simultaneously, the determination of DA exhibits an adjustable determination range and very low detection limit and high sensitivity while eliminating the interference from AA.
2. Experimental
2.1. Chemicals and apparatus
Dopamine hydrochloride and ascorbic acid were purchased from Aladdin Chemistry Co, Ltd. Nafion was obtained from Aldrich. Phosphate buffer solutions (PBS) with various pH values was prepared by sodium phosphate monohydrate (NaH2PO4·H2O) and sodium phosphate dibasic dehydrate (Na2HPO4·2H2O). Freshly-prepared solutions of DA and AA were used in each experiment. All other chemicals used were of analytical grade and used without further purification. High-purity nitrogen was used for the deaeration.The microstructure was observed via a JEOL JEM-2100F (HR) using an accelerating voltage of 200 kV. The valence of nitrogen in carbon nanorods was characterized by X-ray photoelectron spectroscopy (XPS, ESCALab250). The composition of nitrogen was analyzed by inductively coupled plasma-atomic emission spectrometry (Optima 2000DV). Nitrogen sorption isotherm of N-CNRs was measured by a Micrometrics TriStar 3000 Analyzer at 77 K. Electrochemical measurements were carried out with an Ingsens electrochemical workstation with a three-electrode system, which consists of a glassy carbon electrode or a modified electrode as the working electrode, a platinum wire as the auxiliary electrode and an Ag/AgCl (in saturated KCl solution) electrode as the reference electrode, respectively.
2.2. Preparation of the N-CNRs-Nafion modified electrode
N-CNRs were synthesized from the carbonization of PANI nanorods under a highly pure N2 atmosphere from 600 °C for 2 h, as reported in our previous work;15 detailed synthesis procedures are described in the Supporting Information (SI).† Prior to surface modification, glassy carbon electrodes (4 mm in diameter) were mechanically polished by a slurry of 1.0, 0.3 and 0.05 μm aluminum oxide (Al2O3) powder, respectively, then washed and sonicated in deionized water and ethanol in turn. A mixture containing 3 mg as-prepared N-CNRs, 3 mL DMF and Nafion solution (0.5 wt%) was dispersed ultrasonically for 2 h to obtain a well-dispersed ink. 10 μL of above-mentioned ink was dropped on the surface of the pretreated GCE and dried under an infrared lamp for 20 min to obtain the N-CNRs-Nafion/GCE. Before and after each experiment, the modified electrodes were carefully washed with distilled water and reactivated by the above-mentioned method. All the experiments were carried out in 10 mL PBS solutions as the supporting electrolyte at room temperature. Oxygen was evacuated and purged with high-purity nitrogen before each experiment.3. Results and discussion
3.1. Structural characterization of the N-CNRs
As-prepared N-CNRs possess a porous structure; the specific surface area and the pore parameters of the N-CNRs are summarized in Table S1.† The abundant pores are helpful for the diffusion of electrolyte and a relatively high specific surface area (362 m2 g−1) provides more reaction sites. The composition of nitrogen in the N-CNRs is up to 25.8 wt%. The TEM images of N-CNRs are shown in Fig. 1. Fig. 1a shows carbon nanorods with sizes ranging from 55 to 85 nm. Some mesoporous tunnels can be observed. At the same time, the HRTEM image presents abundant micropores with a ∼1.2 nm pore size and the finger of the graphitized carbon can be observed clearly, as shown in Fig. 1b. The porous structure is advantageous in terms of the quick mass transfer of the electrolyte in N-CNRs. Fig. 1c is an SAED pattern, revealing that N-CNRs have a polycrystalline structure. | ||
| Fig. 1 Analyses for the microstructure of N-CNRs: (a) TEM image; (b) HRTEM image; (c) SAED pattern. | ||
