Combined electrophoretic deposition–anodization method to fabricate reduced graphene oxide–TiO2 nanotube films†
Jung-Ho
Yun
a,
Roong Jien
Wong
a,
Yun Hau
Ng
*a,
Aijun
Du
b and
Rose
Amal
*a
aARC Centre of Excellence for Functional Nanomaterials, School of Chemical Engineering, the University of New South Wales, Sydney, NSW 2052, Australia. E-mail: r.amal@unsw.edu.au; yh.ng@unsw.edu.au; Fax: +612-9385-5966; Tel: +612-9385-4361
bCentre for Computational Molecular Science, Australia Institute for Bioengineering and Nanotechnology (AIBN), the University of Queensland, Brisbane, QLD 4072, Australia
First published on 4th July 2012
Abstract
We directly constructed reduced graphene oxide–titanium oxide nanotube (RGO–TNT) film using a single-step, combined electrophoretic deposition–anodization (CEPDA) method. This method, based on the simultaneous anodic growth of tubular TiO2 and the electrophoretic-driven motion of RGO, allowed the formation of an effective interface between the two components, thus improving the electron transfer kinetics. Composites of these graphitic carbons with different levels of oxygen-containing groups, electron conductivity and interface reaction time were investigated; a fine balance of these parameters was achieved.
1. Introduction
Anodization, a powerful electrochemical process to induce anisotropic growth of simple metal oxide, has proven a successful tool in the preparation of various metal oxide semiconductors with unique nanostructures suitable for specific applications.1–6 Among the examples of anodized metal oxide, including the flower-like WO3,3 platelet CuxO,4 porous Al2O3 membranes5 and ZnO nanowires,6 TiO2 nanotube (TNT) arrays are noticeably the most studied anodized semiconductors in light energy conversion applications.7–9 While anodized photo-active semiconductors sometimes yield randomly oriented nanostructures, TNT arrays obtained through anodization afford highly ordered and vertically aligned nanotubes, accompanied with a high surface area and ease of scale-up. In addition to its excellent electron transport properties in separating and directing electrons to the collecting electrode surface, these uniform tubular arrays are also beneficial in improving the light scattering ability within the inner tubes for greater light harvesting. Efforts in further advancing the applicability of TNT arrays have been made through introducing other components for (i) extending the region of sunlight absorption and (ii) enhancing the intrinsic charge separation/transport efficiency.To date, nanotubes of TiO2 have been covered,10 coated,11 filled12 or decorated13 with metal, metal oxide and organic components (e.g. graphitic carbon derivatives). As anodizaton is only effective in producing simple metal oxides, the introduction of other components can only be achieved via other strategies. These are usually associated with additional coupling experiments, for example, successive immersing and alternating TNTs into solutions of reactant precursors (Cd and S), referred to as successive ionic layer adsorption and reaction (SILAR);14 dipping TNTs into solution containing the mixture of precursors (Pb and S) at a certain temperature, concentration and pressure, referred to as chemical bath deposition (CBD);15 reducing metal ions such as Pt4+ and Ag+ on the photoexcited TNTs in a photodeposition process;16,17 and electrodepositing single metal component (Cu) into inner tubes of TNTs.12 Recently we developed a square-wave pulsed-electrodeposition technique to wrap the inner walls of TNTs with chalcogenide-type ternary semiconductor CuInS2.13 All these processes achieve great effectiveness for specific candidate materials, while a universal method suitable for all objects to be deposited onto/into TNTs is still absent. Therefore, searching for new TNT-modifying methods has emerged as one of the major research activities in materials chemistry.
Modification of TiO2 nanoparticles with graphitic carbon to enhance electron shuttling has been extensively studied.18–24 TiO2 nanoparticles were deposited on graphene mat, wrapped by a graphitic hollow sphere, and linked with functionalized carbon using hydrolysis,25 hydrothermal26 and photocatalytic reduction methods19,22,23,27 and photo-induced polymerization process.28–30 However, similar reports on TNTs are rare, which may be due to the restrictive nature of the closely packed tubes and limited contact on the porous surface. Yang et al. reported the formation of graphitic carbon nanotubes inside the tubular structure of TNTs by carbonizing poly(ethylene glycol), which was placed beneath the TNTs in a tube furnace.31 Liu and co-workers employed a cyclic voltammetric reduction process to reduce graphene oxide onto the TNTs.10 Very recently, Song et al.32 demonstrated the deposition of graphene oxide onto the surface of TNT arrays by a simple dip-and-dry method. All reports revealed the positive effect of coupling graphitic carbon to TNTs in the applications of photocatalysis and photoelectrochemistry. These existing techniques share a common ground, i.e. introducing the graphitic carbon to pre-synthesized TNTs. TNTs were first prepared by anodizing the titanium foil. Subsequent to the annealing of the amorphous TNT, graphitic carbons were then attached to TNTs in separate experiments.