XPS has been used to try to elucidate the chemical nature of the structures in which these heteroatoms are included in cokes. Fig. 2 presents the wide scan XPS spectrum of N-CNRs and the high resolution N (1s) spectrum of N-CNRs. In the XPS spectra, the peaks at 284.81, 400.48, and 532.4 eV correspond to the C 1s of sp2 C, N 1s of the doped N, and O 1s of the absorbed oxygen, respectively (Fig. 2a). In Fig. 2b, there are two different electronic states in the N 1s spectrum of the N-CNRs, the binding energy (BE) centered at 398.7, 400.48 eV corresponding to the “pyridinic” N and “pyrrolic” N of N-CNRs, respectively. In the case of pyridinic-N, the nitrogen atom contributes one p-electron to the aromatic π-system and has a lone electron pair in the plane of the ring; as for pyrrolic-N, the nitrogen atom contributes two p-electrons to the π-system, and a hydrogen atom is bound in the plane of the ring.16,17

3.2. Cyclic voltammetric behaviors of DA and AA at the N-CNRs-Nafion/GCE
The electrochemical performance of the N-CNRs is carried out in three different electrolytes involving 0.1 M PBS, 0.5 mM AA and 0.5 mM DA in 0.1 M PBS. Typical voltammetric responses of N-CNRs-Nafion/GCE in pH 4.0 PBS at the scan rate of 50 mV s−1 are shown in Fig. 3c. For comparison, Fig. 3a shows the typical CVs of PBS, AA and DA at the bare GCE, presenting a large broad oxidation peak for AA at 380 mV, and no corresponding reduction peak. DA shows a pair of asymmetrical redox peaks with a separation of the peak potentials (ΔEp) of 242 and 442 mV, respectively; furthermore, PBS produces no Faradaic current from −0.2 V to 0.6 V. As seen in Fig. 3a, the separation of the anodic peak potentials for DA and AA are not enough large to obtain good selectivity at the bare GCE.18,19 The anodic peak potentials of DA and AA usually depend on the experimental conditions, for example the different buffer solutions and various pH values.20–22 It is clear that the electrochemical reactions of these species at the bare GCE are totally indistinguishable and irreversible with a large overpotential, indicating the sluggish electron transfer kinetics at the bare GCE. Therefore, it is impossible to separate DA and AA from each other using the voltammetric peaks of the bare GCE. | ||
| Fig. 3 CVs for the bare GCE (a), Nafion/GCE (b), and N-CNRs-Nafion/GCE (c) in 0.1 M PBS, 0.5 mM AA + 0.1 M PBS and 0.5 mM DA + 0.1 M PBS solutions, respectively. pH = 4.0, Scan rate: 50 mV s−1. | ||
The same characterization was performed for the Nafion/GCE, as shown in Fig. 3b. The CVs similar to the bare GCE present a potential range from −0.2 to 0.6 V, however, a ΔEp of 162 mV for DA on the Nafion/GCE is smaller than 200 mV on the bare GCE. As ΔEp is a function of the rate of electron transfer, the smaller ΔEp, the higher the electron transfer rate. According to Nicholson's method,23 GCE modified by Nafion clearly shows an improved electron transfer rate. Due to its unique ion-exchange, discrimination and biocompatibility properties, Nafion has been extensively used for the modification of electrode surfaces and the construction of biosensors.24
Fig. 3c gives the well-defined redox peaks of DA at 362 and 322 mV, respectively. The separation of anodic and cathodic peak potentials is 40 mV, indicating that the reversibility of DA at N-CNRs-Nafion/GCE is considerably improved, and the anodic peak current is enhanced about 1.7 times in comparison with the anodic peak current on the bare GCE. These experimental results demonstrate that N-CNRs not only enhance the sensitivity of GCE for the detection of DA and AA, but also dramatically enlarge the separation of anodic peaks between DA and AA. It is assumed that the greatly enhanced electrochemical behavior of N-CNRs may be attributed to a large number of exposure edge plane defect sites at the surface of the N-CNRs and the dominant porous structure and the appropriate porous size accessible for quick mass transfer and ion diffusion; also nitrogen-containing functional groups on the surface of carbon nanorods play an important role in the redox process. Therefore, the performance of N-CNRs-Nafion/GCE is visibly superior to the bare GCE and Nafion/GCE. The effect of the N-doped carbon nanorods on the electrochemical performance will be further discussed in the mechanism analysis.
In order to investigate further the electrochemical behavior of DA and AA at the modified electrode, Fig. 4 presents the effect of scan rates ranging from 100 to 500 mV s−1 on 0.5 mM DA and 0.5 mM AA oxidation at the N-CNRs-Nafion/GCE by CV in PBS solution at pH 4.0, respectively. The anodic and cathodic peak currents of DA increase linearly with increasing scan rates, as shown in Fig. 4a, indicating direct electron transfer between DA and the modified electrode. The linearity of the plots is very reasonable, with correlation coefficients of 0.9988 (Ipc,DA(μA) = −0.1517v − 26.2637) on the cathodic current and 0.9977 (Ipa,DA(μA) = 0.2101v + 39.9427) on the anodic current. This result implies that the electrochemical oxidation of DA at the modified electrode is dominated by a surface-adsorption controlled process rather than a diffusion-controlled process.25 The Epa is shifted to more positive values with increasing the scan rate, suggesting that the electron transfer was quasi-reversible.4,26
 | ||
| Fig. 4 CVs for the oxidation of 0.5 mM DA (a) and 0.5 mM AA (b) at the N-CNRs-Nafion/GCE in pH 4.0 PBS. Scan rates: 100, 200, 300, 400, 500 mV s−1. The inset is the plots of the peak currents vs. scan rates. | ||
The anodic peak current (Ipa,AA) of AA at N-CNRs-Nafion/GCE is also proportional to the scan rate at the range of 100–500 mV s−1 with a linear regression equation of Ipa,AA(μA) = 0.03531v + 11.7390 (R = 0.9987), showing a typical surface-adsorption kinetics (Fig. 4b).
3.3. Effects of the pH value
The pH of the supporting electrolyte has a significant influence on the DA and AA oxidation at the N-CNRs-Nafion/GCE by affecting both the peak current and the peak potential because the protons participate in the electrode reaction process of DA and AA.27 DPV has much higher current sensitivity and better resolution than CV, thus it is usually used in the determination of DA and AA concentration to deduce the lower limit of detection.The peak current of DA presents an ‘M’ shape because of the effect of the pH value, as shown in Fig. 5a. When the pH < 4.0, the peak current quickly increases and reaches a maximum value at pH = 4.0, whereas the second maximum value is at pH = 6.0. This result assures the change in electrostatic interaction between DA and N-CNRs and clarifies the electrode reaction depending on the pH value of the solution. As for AA, the peak current also reaches a maximum value at a pH value of 4.0. However, it gradually decreases with the increase of pH value. This result is in good agreement with Habibi and Pournaghi-Azar's report.28 In fact, AA with a pKa of 4.17 mainly exists as a neutral form in solutions under a pH value of 4.0, while at pH ≥ 5.0, the anionic form dominates and AA could be repulsed by the sulfonic acid groups of Nafion with the same electric charge at the modified electrode.
 | ||
| Fig. 5 Effect of pH on the peak current (a), the peak potential (b) and the separation of the peak potentials ΔEp (c) for the oxidation of 0.5 mM AA and 5 μM DA in 0.1 M PBS at the N-CNRs-Nafion/GCE. DPV conditions: potential incremental, 1 mV; amplitude, 50 mV; pulse width, 50 mV; period, 75 ms. | ||
The effect of pH on the anodic peak potentials the DA and AA oxidation at N-CNRs-Nafion/GCE are shown in Fig. 5b. All the peak potentials of DA and AA give a similar trend and increase by a range of 2.0–8.0, indicating that the protons participate directly in the determination step of all the oxidation processes.29 The equations of Epavs. pH value are expressed as: Epa = 0.4893–0.05236 pH for DA with a correlation coefficient of 0.9972 and Epa = 0.3617–0.05284 pH for AA with a correlation coefficient of 0.9882. The peak potentials for DA and AA show a linear variation with a slope of about 60 mV pH−1, corresponding to a 2e−/2H+ transfer process.10 Consequently, the overall process is proton dependent and the electron-transfer step is preceded by a protonation with an equal number of protons and electrons involved in DA and AA oxidation. Fig. 5c shows the effect of pH on the separation of the anodic peak potentials of DA and AA. The maximum separation of peak potentials for DA-AA is observed at pH 4.0. Therefore, for selective determination of DA and AA, pH 4.0 is optimum for electrochemical detection of DA and AA, assured to obtain high sensitivity and good selectivity.