In this paper, we present a combined electrophoretic deposition–anodization (CEPDA) approach, which only requires a single step to prepare a reduced graphene oxide–TNT (RGO–TNT) film. CEPDA is an electrochemical process that allows the simultaneous anodic growth of TiO2 nanotubes and the electrophoretic-driven attachment of RGO. The electric field applied between Ti foil and Pt electrode induced the oxidative formation of TiO2 and the instantaneous electrophoretic motion of surface charged RGO in the electrolyte resulted in the accumulation of rigid RGO flakes deposited on the TiO2 electrode. The formation mechanism in the CEPDA approach was studied systematically by varying the applied potential and reaction time. The physicochemical properties of the composites were then examined with a combination of physical and photoelectrochemical characterization techniques. In addition, the influence of RGO sizes (micron-size RGO (mRGO) and nano-size RGO (nRGO)) and RGO functional groups on the photoelectrochemical properties was investigated.
2. Experimental
Synthesis of graphene oxide (GO) and reduced graphene oxide (RGO) as precursors for RGO–TNT films
Two different synthetic graphite sources were used to prepare GO and RGO: micron-sized graphite powder (mG) (1–2 μm, Aldrich) and nano-sized graphitic carbon powder (nG) (<500 nm, Aldrich). GO was synthesized using Hummers' method by reacting the commercial graphite powder with a mixture of H2SO4 (Aldrich, 99%), NaNO3 (Aldrich), and KMnO4 (Aldrich) followed by the addition of H2O2 (Aldrich, 30%).33 The solid product was filtered and washed repeatedly with 1 M HCl and deionized water, then dried under a vacuum at room temperature. The resultant brown GO platelet was ground into a fine powder before being subject to chemical reduction. In a chemical reduction process to reduce GO to RGO, 300 mg of GO powder was dispersed in 600 ml of Milli-Q water under an ultrasonic treatment for 15 min. A concentration of 0.5 mg ml−1 was selected; it represented the highest concentration of GO suspension that was still effectively dispersed (concentration >0.5 mg ml−1 resulted in settlement of GO powder at the bottom of reaction flask, which restricts the effective chemical reduction). 3 ml of hydrazine hydrate solution (Aldrich, 80%) was added to the GO solution and the suspension was heated to 85 + 5 °C for 6 h under vigorous stirring (approximately 600 rpm). Subsequently, the resultant RGO was retrieved from the suspension by a centrifuge (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm × 3 times) and washed thoroughly with Milli-Q water. The prepared GO and RGO were labelled in accordance with the size of the parent graphite materials: the micron-size graphite yielded mGO and mRGO, while nGO and nRGO refer to those obtained from nano-size graphite.
000 rpm × 3 times) and washed thoroughly with Milli-Q water. The prepared GO and RGO were labelled in accordance with the size of the parent graphite materials: the micron-size graphite yielded mGO and mRGO, while nGO and nRGO refer to those obtained from nano-size graphite.
CEPDA preparation of RGO–TNT films
The Ti foils (Aldrich, 99.7% metal basis, 0.127 mm thick) were first cleaned with acetone under mild sonication in Milli-Q water followed by vacuum drying. An in-house designed sealed cylindrical Teflon® cell was used as the CEPDA reactor. Ti foil of 20 × 20 × 0.127 mm in size was used as the working electrode and was located at the bottom of the reactor with an area of 6.25 cm2 constantly exposed to the electrolyte. A platinum plate used as the counter electrode was fixed at 4 cm perpendicularly above the Ti foil. The electrolytes were prepared by mixing ethylene glycol (Aldrich, >99%), 0.5 wt% of NaF (Ajax Chemicals, 99%), 5 wt% Milli-Q water and 5 mg of graphitic carbon materials (m/nGO, m/nRGO or m/nG).9 The electrolyte was ultrasonicated and pre-heated to 30 °C before being transferred to the CEPDA reactor. A programmable DC power supply (PST-3201 GW Instek) interfaced with a computer was used to supply bias to the system. Two series of samples were obtained: in the first series (1) a constant duration of 24 h was applied to samples prepared under 10, 20 and 40 V; and in the second series (2) a constant length of RGO–TNT arrays (measured by cross-section scanning electron micrographs) was achieved by manipulating both the bias and the reaction duration. In the second series, three RGO–TNT arrays with a constant 5 μm length were synthesized at (i) 20 V for 24 h, (ii) 40 V for 6 h, and (iii) 60 V for 80 min. After the CEPDA reaction, the RGO–TNT arrays were rinsed with water followed by calcinations at 450 °C for 1 h (ramping 5 °C min−1).Physical and photoelectrochemical characterizations