3.4. Selective determination of DA in the presence of AA at the N-CNRs-Nafion/GCE
It is well known that AA coexists with DA in the extracellular fluid of the central nervous system and its oxidation peak potential is close to DA on the bare GCE, thus AA is the major interferent for DA detection. Based on this drawback, the excellent catalytic activity of the N-CNRs remarkably facilitates the electron transfer of DA (see Fig. 3c). An intrinsic property of the N-CNRs substantially differentiates between the oxidation processes of DA and AA and avoids the co-oxidation of AA redox-mediated by DA, which has been observed in MWNTs with frequently used GCEs.10 We can exploit the electrostatic attraction of the Nafion film30 and nitrogen-containing functional groups on the surface of carbon nanorods toward DA, and the electrostatic repulsion against AA to separate two overlapping anodic peaks via DPV analysis. These demonstrations, coupled to the improved reversibility of the redox process of DA at the N-CNRs-Nafion/GCE, essentially make it possible to determine DA sensitively and selectively without the interference of AA. The interference from AA is investigated and determination of DA concentration is performed with the DPV technique in a 0.1 M deaerated PBS solution at pH 4.0. Compared with the CV of AA in Fig. 3c, DPV of AA shows that the oxidative potential is shifted negatively from 182 mV to 103 mV at the modified N-CNRs-Nafion/GCE, and that the anodic peak current is increased.Fig. 6 shows typical DPV curves at the N-CNRs-Nafion/GCE with concentrations from 0.008 μM to 15.0 μM DA in the presence of 0.5 mM AA, and two well-distinguished peaks at about 103 and 337 mV are obtained, respectively. It could be clearly observed that the DPV peak current of the DA is linearly related to its concentration, whereas the current of the anodic peaks of AA almost keeps constant, revealing that oxidation of DA and AA have no mutual influence on the N-CNRs-Nafion/GCE. The linear regression equation is expressed as: Ip,DA(μA) = 0.03531CDA + 11.74 with a correlation coefficient of 0.9983 (the inset in Fig. 6). The slope of the above equation is 0.03531 μA μM−1, and the detection limit of 8.9 × 10−9 M could be estimated (S/N = 3).31 Furthermore, the relative standard deviation (RSD%) of the modified electrode in 10 successive scans was 1.8% for 15.0 μM DA in the presence of 0.5 mM AA, confirming the excellent reproducibility of this modified electrode.
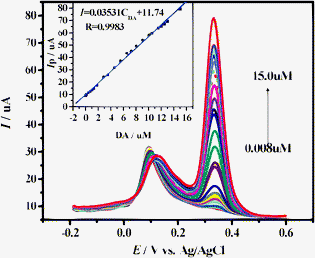 | ||
| Fig. 6 DPV for 0.008, 0.02, 0.05, 0.1, 0.4, 0.7, 1.0, 1.3, 1.8, 2.8, 3.5, 4.5, 5.5, 6.5, 7.2, 8.0, 9.0, 10.0, 10.5, 11.5, 12.0, 12.5, 13.0, and 15.0 μM DA in the presence of 0.5 mM AA at the N-CNRs-Nafion/GCE in PBS solution at pH 4.0. DPV conditions: potential incremental, 1 mV; amplitude, 50 mV; pulse width, 50 mV; period, 75 ms. The insert is the relationship between the peak current and the concentration of DA. | ||
The measurement results indicate that the N-CNRs-Nafion/GCE gives excellent performance for the reliable determination of DA concentration free from AA interference, even at excessive AA concentrations. Meanwhile, a peak separation of 234 mV between AA and DA allows to distinguish DA selectively by using DPV. The response characteristics for the determination of DA and AA of N-CNRs compared to the other reported modified electrodes are listed in Table 1. It is found that the N-CNRs-Nafion modified electrode is more sensitive to DA with a low detection limit of 8.9 × 10−9 M reported for the first time, which is superior to the previous results on other modified electrodes. Thus, this modified electrode exhibits high sensitivity and excellent selectivity to be of great utility for making DA sensor for the detection of neurotransmitters.