The morphology of the RGO–TNT arrays was characterized using a scanning electron microscope (SEM-S900 Hitachi). Chemical states and reduction degree of the pure TNT, RGO–TNT, GO and RGO were analyzed using X-ray photoelectron spectroscopy (XPS ESCALab220i-XL, VG Scientific) with monochromated Al Kα radiation (hν = 1486.6 eV) and a source power of 120 W. Raman spectra of graphite, GO and RGO were obtained from a Renishaw inVia spectrometer using a 514 nm argon ion laser with 1800 l mm−1 grating. The ζ-potential of RGO was measured by phase analysis light scattering (PALS, Brookhaven BI-90 PALS). The powdery RGO (0.1 mg ml−1) was ultrasonicated in a 0.5 mM NaCl aqueous solution with a pH adjustment between 2 and 10.Photoelectrochemical (PEC) characterizations were performed using a three-electrode PEC system consisting of the prepared CEPDA sample as a working electrode, a Pt counter electrode and a Ag/AgCl reference electrode. 0.5 M Na2SO4 was used as the electrolyte and a 300 W xenon lamp (PECCELL) was used as the lamp source. The incident photon-to-current efficiency (IPCE) measurements were performed using a monochromator (Newport 74125) coupled with a 280 W xenon lamp (Newport 66902). Current–voltage profiles and amperometric responses were obtained using a potentiostat (PGSTAT302N, Autolab), which is connected to a computer with interfacing software of GPES. Film conductance derived from electrochemical impedance spectroscopy (EIS) was obtained on an INPHAZE impedance spectrometer using a similar three-electrode system as the PEC system. The electrolyte was 0.1 M Na2SO4 and the frequency range from 10 mHz to 1 MHz was applied.
3. Results and discussion
CEPDA-derived mRGO–TNT films
Scheme 1 illustrates the working principle of the combined electrophoretic deposition–anodization (CEPDA) method. In a typical anodization process, a metal foil (Ti in this study) is employed as a working electrode, while platinum (Pt) is usually used as the counter electrode. When a positive bias is constantly applied between the electrodes, electrons are withdrawn from the Ti foil to form Ti4+, while O2− ions are simultaneously incorporated into the Ti film. Owing to the nature of the high positive field mechanism, Ti4+ cations migrate outwards to the metal–electrolyte interface and are solvated by F− ions to form water-soluble TiF62− complexes.9 In addition, O2− ions move towards the bulk Ti. The opposite migration direction of Ti4+ cations and O2− anions, induced by the applied electric field, forms the fundamental principle of the CEPDA technique. While having O2− and F− ions from the electrolyte to trigger the oxidative formation and chemical dissolution of Ti-oxide, respectively, we introduced the negatively surface-charged RGO particles in the electrolyte to allow its electrophoretic-driven migration towards the Ti working electrode. Electrophoretic deposition (EPD) has proven an effective technique to form a homogeneous and rigid deposit of various kinds of carbon materials on the flat electrodes such as on indium or fluorine doped tin oxide.34,35 EPD is achieved through the motion of homogeneously dispersed and charged particles towards an electrode under an applied electric field. Therefore, under the current working system, the graphitic carbon used must possess a negative surface charge in order to be attracted to the positive Ti anode, as illustrated in Scheme 1. | ||
| Scheme 1 Formation mechanism of RGO–TNT array via a CEPDA method. | ||
Treatment with a combination of strong acids and oxidant (H2SO4, KMnO4 and HCl) during synthesis introduced various oxygen functional groups to RGO.36 Among all oxygen-containing groups, the presence of carboxylic acid (CO(O)H) electrostatically stabilized the RGO in aqueous or other polar liquids by giving RGO a negative surface charge together with a certain extent of hydrophilicity. These two factors led to a significant improvement in the stability of RGO suspension in both the aqueous solution and the polar ethylene glycol solvent, making it suitable for electrophoretic deposition simultaneous with the anodic growth of TNTs. In comparison, unmodified graphite was barely dispersed in these media. Fig. 1a shows the surface charge and stability of the mRGO in an aqueous suspension, determined by the measurement of its ζ-potential over a range of pH 2–10. Although the magnitude of the ζ-potential was pH dependent, due to the strong pH-dependency of the ionization of carboxylic groups, all ζ-potentials were in the negative region, indicating the negative surface charge of RGO. It suggested that the negatively charged RGO will migrate to the positive Ti electrode during CEPDA reaction. When the pH was greater than 4, the ζ-potential reached −40 mV.37 As established in colloidal chemistry, a suspension with a ζ-potential greater than 30 mV (absolute value regardless of positive or negative) is generally considered a stable dispersion because of the presence of sufficient mutual repulsion among the particles.38
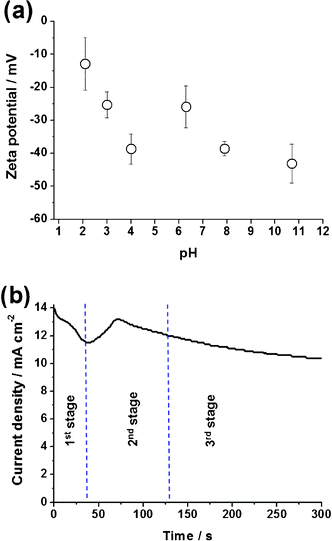 | ||
| Fig. 1 (a) ζ-potential of mRGO as a function of pH and (b) current density profile during the CEPDA process of Ti foil in ethylene glycol-based electrolyte containing NaF and mRGO. | ||
After recognizing the feasibility of driving the negatively charged RGO towards the positive Ti working electrode, its influence on the anodic growth of TiO2 was monitored through the current density profile recorded during the CEPDA reaction (Fig. 1b). Anodic growth of TNTs was clearly defined into three typical stages as it progressed: formation of a compact oxide layer (1st stage), chemical dissolution of the oxide (2nd stage), and the equilibrium achieved between the formation and dissolution of the oxide (3rd stage).9 Initial formation of a thin TiO2 layer covered the exposed Ti foil and resulted in a lower conductivity of the film, thus resulting in the current decay. Subsequently, the migration of F− anions towards the TiO2 film induced the chemical dissolution of TiO2 (formation of TiF62− complexes), allowing more Ti to undergo anodic oxidation. A temporary rebound in current was therefore observed. The competitive reactions between formation and dissolution of TiO2 eventually led to an equilibrium state of the current behaviour. Note that oxidation occurred slightly faster than dissolution by manipulating the water content in ethylene glycol to maintain a continuous growth of nanotubes. The growing of TNTs slowed continually throughout the process as the electric field within the TNT layer was progressively reduced when the thickness of the oxide increased. The presence of the mRGO in the electrolyte and its electrophoretic motion towards the Ti foil under a constant applied voltage did not affect the anodic mechanism of the Ti foil.
A tilted SEM image in Fig. 2 shows the morphological feature of the mRGO–TNT arrays fabricated by CEPDA method at 10 V for 24 h. The TNT arrays were well aligned and the length was reasonably uniform with a clear deposit of translucent mRGO sheets on top of the nanotubes. Nanotubes of TiO2 can be seen located beneath the mRGO sheets in the magnified SEM image (Fig. S1 in the supporting information†). The inset SEM image reveals the typical appearance of pure TNT arrays in which the top surface is uncovered.
 | ||
| Fig. 2 SEM image of mRGO–TNT array by the CEPDA method at 10 V for 24 h (inset: pure TNT array by the anodization at 10 V for 24 h). | ||
XRD diffractograms in Fig. 3a confirm the successful preparation of anatase TiO2 in both samples with comparable crystallinity regardless of the presence of mRGO. Because XRD is a bulk-phase analysis method, the graphitic carbon peak was not observed due to the relatively trace amount compared with TiO2, although the SEM indicated its significant existence on the surface. The XPS technique was used to probe the surface chemistry from the outermost 5–10 nm of the samples. Fig. 3(b) and (c) shows the C 1s and Ti 2p spectra of the TNTs and mRGO–TNT. Compared to the C 1s spectra, the primary difference is the increase in intensity in all carbon species regions (284.5, 286, and 288.5 eV) due to the attached mRGO. A peak at 284.5 eV is always observed in pure TiO2 (and other oxides) as it readily adsorbs CO2 and other organic substances from the ambient atmosphere, unless the whole process of synthesis and characterization are performed under vacuum conditions. Another noticeable feature of the C 1s spectra is that the atomic ratio of oxygen-bound carbon (C–O at 286 eV; O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 288.5 eV) of mRGO–TNT is 31%, a triple raise from 11% of pure TNT. This confirms the presence of mRGO on the surface of nanotubes because mRGO is known to contain the carbonyl, epoxy and carboxylic groups. The XPS spectra of Ti 2p in Fig. 3c indicates two peaks centred at 458–458.5 eV and 463.5–464 eV, corresponding to Ti 2p3/2 and Ti 2p1/2, respectively. Compared to the binding energy of pure TNT array, the mRGO–TNT presented a shift of 0.5 eV from 458.6 to 458.1 eV. This negative shift of binding energy suggests the formation of Ti–O–C bonds between the surface TiO2 and RGO,39 or the creation of Ti3+ species similar with those of carbon-doped titania.40 Hence, unlike the recently reported dip-drying method, which most likely yielded a physically attached RGO on titania nanotubes, this CEPDA method enables a stronger interaction between the TiO2 and RGO. In fact, the prepared mRGO–TNT was observed to remain intact even under vigorous agitation and mild ultrasonication.