| Type of electrode | Technique | Linear range/μM | Detection limit/μM | The peak separation between DA and AA/mV | Reference |
|---|---|---|---|---|---|
| MWCNT modified carbon-ceramic electrode | DPV | 0.5–100 | 0.31 | 205 | 28 |
| Gold nanocluster/polypyrrole electrode | DPV | 0.075–20 | 0.015 | 190 | 4 |
| Phytic acid/SWCNT electrode | DPV | 0.2–10 | 0.08 | 170 | 32 |
| Overoxidized polypyrrole film/SWCNT electrode | DPV | 1–50 | 0.38 | 150 | 33 |
| OMC/Nafion/GC electrode | DPV | 1–90 | 0.5 | 155 | 34 |
| RuO2/MWCNT electrode | I-T | 0.6–3600 | 0.06 | — | 35 |
| MWCNT/IL gel/GC electrode | DPV | 1–100 | 0.1 | 200 | 36 |
| N-CNRs-Nafion/GC electrode | DPV | 0.008–15 | 0.0089 | 234 | This work |
3.5. The reaction mechanism of the electrochemical oxidation of DA
According to the previous researches,37,38 the electrocatalytic oxidation of DA in aqueous solution occurs as a two-electron process as follows: Firstly, DA is initially oxidized to form o-dopaminoquinone (o-DQ) undergoing a two electron oxidation (E step), and two protons of the hydroxyl groups, accompanied by two electrons, are lost. At pH 4.0, sufficient protonated o-dopaminoquinone is available to allow the cyclization reaction to take place. Subsequently, when the amine is deprotonated, the molecule can undergo a proton-assisted intramolecular 1,4-Michael addition, which results in a cyclization reaction (C step), yielding leucodopaminochrome (LDC). Secondly, LDC is more easily oxidized than the parent DA and can experience a further two-electron oxidation to form dopaminochrome (DC) (E step). The general mechanisms of the electrochemical behavior of DA can be represented in Scheme 1.39,40 | ||
| Scheme 1 The general mechanisms of the electrochemical behavior of DA. | ||
The sensitivity and selectivity of the electrochemical sensor for determination of DA depends on the chemical reaction between the modified electrodes and the electrolyte. Therefore, the probable reaction mechanisms are as follows: firstly, the porosity of carbon nanorods provides tunnels for electrolyte ions to cause quick mass transfer. The graphite structure and the abundant surface active site assure faster electron transfer in electrochemical reaction. Secondly, the IR results in ref. 15 and XPS spectra verify that “pyridinic” N and “pyrrolic” N in the N-CNRs surface as the electron donors contain a larger stable π-conjugated delocalized system, which are very active towards electrophiles. We considered the electron-rich N-CNRs an excellent nucleophile, which will attack the electron-deficient o-DQ in solution by 1,4-Michael addition reactions. In the electrochemical step, these products are easily oxidized to form LDC at lower oxidation potential than DA. The most severely interfering compound, AA, does not undergo 1,4-Michael addition with the N-CNRs, therefore does not interfere with the detection of DA. Finally, N-CNRs with added Nafion as a perfluorosulfonated cation-exchange polymer could eliminate the interference of AA and retain high sensitivity and good selectivity. The N-CNRs are electronegative because Nafion is a cation-exchange polymer, repelling AA and providing a transport channel solely for cations, such as dopamine. DA has cation groups (pKaca. 8.87) in 0.1 M PBS solution at pH 4.0 and its detection is enhanced by the electrostatic attraction between N-CNRs-Nafion and DA, whereas AA has neutral or negative groups and is repulsed making it difficult for it to gain access to the surface of N-CNRs-Nafion. Scheme 2 illustrates this effect of the selectivity and sensitivity on the DA determination in the presence of AA.
 | ||
| Scheme 2 The reaction mechanism for DA determination in the presence of AA on the Nafion-N-CNRs modified electrode. | ||
4. Conclusions
The N-CNRs-Nafion modified electrode shows outstanding electrochemical and electrocatalytic behaviors for the selective determination of DA during interference from high-concentrations of AA in PBS at pH 4.0. Visible current enhancement, fast electron transfer kinetics and low overpotentials are observed. The excellent electrocatalytic performance of N-CNRs is contributed to by the exposure of edge plane defects and active groups, like nitrogen-containing functional groups, on the surface of the carbon nanorods. The selective determination of DA is achieved with a detection limit of 8.9 × 10−9 M (S/N = 3) by DPV. The N-CNRs-Nafion modified electrode exhibits a wide linear range, a very low detection limit and anti-interference ability. In further work, the modified electrode will be exploited for potential applications in electrochemical sensors.Acknowledgements
The authors wish to acknowledge financial support from the National Natural Science Foundation of China (21031001 and 20876067) and the Fundamental Research Funds for the Central Universities (21609203).References
- C. Guzmán, G. Orozco, Y. Verde, S. Jiménez, G. A. Luis, E. Juaristi and E. Bustos, Electrochim. Acta, 2009, 54, 1728 CrossRef.
- Y. Zhang and J. B. Zheng, Electrochim. Acta, 2007, 52, 7210 CrossRef CAS.
- C. S. Shan, H. E. Yang, D. X. Han, Q. X. Zhang, A. Ivaskac and L. Niu, Biosens. Bioelectron., 2010, 25, 1504 CrossRef CAS.
- J. Li and X. Q. Lin, Sens. Actuators, B, 2007, 124, 486 CrossRef.
- Z. Y. Wang, S. N. Liu, P. Wu and C. X. Cai, Anal. Chem., 2009, 81, 1638 CrossRef CAS.
- M. A. Dennis, K. Michael, M. M. Khodabakhsh, N. Stuart and W. Michael, Anal. Chem., 1989, 61, 2603 CrossRef.
- B. J. Christopher, P. M. Jennifer and V. B. Jill, Anal. Chim. Acta, 2010, 622, 105 Search PubMed.
- S. Kapur and D. Mamo, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2003, 27, 1081 CrossRef CAS.
- N. D. Volkow, J. S. Fowler and G. J. Wang, Behav. Pharmacol., 2002, 13, 355 CrossRef CAS.
- M. N. Zhang, K. P. Gong, H. W. Zhang and L. Q. Mao, Biosens. Bioelectron., 2005, 20, 1270 CrossRef CAS.
- S. Q. Song, Q. M. Gao, K. S. Xia and L. Gao, Electroanalysis, 2008, 20, 1159 CrossRef CAS.
- R. Hoffmann, A. Kriele, H. Obloh, N. Tokuda, W. Smirnov, N. J. Yang and C. E. Nebel, Biomaterials, 2011, 32, 7325 CrossRef CAS.
- D. A. C. Brownson, L. J. Munro, D. K. Kampouris and C. E. Banks, RSC Adv., 2011, 1, 978 RSC.
- Z. A. Alothman, N. Bukhari, S. M. Wabaidur and S. Haider, Sens. Actuators, B, 2010, 146, 314 CrossRef.
- D. S. Yuan, T. X. Zhou, S. L. Zhou, W. J. Zou, S. S. Mo and N. N. Xia, Electrochem. Commun., 2011, 13, 242 CrossRef CAS.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641 CrossRef CAS.