O at 288.5 eV) of mRGO–TNT is 31%, a triple raise from 11% of pure TNT. This confirms the presence of mRGO on the surface of nanotubes because mRGO is known to contain the carbonyl, epoxy and carboxylic groups. The XPS spectra of Ti 2p in Fig. 3c indicates two peaks centred at 458–458.5 eV and 463.5–464 eV, corresponding to Ti 2p3/2 and Ti 2p1/2, respectively. Compared to the binding energy of pure TNT array, the mRGO–TNT presented a shift of 0.5 eV from 458.6 to 458.1 eV. This negative shift of binding energy suggests the formation of Ti–O–C bonds between the surface TiO2 and RGO,39 or the creation of Ti3+ species similar with those of carbon-doped titania.40 Hence, unlike the recently reported dip-drying method, which most likely yielded a physically attached RGO on titania nanotubes, this CEPDA method enables a stronger interaction between the TiO2 and RGO. In fact, the prepared mRGO–TNT was observed to remain intact even under vigorous agitation and mild ultrasonication.
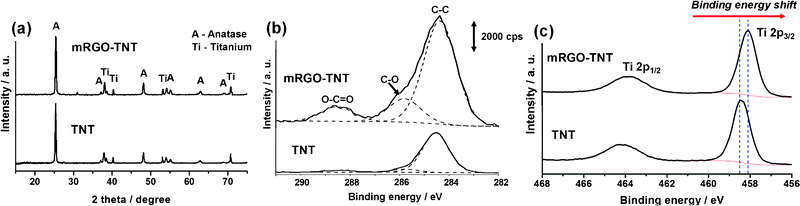 | ||
| Fig. 3 (a) XRD spectra, (b) C 1s XPS spectra and (c) Ti 2p XPS spectra for pure TNT and mRGO–TNT. | ||
We compared the photoelectrochemical properties of the pure TNT and the mRGO–TNT arrays by examining their incident photon-to-current efficiency (IPCE) in a three-electrode electrochemical cell using a monochromatic excitation source (Fig. 4). IPCE was calculated by normalizing the photocurrent to the incident light energy and intensity using eqn (1),
| IPCE (%) = 100 × 1240 × Isc/(P × λ) | (1) |
 | ||
| Fig. 4 IPCE measurement of the TNT array and mRGO–TNT array at 40 V for 24 h (inset image: wavelength onset of the TNT array and mRGO–TNT array). | ||
Properties of G-, GO- and RGO–TNT films with micro/nano graphitic carbon
In this section, the CEPDA method was extended to prepare various graphitic carbon–TNT arrays with different oxygen functional groups and electronic properties: mG–TNT, mGO–TNT and mRGO–TNT films. These films were examined in a three-electrode type photoelectrochemical cell. Fig. 5a represents the typical current–voltage profile for all TNT series during the repeating ON–OFF illumination cycles. All samples exhibited rapid and reproducible photocurrent, while the photocurrent density increased with the applied potential until the saturated photocurrent was achieved at ∼0.7–0.8 V. Magnitudes of the saturated photocurrent for all samples were presented in Fig. 5b. By using the pure TNT arrays as the reference, only the mRGO–TNT samples showed improvement in photocurrent generation, but both mGO- and mG- samples demonstrated a detrimental effect on TNT. Although all mG, mGO and mRGO consist of mainly graphitic carbon, the oxygen-containing groups and the electronic properties of each of them influenced the binding between the carbon and the surface of TiO2, light obstruction, and conductivity of the films. | ||
| Fig. 5 (a) I–V measurement of TNT array and (b) saturated photocurrent density of TNT, mG–TNT, mGO–TNT, and mRGO–TNT arrays (prepared at 40 V for 24 h) obtained at 0.75 V vs. Ag/AgCl in 0.5 M Na2SO4 electrolyte under 300 W Xe lamp. | ||
The oxygen-containing groups and the conductance of the samples are shown in Fig. 6. The deconvoluted C 1s XPS spectra of mG, mGO and mRGO in Fig. 6a indicated the presence of non-oxygenated C–C bond (284.5–285.0 eV), the C–N bond (285.5–286.4 eV), the C–O (epoxy and hydroxyl) (286.5 eV), and the O–CO from carboxylic acid (288.7 eV). Comparing the atomic ratio of the oxygen-containing groups (C–O and O–CO) against the total carbon (graphitic carbon and all functionalized carbon) allows us to deduce the relative elemental composition of the samples; this approach tells us that mGO contains ∼56.6% oxygen functional groups, mRGO contains ∼8.3%, and mG contains 0%, while carboxylic group percentages were ∼4.2, ∼1.8, and 0%, respectively. It is believed that the affinity of the carboxylic groups toward the surface hydroxyl groups of TiO2 strengthens their interaction. Therefore, it is assumed that mGO and mRGO were able to form an effective interface with TiO2, while mG was merely driven by the electrophoretic motion without being firmly attached. Furthermore, a larger amount of the carboxylic groups, which carry the negative surface charge, also contributed to the better attachment on TNTs under the constant positive bias across the electrodes. However, a strong interaction between graphitic carbon and TNTs was not the sole factor in facilitating efficient electron transportation. This can be seen from the saturated photocurrent density of mGO-TNT being lower than that of the pure TNT. Conductance of the films is another crucial aspect in determining the final performance of the electrode.
 | ||
| Fig. 6 (a) C 1s XPS spectra of mG, mRGO and mGO and (b) electrochemical impedance spectra (EIS) conductance plots of mRGO–TNT, mG–TNT, mGO–TNT, and TNT electrodes in 0.1 Na2SO4 electrolyte and frequency range from 10 mHz to 1 MHz. | ||
Fig. 6b shows EIS conductance as a function of frequency for pure TNT, mGO–TNT, mG–TNT and mRGO–TNT electrodes.
The response at high frequencies (×102 Hz and above) represents the bulk conductance of the electrolyte in series with the thin film, therefore all films indicated comparable conductance. Details of the films themselves were obtained from the region between 10 mHz and 100 Hz. The mRGO–TNT and mG–TNT are significantly more conductive than that of the pure TNT and mGO–TNT. The intrinsic conjugated π-bond in graphite and the conductivity restoration in mRGO led to their superior film conductance. mGO with considerable oxygen functional groups has disrupted the delocalization of electrons, thus resulting in a poorer film conductance. Note that under no bias condition in a four-point probe resistivity measurement (data not shown), mGO was found to be insulating while mG and mRGO were conducting. Combined XPS and EIS conductance analyses show that a good balance between the conductivity enhancement and the attachment quality of graphitic carbon is of great importance in achieving a beneficial effect on the TNT photoelectrodes. Highly functionalized graphene (i.e. GO) provides a quality platform for attachment but sacrifices the electrode conductance and vice versa. Therefore, the RGO, which possesses both carboxylic groups and delocalized electron pools successfully demonstrated the constructive effects in the TNT photoelectrochemical performances.42
Fig. 7 shows the comparison of saturated photocurrent density for pure TNT, GO–TNT and RGO–TNT with different graphitic sizes (micro (1–2 μm) and nano (<500 nm)) prepared at 10, 20 and 40 V. All samples were prepared with 24 h CEPDA reaction time and the lengths of the nanotubes (measured from the cross-sectional SEM images) were in accordance with the applied voltage. The photocurrent generated by the films, regardless of the modification with graphitic carbon, corresponded to the applied voltage. This is attributed to the increased accumulation of TiO2 components in the film as the reaction proceeds with time. Noticeably, the nano-size GO influenced the photoelectrochemical performances of TiO2 in a similar way to its micron-size counterparts but to a different extent: a constructive effect on m/n-RGO and detrimental influence on m/n-GO. In general, nano-size graphitic carbon–TNTs generated only a slightly higher photocurrent density than that of microsize graphitic carbon–TNT. Since the oxidation of graphite into GO takes place at the edges of graphite sheets (i.e. oxygen functional groups always located at the edges of GO), nG carbon has relatively denser edges and is resistant to greater oxidation. This was proven by the XPS C 1s spectra for both nGO and mGO (data not shown). mGO held ∼56.6% of oxygen-containing carbon, while nGO only contained ∼42%. Thus, the conjugated π bonds in nGO/nRGO were less disrupted and afforded better electron conductivity than mGO/mRGO.
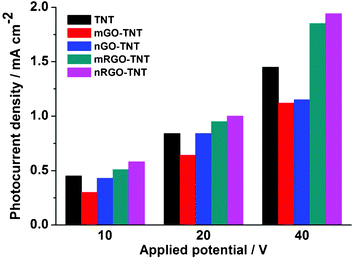 | ||
| Fig. 7 Saturated photocurrent density of TNT, mGO–TNT, nGO–TNT, mRGO–TNT, and nRGO–TNT arrays prepared at 10 V, 20 V, and 40 V for 24 h. Measurements performed using 0.75 V vs. Ag/AgCl in 0.5 M Na2SO4. | ||
While the negatively charged RGO is driven towards the Ti working electrode, there are two interactions between the RGO and the Ti: (i) physically attached RGO driven by the electrophoretic phenomenon, and (ii) the chemical interaction between the carboxylic groups (oxygen functional groups of RGO) and the surface hydroxyl groups of TiO2. The rapidness of the TiO2 formation (applied bias-dependent) determined the interaction time between the surface hydroxyl and the carboxylic groups. Compared with TNT anodized under more rapid conditions, a slower growth of TNTs under a moderate voltage allowed more time for the formation of an effective interface between the RGO and the TiO2 and thus the electron-transfer kinetics. Fig. 8 shows the nRGO–TNT arrays with a comparable length of 5 μm prepared with different applied voltages (smaller applied voltage resulted in a longer CEPDA time to achieve the same tube length) and their enhancement factor in generating photocurrent. It is clear that, with the identical tube length as determined by the SEM images, nRGO–TNT photoelectrode prepared at 20 V for 24 h produced a greater enhancement (19%) than those prepared at 40 V for 6 h (8.8%) and 60 V for 80 min (0%). Similar to the results of nRGO–TNT photoelectrodes, mRGO–TNT photoelectrodes with identical tube lengths also presented greater enhancement with an increase in reaction time. It indicates that sufficient relaxation time for the newly formed TiO2 to make contact with the RGO is necessary.