- C. Jordi, M. R. Josep, R. Jaime, I. Francesc and M. J. Juan, J. Am. Chem. Soc., 1996, 118, 8071 CrossRef.
- A. Safavi, N. Maleki, O. Moradlou and F. Tajabadi, Anal. Biochem., 2006, 359, 224 CrossRef CAS.
- S. B. Hocevar, J. Wang, K. P. Deo, M. Musameh and B. Ogorevc, Electroanalysis, 2005, 17, 417 CrossRef CAS.
- L. Lin, J. Chen, H. Yao, Y. Chen, Y. Zheng and X. Lin, Bioelectrochemistry, 2008, 73, 11 CrossRef CAS.
- K. Miyazaki, G. Matsumoto, M. Yamada, S. Yasui and H. Kaneko, Electrochim. Acta, 1999, 44, 3809 CrossRef CAS.
- J. Mathiyarasu, S. Senthilkumar, K. L. N. Phani and V. Yegnaraman, J. Appl. Electrochem., 2005, 35, 513 CrossRef CAS.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351 CrossRef CAS.
- J. Wang, M. Musameh and Y. Lin, J. Am. Chem. Soc., 2003, 125, 2408 CrossRef CAS.
- C. Y. Deng, J. H. Chen, M. D. Wang, C. H. Xiao, Z. Nie and S. Z. Yao, Biosens. Bioelectron., 2009, 24, 2091 CrossRef CAS.
- L. D. Zhu, C. Y. Tian, D. X. Yang, X. Y. Jiang and R. L. Yang, Electroanalysis, 2008, 20, 2518 CrossRef CAS.
- J. S. Huang, Y. Liu, H. Q. Hou and T. Y. You, Biosens. Bioelectron., 2008, 24, 632 CrossRef CAS.
- B. Habibi and M. H. Pournaghi-Azar, Electrochim. Acta, 2010, 55, 5492 CrossRef CAS.
- J. S. Ye, Y. Wen, W. D. Zhang, M. G. Leong, G. Q. Xu and F. S. Shen, Electroanalysis, 2003, 15, 1693 CrossRef CAS.
- S. R. Ali, Y. F. Ma, R. R. Parajuli, Y. Balogun, W. Y. C. Lai and H. X. He, Anal. Chem., 2007, 79, 2583 CrossRef CAS.
- J. W. Mo and B. Ogorevc, Anal. Chem., 2001, 73, 1196 CrossRef CAS.
- S. Jo, H. Jeong, S. R. Bae and S. Jeon, Microchem. J., 2008, 88, 1 CrossRef CAS.
- Y. X. Li, P. Wang, L. Wang and X. Q. Lin, Biosens. Bioelectron., 2007, 22, 3120 CrossRef CAS.
- D. Zheng, J. S. Ye, L. Zhou, Y. Zhang and C. Z. Yu, J. Electroanal. Chem., 2009, 625, 82 CrossRef CAS.
- L. C. Jiang and W. D. Zhang, Electroanalysis, 2009, 21, 1811 CrossRef CAS.
- Y. F. Zhao, Y. Q. Gao, D. P. Zhan, H. Liu, Q. Zhao, Y. Kou, Y. H. Shao, M. X. Li, Q. K. Zhuang and Z. W. Zhu, Talanta, 2005, 66, 51 CrossRef CAS.
- A. Ciszewski and G. Milczarek, Anal. Chem., 1999, 71, 1055 CrossRef CAS.
- H. T. Deng, J. Gary and V. Berkel, Electroanalysis, 1999, 11, 857 CrossRef CAS.
- M. D. Hawley, S. V. Tatawawadi, S. Piekarski and R. N. Adarns, J. Am. Chem. Soc., 1967, 89, 447 CrossRef CAS.
- N. J. Ke, S. S. Lu and S. H. Cheng, Electrochem. Commun., 2006, 8, 1514 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: [DETAILS]. See DOI: 10.1039/c2ra21041j |
| This journal is © The Royal Society of Chemistry 2012 |