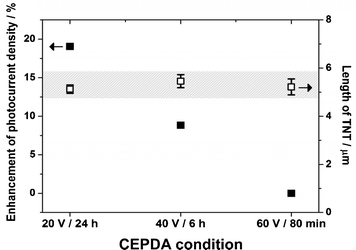 | ||
| Fig. 8 Enhancement of photocurrent density of nRGO–TNT arrays with different CEPDA conditions. | ||
4. Conclusions
A new CEPDA technique, based on the simultaneous anodic growth of TiO2 and the electrophoretic-driven negatively charged RGO, has been developed to prepare TNT-based composites. It offers an alternative method for the fabrication of composite electrodes without the necessity of other coating methods such as spin-/drop-/dip-casting, thus affording higher film stability in the reaction media in an agitated environment. Various characterization tools confirm the feasibility of this CEPDA technique to synthesize the RGO–TNT composites. The SEM image revealed the presence of thin and translucent RGO sheets on top of the tubular TiO2, while the XPS analysis showed the surface interaction of these two components. The highest photoresponse observed for RGO–TNT indicated the importance to balance the degree of oxidation and conductivity of the graphitic carbon. Graphitic carbon with extensive oxygen groups (i.e. GO) disrupted the electron conductivity; unmodified graphite, though having superior conductivity, has weak interaction with TiO2 due to the absence of oxygen-containing groups. Therefore, RGO with the right balance of oxygen functional group and electron conductivity was found to be constructive in the photoelectrochemical performance of TiO2. A longer reaction time for functional groups on RGO and TiO2, provided by milder anodic conditions, was also found to be beneficial for the formation of a more effective interface between the two components. This CEPDA method offers an alternative, simplified way to prepare the composite electrode with the possibility to fine-tune properties and is worthy of further investigation.Acknowledgements
The work was supported by the ARC Centre of Excellence for Functional Nanomaterials. The authors would also like to acknowledge the UNSW Mark Wainwright Analytical Centre, and particularly thank Dr Bill Gong for his generous help in the XPS analysis.References
- D. Gong, C. A. Grimes, O. K. Varghese, W. Hu, R. S. Singh, Z. Chen and E. C. Dickey, J. Mater. Res., 2001, 16, 3331–3334 CrossRef CAS.
- J. M. Macak, H. Tsuchiya, L. Taveira, A. Ghicov and P. Schmuki, J. Biomed. Mater. Res., Part A, 2005, 75A, 928–933 CrossRef CAS.
- C. Ng, Y. H. Ng, A. Iwase and R. Amal, Phys. Chem. Chem. Phys., 2011, 13, 13421–13426 RSC.
- D. P. Singh, N. R. Neti, A. S. K. Sinha and O. N. Srivastava, J. Phys. Chem. C, 2007, 111, 1638–1645 CAS.
- O. Jessensky, F. Muller and U. Gosele, Appl. Phys. Lett., 1998, 72, 1173–1175 CrossRef CAS.
- Z. Hu, Q. Chen, Z. Li, Y. Yu and L.-M. Peng, J. Phys. Chem. C, 2010, 114, 881–889 CAS.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2006, 6, 215–218 CrossRef CAS.
- J. M. Macak, H. Tsuchiya, A. Ghicov, K. Yasuda, R. Hahn, S. Bauer and P. Schmuki, Curr. Opin. Solid State Mater. Sci., 2007, 11, 3–18 CrossRef CAS.
- J.-H. Yun, Y. H. Ng, C. Ye, A. J. Mozer, G. G. Wallace and R. Amal, ACS Appl. Mater. Interfaces, 2011, 3, 1585–1593 CAS.
- C. Liu, Y. Teng, R. Liu, S. Luo, Y. Tang, L. Chen and Q. Cai, Carbon, 2011, 49, 5312–5320 CrossRef CAS.
- W.-T. Sun, Y. Yu, H.-Y. Pan, X.-F. Gao, Q. Chen and L.-M. Peng, J. Am. Chem. Soc., 2008, 130, 1124–1125 CrossRef CAS.
- J. M. Macak, B. G. Gong, M. Hueppe and P. Schmuki, Adv. Mater., 2007, 19, 3027–3031 CrossRef CAS.
- J.-H. Yun, Y. H. Ng, S. Huang, G. Conibeer and R. Amal, Chem. Commun., 2011, 47, 11288–11290 RSC.
- D. R. Baker and P. V. Kamat, Adv. Funct. Mater., 2009, 19, 805–811 CrossRef CAS.
- C. Ratanatawanate, C. Xiong and K. J. Balkus, ACS Nano, 2008, 2, 1682–1688 CrossRef CAS.
- Y.-Y. Song, Z.-D. Gao and P. Schmuki, Electrochem. Commun., 2011, 13, 290–293 CrossRef CAS.
- I. Paramasivam, J. M. Macak, A. Ghicov and P. Schmuki, Chem. Phys. Lett., 2007, 445, 233–237 CrossRef CAS.
- W. C. Oh, A. R. Jung and W. B. Ko, J. Ind. Eng. Chem., 2007, 13, 1208–1214 CAS.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. Lett., 2009, 1, 520–527 CrossRef.
- O. Akhavan, M. Abdolahad, A. Esfandiar and M. Mohatashamifar, J. Phys. Chem. C, 2010, 114, 12955–12959 CAS.
- Y. H. Ng, I. V. Lightcap, K. Goodwin, M. Matsumura and P. V. Kamat, J. Phys. Chem. Lett., 2010, 1, 2222–2227 CrossRef CAS.
- N. J. Bell, Y. H. Ng, A. Du, H. Coster, S. C. Smith and R. Amal, J. Phys. Chem. C, 2011, 115, 6004–6009 CAS.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741–772 CrossRef CAS.
- M. Inagaki, Y. Hirose, T. Matsunaga, T. Tsumura and M. Toyoda, Carbon, 2003, 41, 2619–2624 CrossRef CAS.
- Q. C. Xu, D. V. Wellia, Y. H. Ng, R. Amal and T. T. Y. Tan, J. Phys. Chem. C, 2011, 115, 7419–7428 CAS.
- G.-h. Moon, Y. Park, W. Kim and W. Choi, Carbon, 2011, 49, 3454–3462 CrossRef CAS.
- Y. H. Ng, S. Ikeda, T. Harada, S. Higashida, T. Sakata, H. Mori and M. Matsumura, Adv. Mater., 2007, 19, 597–601 CrossRef CAS.
- Y. H. Ng, S. Ikeda, T. Harada, S. Park, T. Sakata, H. Mori and M. Matsumura, Chem. Mater., 2008, 20, 1154–1160 CrossRef CAS.
- Y. H. Ng, S. Ikeda, T. Harada, T. Sakata, H. Mori, A. Takaoka and M. Matsumura, Langmuir, 2008, 24, 6307–6312 CrossRef CAS.
- L. Yang, S. Luo, S. Liu and Q. Cai, J. Phys. Chem. C, 2008, 112, 8939–8943 CAS.
- P. Song, X. Y. Zhang, M. X. Sun, X. L. Cui and Y. H. Lin, Nanoscale, 2012, 4, 1800–1804 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- S. Bittolo Bon, L. Valentini, J. M. Kenny, L. Peponi, R. Verdejo and M. A. Lopez-Manchado, Phys. Status Solidi A, 2010, 207, 2461–2466 CrossRef CAS.
- S. J. An, Y. Zhu, S. H. Lee, M. D. Stoller, T. Emilsson, S. Park, A. Velamakanni, J. An and R. S. Ruoff, J. Phys. Chem. Lett., 2010, 1, 1259–1263 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- It was difficult to accurately measure the pH of ethylene glycol (electrolyte for CEPDA reaction). However, both theoretical calculation and previous experimental measurements suggested the polar ethylene glycol (with 5–10 wt% water) to have a pH range of 4.5–5: K. Sandengen, B. Kaasa and T. Østvold, Ind. Eng. Chem. Res., 2007, 46, 4734–4739 CrossRef CAS.
- R. J. Hunter, Zeta potential in colloid science: Principles and applications, Academic Press, London and New York, 1981 Search PubMed.
- M. A. De Maeztu, J. I. Alava and C. Gay-Escoda, Clin. Oral Implants Res., 2003, 14, 57–62 CrossRef.
- Y. Li, D.-S. Hwang, N. H. Lee and S.-J. Kim, Chem. Phys. Lett., 2005, 404, 25–29 CrossRef CAS.
- A. Du, Y. H. Ng, N. J. Bell, Z. Zhu, R. Amal and S. C. Smith, J. Phys. Chem. Lett., 2011, 2, 894–899 CrossRef CAS.
- An accurate optimization of the oxygen-containing groups and the film conductivity is not covered in this study, but it would provide useful insights into this photoelectrochemical system and be worth investigating in the future..
Footnote |
| † Electronic Supplementary Information (ESI) available: SEM image of mRGO–TNTs by the CEPDA method. See DOI: 10.1039/c2ra20827j/ |
| This journal is © The Royal Society of Chemistry 2012 |